Genome-Wide Co-Expression Distributions as a Metric to Prioritize Genes of Functional Importance

 , , ,
, , ,
Abstract
1. Introduction
2. Material & Methods
2.1. Algorithm
2.2. Assessing Biological Relevance
2.3. Data Resources
3. Results and Discussion
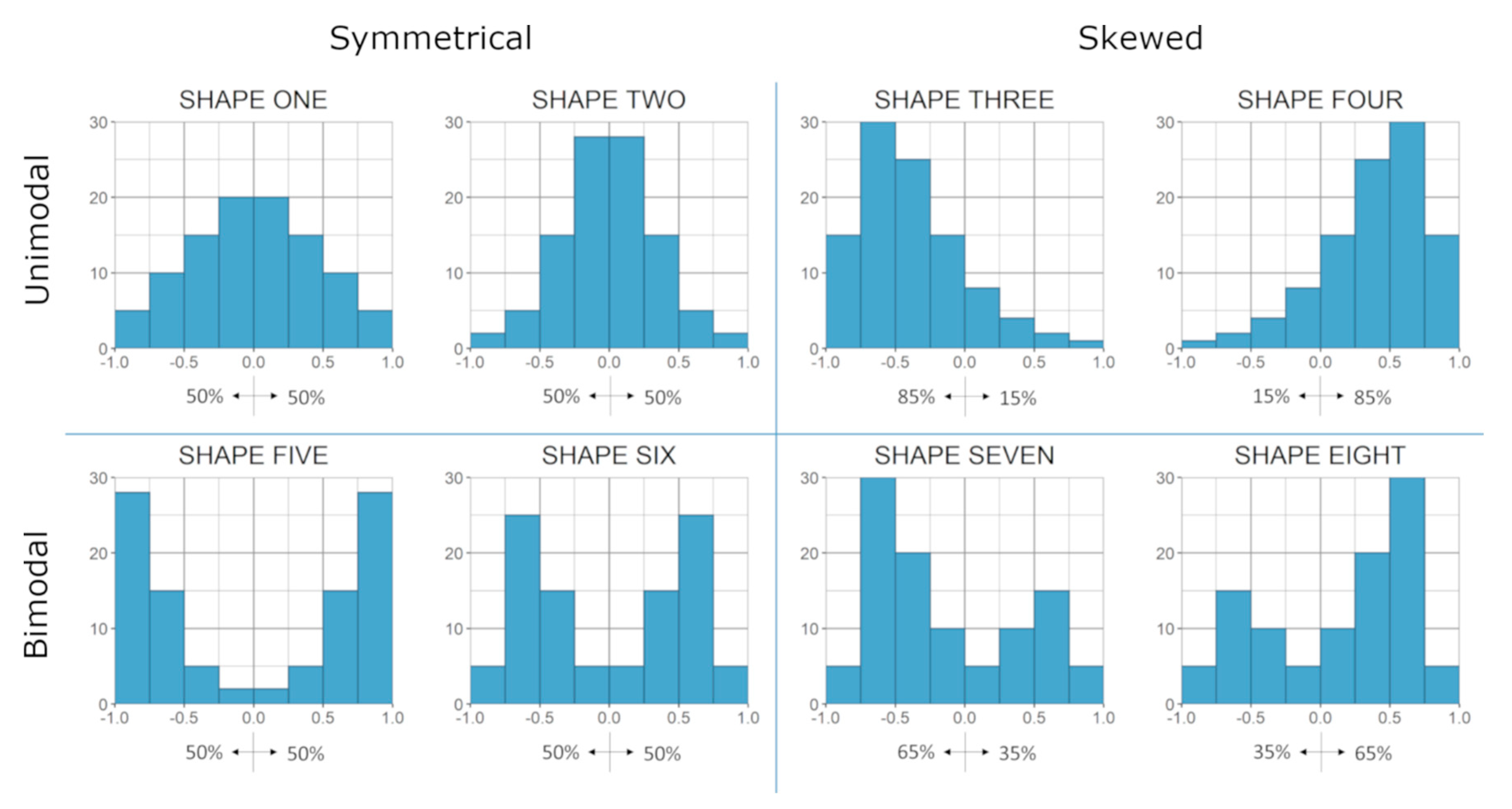
3.1. Overall Co-Expression Distribution
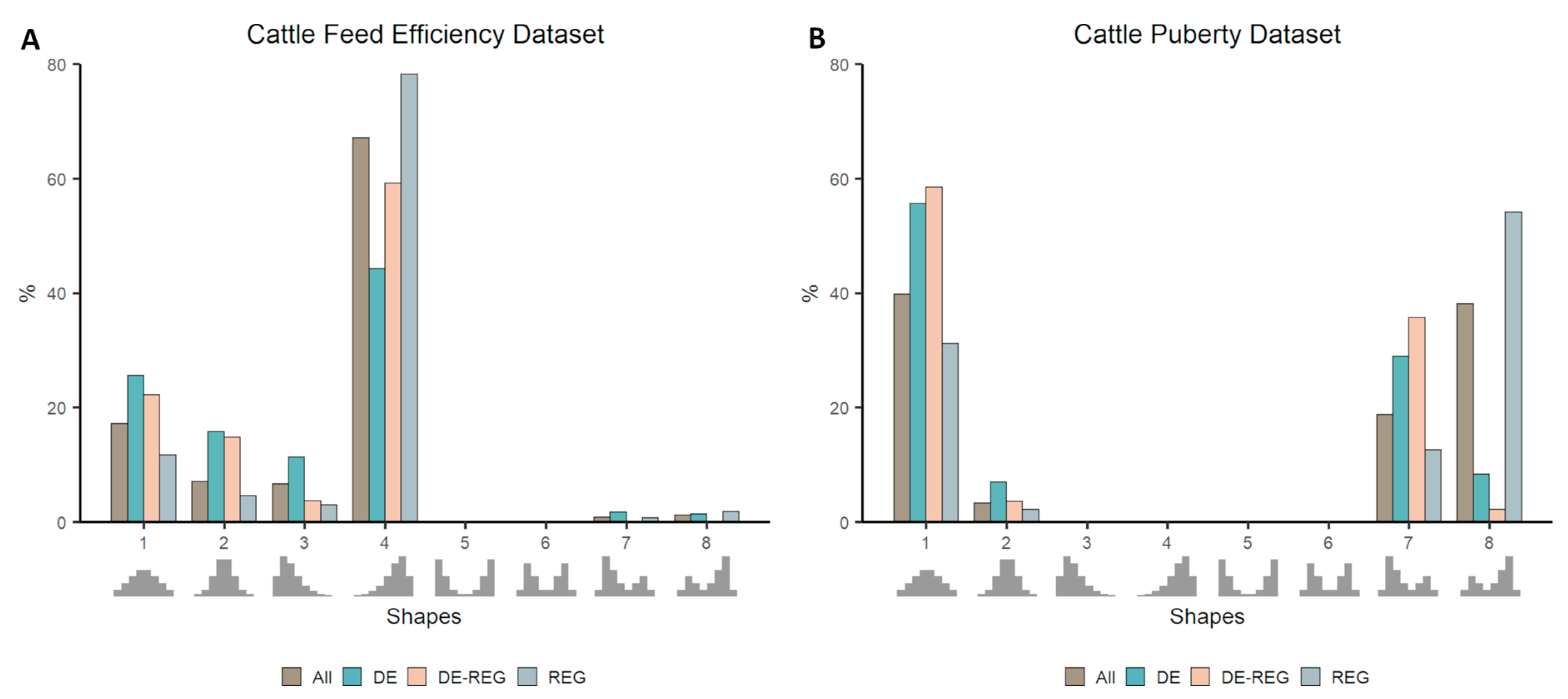
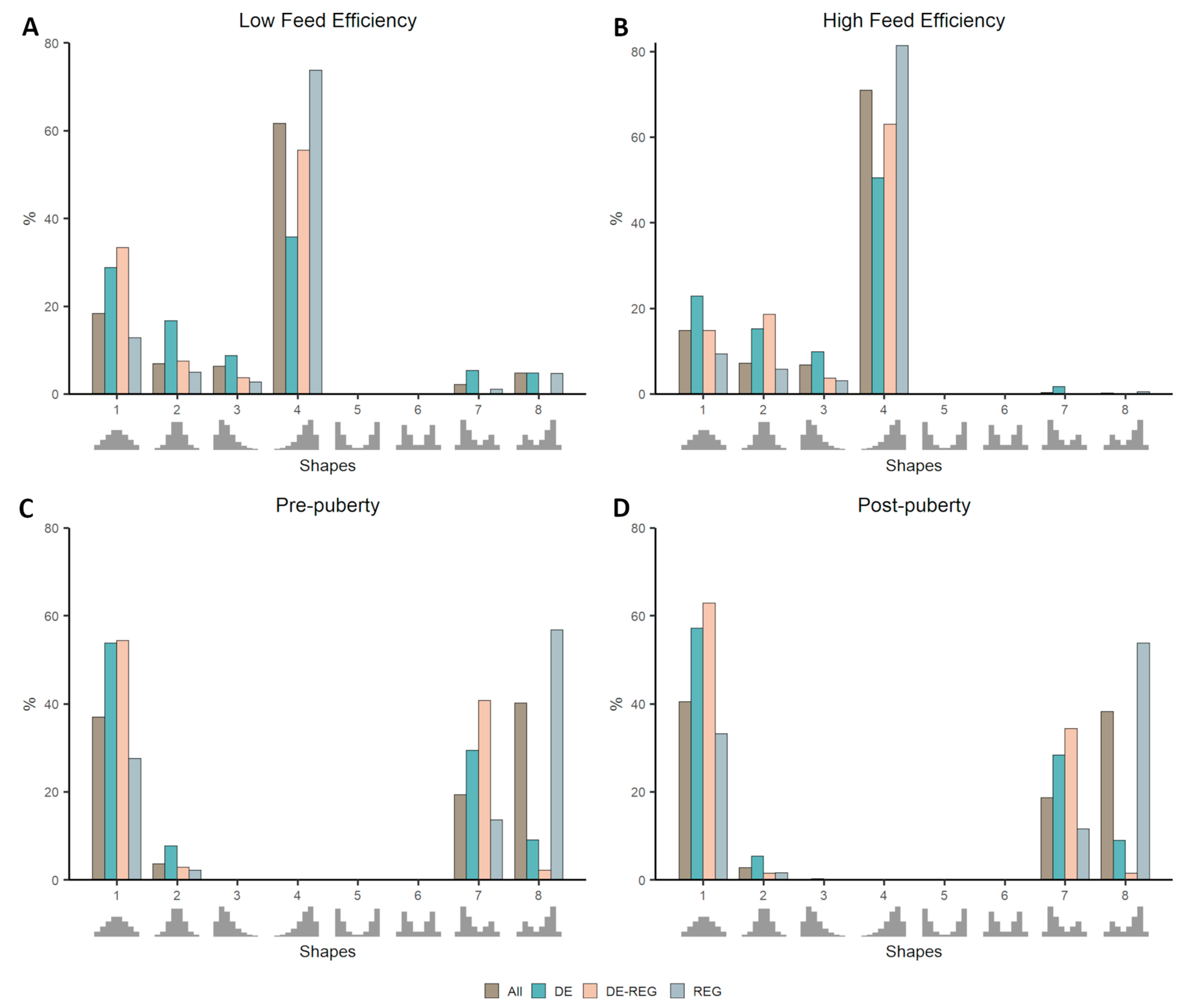
3.2. Co-Expression Distribution in Datasets With Contrasting Phenotypes
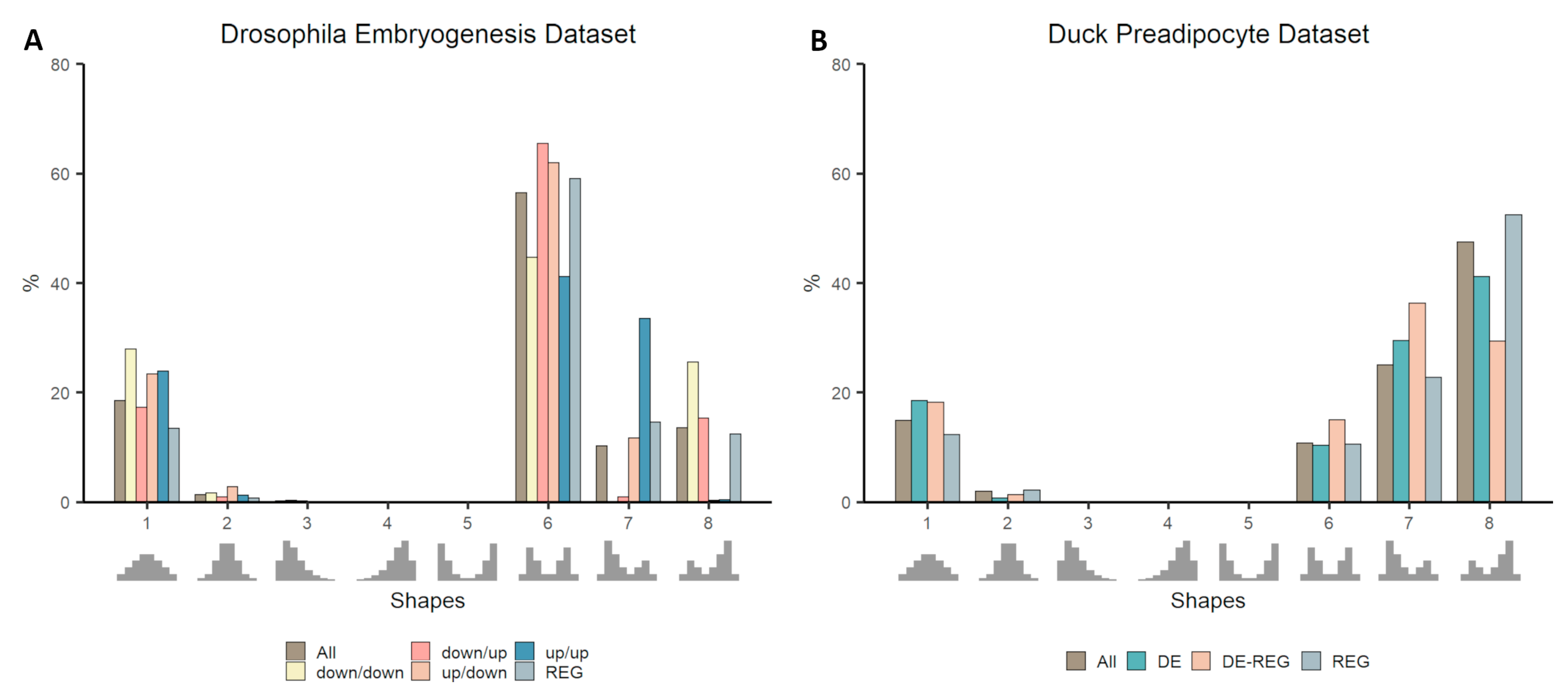
3.3. Co-Expression Distribution in Time-Series Datasets
3.4. Co-Expression Distribution in a Physiological Baseline Dataset
3.5. Relationship between Gene Categories and Distribution Shapes
3.6. Relationship between Gene Degree and Distribution Shapes
3.7. Functional Enrichment within Distribution Shape
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Availability of Data and Materials
References
- Swami, M. Networking complex traits. Nat. Rev. Genet. 2009, 10, 2566. [Google Scholar] [CrossRef]
- Hudson, N.J.; Dalrymple, B.P.; Reverter, A. Beyond differential expression: The quest for causal mutations and effector molecules. BMC Genom. 2012, 13, 356. [Google Scholar] [CrossRef] [PubMed]
- Mar, J.C.; Matigian, N.A.; Mackay-Sim, A.; Mellick, G.D.; Sue, C.M.; Silburn, P.A.; McGrath, J.J.; Quackenbush, J.; Wells, C.A. Variance of Gene Expression Identifies Altered Network Constraints in Neurological Disease. PLoS Genet. 2011, 7, e1002207. [Google Scholar] [CrossRef] [PubMed]
- Barabási, A.-L.; Oltvai, Z.N. Network biology: Understanding the cell’s functional organization. Nat. Rev. Genet. 2004, 5, 101–113. [Google Scholar] [CrossRef]
- Hudson, N.J.; Reverter, A.; Wang, Y.H.; Greenwood, P.L.; Dalrymple, B.P. Inferring the Transcriptional Landscape of Bovine Skeletal Muscle by Integrating Co-Expression Networks. PLoS ONE 2009, 4, e7249. [Google Scholar] [CrossRef]
- Remondini, D.; O’Connell, B.; Intrator, N.; Sedivy, J.M.; Neretti, N.; Castellani, G.C.; Cooper, L.N. Targeting c-Myc-activated genes with a correlation method: Detection of global changes in large gene expression network dynamics. Proc. Natl. Acad. Sci. USA 2005, 102, 6902–6906. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Reverter, A.; Chan, E.K.F. Combining partial correlation and an information theory approach to the reversed engineering of gene co-expression networks. Bioinformatics 2008, 24, 2491–2497. [Google Scholar] [CrossRef]
- Alexandre, P.A.; Naval-Sanchez, M.; Porto-Neto, L.R.; Ferraz, J.B.S.; Reverter, A.; Fukumasu, H. Systems Biology Reveals NR2F6 and TGFB1 as Key Regulators of Feed Efficiency in Beef Cattle. Front. Genet. 2019, 10, 230. [Google Scholar] [CrossRef]
- Alexandre, P.A.; Kogelman, L.J.; Santana, M.H.; Passarelli, D.; Pulz, L.H.; Fantinato-Neto, P.; Silva, P.L.; Leme, P.R.; Strefezzi, R.F.; Coutinho, L.L.; et al. Liver transcriptomic networks reveal main biological processes associated with feed efficiency in beef cattle. BMC Genom. 2015, 16, 1073. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Reverter, A.; Cánovas, A.; Venus, B.; Anderson, S.T.; Islas-Trejo, A.; Dias, M.M.; Crawford, N.F.; Lehnert, S.A.; Medrano, J.F.; et al. STAT6, PBX2, and PBRM1 Emerge as Predicted Regulators of 452 Differentially Expressed Genes Associated with Puberty in Brahman Heifers. Front. Genet. 2018, 9, 87. [Google Scholar] [CrossRef]
- Fortes, M.R.S.; Zacchi, L.F.; Nguyen, L.T.; Raidan, F.; Weller, M.M.D.C.A.; Choo, J.J.Y.; Reverter, A.; Rego, J.P.A.; Boe-Hansen, G.B.; Porto-Neto, L.R.; et al. Pre- and post-puberty expression of genes and proteins in the uterus of Bos indicus heifers: The luteal phase effect post-puberty. Anim. Genet. 2018, 49, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Reverter, A.; Cánovas, A.; Venus, B.; Islas-Trejo, A.; Porto-Neto, L.R.; Lehnert, S.A.; Medrano, J.F.; Moore, S.S.; Fortes, M.R.S. Global differential gene expression in the pituitary gland and the ovaries of pre- and postpubertal Brahman heifers. J. Anim. Sci. 2017, 95, 599–615. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fortes, M.R.S.; Nguyen, L.T.; Weller, M.M.D.C.A.; Cánovas, A.; Islas-Trejo, A.; Porto-Neto, L.R.; Reverter, A.; Lehnert, S.A.; Boe-Hansen, G.B.; Thomas, M.G.; et al. Transcriptome analyses identify five transcription factors differentially expressed in the hypothalamus of post-versus prepubertal Brahman heifers. J. Anim. Sci. 2016, 94, 3693–3702. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yin, Z.-T.; Zhang, F.; Li, X.-Q.; Chen, S.-R.; Yang, N.; Porter, T.E.; Hou, Z.-C. Dynamics of transcriptome changes during subcutaneous preadipocyte differentiation in ducks. BMC Genom. 2019, 20, 668. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Miao, Y.-R.; Jia, L.-H.; Yu, Q.-Y.; Zhang, Q.; Guo, A.-Y. AnimalTFDB 3.0: A comprehensive resource for annotation and prediction of animal transcription factors. Nucleic Acids Res. 2018, 47, D33–D38. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.; Bluhm, A.; Casas-Vila, N.; Dinges, N.; DeJung, M.; Sayols, S.; Kreutz, C.; Roignant, J.-Y.; Butter, F.; Legewie, S. Quantifying post-transcriptional regulation in the development of Drosophila melanogaster. Nat. Commun. 2018, 9, 4970. [Google Scholar] [CrossRef]
- Rhee, D.Y.; Cho, D.Y.; Zhai, B.; Slattery, M.; Ma, L.; Mintseris, J.; Wong, C.Y.; White, K.P.; Celniker, S.E.; Przytycka, T.M.; et al. Transcription Factor Networks in Drosophila melanogaster. Cell Rep. 2014, 8, 2031–2043. [Google Scholar] [CrossRef]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Crow, M.; Lim, N.; Ballouz, S.; Pavlidis, P.; Gillis, J. Predictability of human differential gene expression. Proc. Natl. Acad. Sci. USA 2019, 116, 6491–6500. [Google Scholar] [CrossRef]
- Lee, H.K.; Hsu, A.K.; Sajdak, J.; Qin, J.; Pavlidis, P. Coexpression Analysis of Human Genes Across Many Microarray Data Sets. Genome Res. 2004, 14, 1085–1094. [Google Scholar] [CrossRef] [PubMed]
- Reverter, A.; Hudson, N.J.; Nagaraj, S.H.; Pérez-Enciso, M.; Dalrymple, B.P. Regulatory impact factors: Unraveling the transcriptional regulation of complex traits from expression data. Bioinformatics 2010, 26, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Cánovas, A.; Reverter, A.; DeAtley, K.L.; Ashley, R.L.; Colgrave, M.L.; Fortes, M.R.S.; Islas-Trejo, A.; Lehnert, S.; Porto-Neto, L.; Rincón, G.; et al. Multi-Tissue Omics Analyses Reveal Molecular Regulatory Networks for Puberty in Composite Beef Cattle. PLoS ONE 2014, 9, e102551. [Google Scholar] [CrossRef] [PubMed]
- Mota, L.F.M.; Bonafé, C.M.; Alexandre, P.A.; Santana, M.H.; Novais, F.J.; Toriyama, E.; Pires, A.V.; da Luz Silva, S.; Leme, P.R.; Ferraz, J.B.S.; et al. Circulating leptin and its muscle gene expression in Nellore cattle with divergent feed efficiency. J. Anim. Sci. Biotechnol. 2017, 8, 71. [Google Scholar] [CrossRef]
- Fonseca, L.D.; Eler, J.P.; Pereira, M.A.; Rosa, A.F.; Alexandre, P.A.; Moncau, C.T.; Salvato, F.; Rosa-Fernandes, L.; Palmisano, G.; Ferraz, J.B.S.; et al. Liver proteomics unravel the metabolic pathways related to Feed Efficiency in beef cattle. Sci. Rep. 2019, 9, 5364. [Google Scholar] [CrossRef]
- Novais, F.J.; Pires, P.R.L.; Alexandre, P.A.; Dromms, R.A.; Iglesias, A.H.; Ferraz, J.B.S.; Styczynski, M.P.-W.; Fukumasu, H. Identification of a metabolomic signature associated with feed efficiency in beef cattle. BMC Genom. 2019, 20, 8. [Google Scholar] [CrossRef]
- Alexandre, P.A.; Reverter, A.; Berezin, R.B.; Porto-Neto, L.R.; Ribeiro, G.; Santana, M.H.A.; Ferraz, J.B.S.; Fukumasu, H. Exploring the Regulatory Potential of Long Non-Coding RNA in Feed Efficiency of Indicine Cattle. Genes 2020, 11, 997. [Google Scholar] [CrossRef]
- Wree, A.; Kahraman, A.; Gerken, G.; Canbay, A. Obesity Affects the Liver—The Link between Adipocytes and Hepatocytes. Digestion 2010, 83, 124–133. [Google Scholar] [CrossRef]
- Fernández-Sánchez, A.; Madrigal-Santillán, E.; Bautista, M.; Esquivel-Soto, J.; Morales-González, A.; Esquivel-Chirino, C.; Durante-Montiel, I.; Sánchez-Rivera, G.; Valadez-Vega, C.; Morales-González, J.A. Inflammation, Oxidative Stress, and Obesity. Int. J. Mol. Sci. 2011, 12, 3117–3132. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nat. Cell Biol. 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Noble, D. Systems biology and the heart. Biosystems 2006, 83, 75–80. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
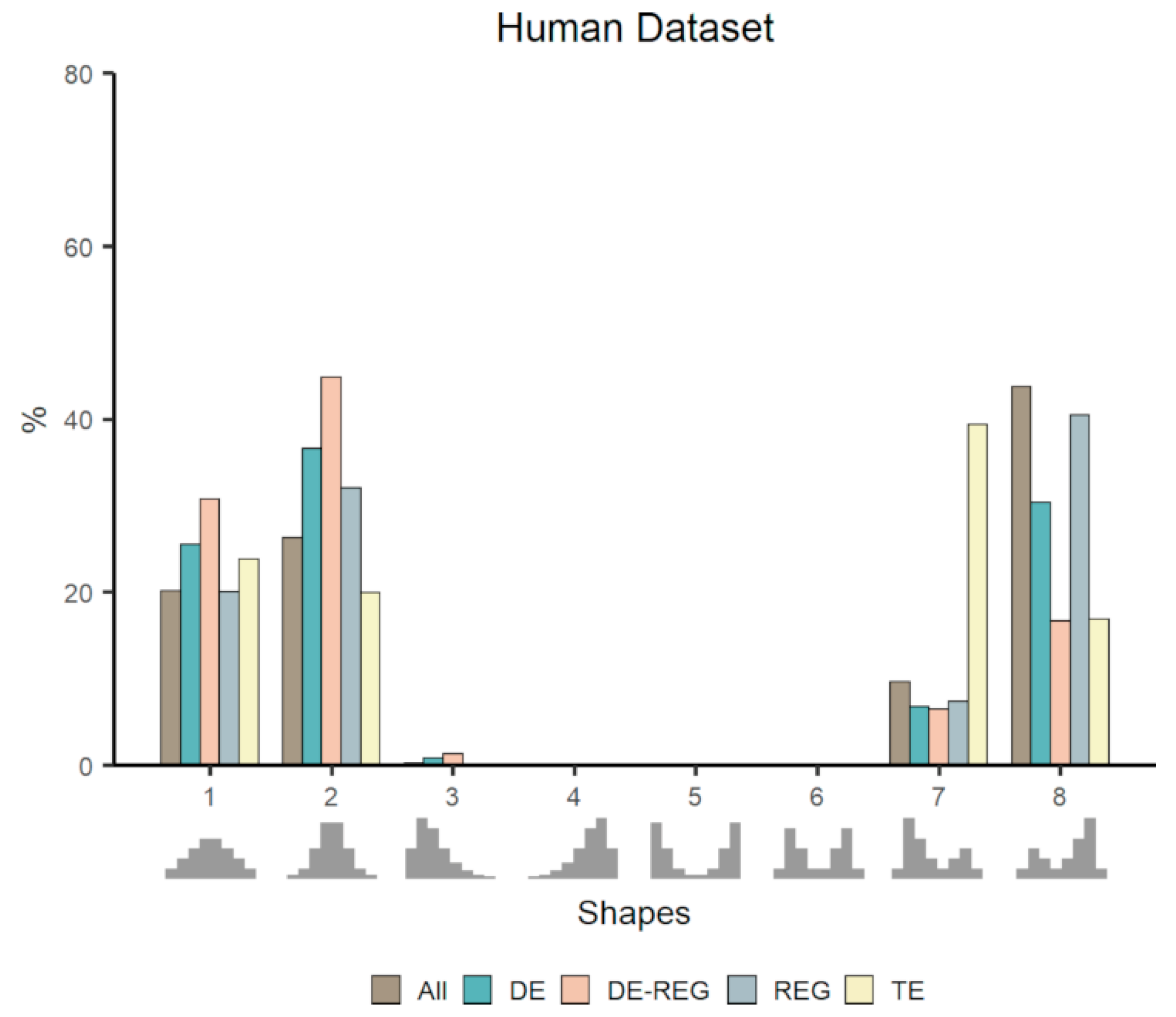{kind=link}
| Feed Efficiency | Puberty | Drosophila | Duck | Human | |
|---|---|---|---|---|---|
| Shape 1 | Ribosomal unit (7.84 × 10−15) Mitochondrial part (1.61 × 10−14) | Membrane part (9.17 × 10−32) | Cytoplasmic part (1.67 × 10−8) Mitochondrion (1.89 × 10−4) | Condensed chromatin outer kinetochore (3.42 × 10−3) | Plasma membrane part (1.95 × 10−5) |
| Shape 2 | Mitochondrial part (3.82 × 10−13) ribosomal unit (1.09 × 10−3) | Condensed chromatin outer kinetochore (3.91 × 10−2) | No functional enrichment | No functional enrichment | Immunoglobulin complex (2.98 × 10−21) |
| Shape 3 | Contractile fiber (7.55 × 10−24) | No functional enrichment | No functional enrichment | No functional enrichment | No functional enrichment |
| Shape 4 | Nucleoplasm (2.31 × 10−27) | No functional enrichment | No functional enrichment | ||
| Shape 5 | - | - | - | - | - |
| Shape 6 | - | - | Nuclear part (7.64 × 10−26) | Proteasome complex (6.71 × 10−3) | - |
| Shape 7 | No functional enrichment | - | Cytosolic part (9.75 × 10−27) | Ribosomal subunit (5.04 × 10−16) | Peroxisomal matrix (3.83 × 10−8) |
| Shape 8 | Neuron projection (2.51 × 10−2) | Nucleoplasm (1.51 × 10−153) | Cytoplasm (1.45 × 10−6) | Golgi apparatus (2.4 × 10−2) | Intracellular organelle part (1.29 × 10−108) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alexandre, P.A.; Hudson, N.J.; Lehnert, S.A.; Fortes, M.R.S.; Naval-Sánchez, M.; Nguyen, L.T.; Porto-Neto, L.R.; Reverter, A. Genome-Wide Co-Expression Distributions as a Metric to Prioritize Genes of Functional Importance. Genes 2020, 11, 1231. https://doi.org/10.3390/genes11101231
Alexandre PA, Hudson NJ, Lehnert SA, Fortes MRS, Naval-Sánchez M, Nguyen LT, Porto-Neto LR, Reverter A. Genome-Wide Co-Expression Distributions as a Metric to Prioritize Genes of Functional Importance. Genes. 2020; 11(10):1231. https://doi.org/10.3390/genes11101231
Chicago/Turabian StyleAlexandre, Pâmela A., Nicholas J. Hudson, Sigrid A. Lehnert, Marina R. S. Fortes, Marina Naval-Sánchez, Loan T. Nguyen, Laercio R. Porto-Neto, and Antonio Reverter. 2020. "Genome-Wide Co-Expression Distributions as a Metric to Prioritize Genes of Functional Importance" Genes 11, no. 10: 1231. https://doi.org/10.3390/genes11101231
APA StyleAlexandre, P. A., Hudson, N. J., Lehnert, S. A., Fortes, M. R. S., Naval-Sánchez, M., Nguyen, L. T., Porto-Neto, L. R., & Reverter, A. (2020). Genome-Wide Co-Expression Distributions as a Metric to Prioritize Genes of Functional Importance. Genes, 11(10), 1231. https://doi.org/10.3390/genes11101231