Genomic Characterization of Methicillin-Resistant Staphylococcus aureus (MRSA) by High-Throughput Sequencing in a Tertiary Care Hospital

 ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Bacterial Identification and Drug Susceptibility Testing
2.3. High-Throughput Sequencing (HTS) on Illumina MiSeq Platform
2.4. Bioinformatics Analysis
2.5. Statistical Analysis
2.6. Data Accessibility
3. Results
3.1. Nature and Source of the Isolates
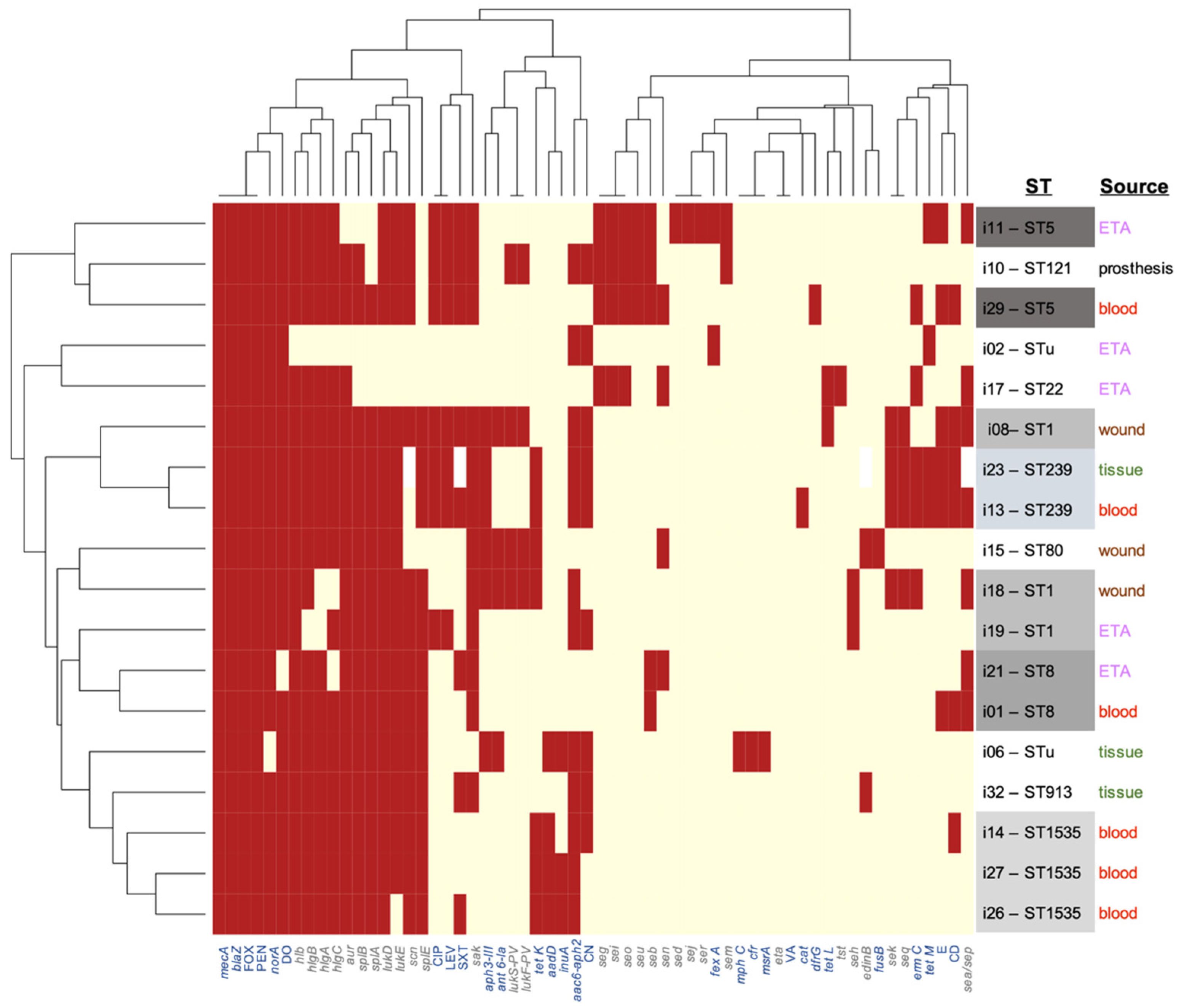
3.2. Virulome Analysis
3.3. Resistome Analysis
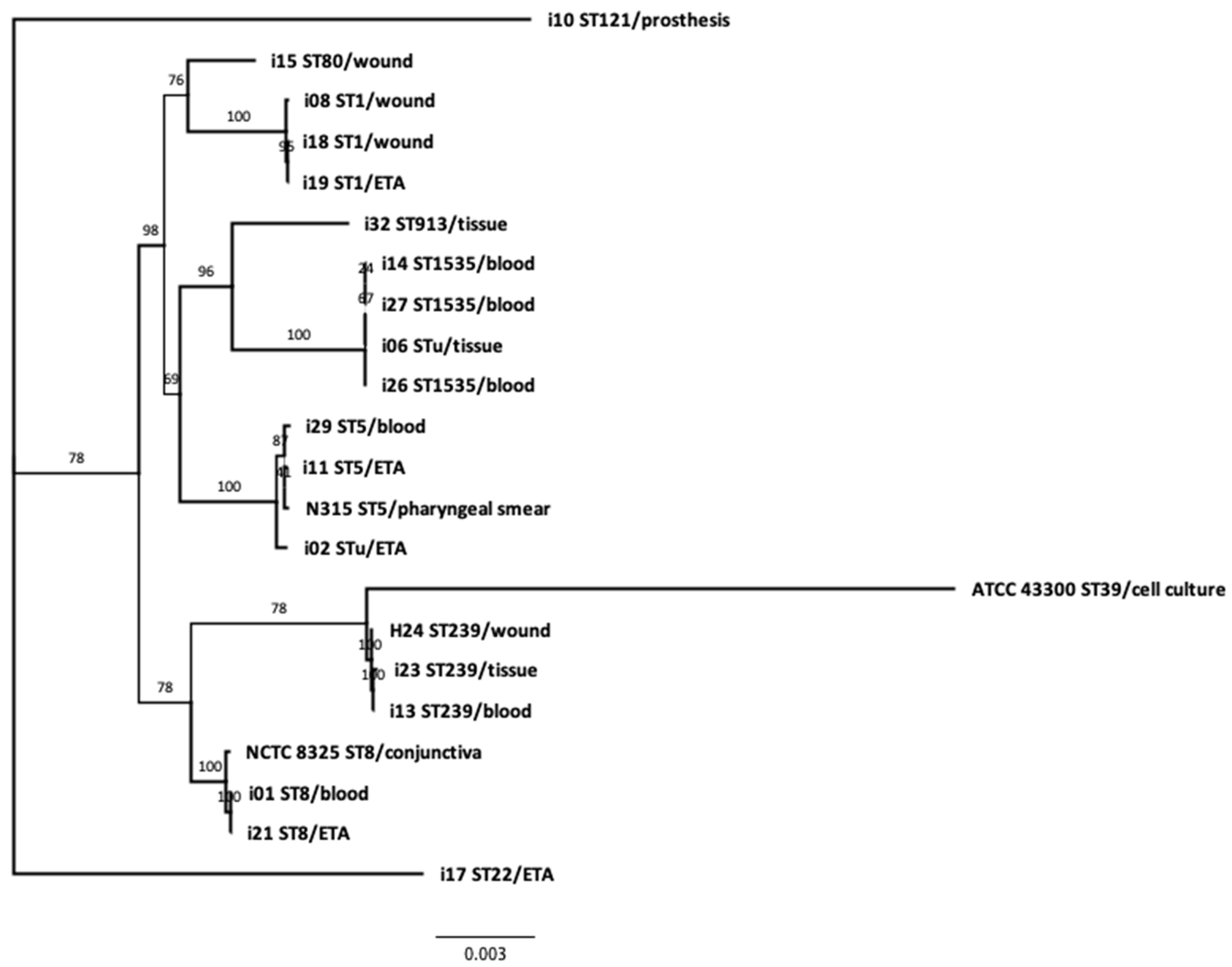
3.4. Phylogeny and Genetic Relatedness of the Sequenced Isolates:
3.5. Clinicoepidemiological–Genotype Correlations
3.6. Phenotype-Genotype Correlations
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gordon, N.C.; Price, J.R.; Cole, K.; Everitt, R.; Morgan, M.; Finney, J.; Kearns, A.M.; Pichon, B.; Young, B.; Wilson, D.J.; et al. Prediction of Staphylococcus aureus antimicrobial resistance by whole-genome sequencing. J. Clin. Microbiol. 2014, 52, 1182–1191. [Google Scholar] [CrossRef] [PubMed]
- Koeser, C.U.; Holden, M.T.G.; Ellington, M.J.; Cartwright, E.J.P.; Brown, N.M.; Ogilvy-Stuart, A.L.; Hsu, L.Y.; Chewapreecha, C.; Croucher, N.J.; Harris, S.R.; et al. Rapid whole-genome sequencing for investigation of a neonatal MRSA outbreak. N. Engl. J. Med. 2012, 366, 2267–2275. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Maksoud, M.; El-Shokry, M.; Ismail, G.; Hafez, S.; El-Kholy, A.; Attia, E.; Talaat, M. Methicillin-resistant staphylococcus aureus recovered from healthcare- and community-associated infections in Egypt. Int. J. Bacteriol. 2016, 2016, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Price, J.R.; Golubchik, T.; Cole, K.; Wilson, D.J.; Crook, D.W.; Thwaites, G.E.; Bowden, R.; Walker, A.S.; Peto, T.E.A.; Paul, J.; et al. Whole-genome sequencing shows that patient-to-patient transmission rarely accounts for acquisition of staphylococcus aureus in an intensive care unit. Clin. Infect. Dis. 2013, 58, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Nizet, V. Pathogen microevolution in high resolution. Sci. Transl. Med. 2010, 2, 16ps4. [Google Scholar] [CrossRef]
- Murray, P.R.; Baron, E.J. Manual of Clinical Microbiology, 6th ed.; ASM Press: Washington, DC, USA, 2007. [Google Scholar]
- CLSI. CLSI Supplement M100: Performance Standards for Antimicrobial Susceptibility Testing, 28th ed.; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2018. [Google Scholar]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 1 August 2019).
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Carattoli, A.; Zankari, E.; Garcia-Fernandez, A.; Voldby Larsen, M.; Lund, O.; Villa, L.; Aarestrup, F.M.; Hasman, H. PlasmidFinder and pMLST: In silico detection and typing of plasmids. Antimicrob. Agents Chemother. 2014. [Google Scholar] [CrossRef]
- Bartels, M.D.; Petersen, A.; Worning, P.; Nielsen, J.B.; Larner-Svensson, H.; Johansen, H.K.; Andersen, L.P.; Jarløv, J.O.; Boye, K.; Larsen, A.R.; et al. Comparing whole-genome sequencing with sanger sequencing for spa typing of methicillin-resistant Staphylococcus aureus. J. Clin. Microbiol. 2014, 52, 4305–4308. [Google Scholar] [CrossRef]
- Joensen, K.G.; Scheutz, F.; Lund, O.; Hasman, H.; Kaas, R.S.; Nielsen, E.M.; Aarestrup, F.M. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J. Clin. Microbiol. 2014, 52, 1501–1510. [Google Scholar] [CrossRef]
- Davis, J.J.; Wattam, A.R.; Aziz, R.K.; Brettin, T.; Butler, R.; Butler, R.M.; Chlenski, P.; Conrad, N.; Dickerman, A.; Dietrich, E.M.; et al. The PATRIC Bioinformatics Resource Center: Expanding data and analysis capabilities. Nucleic Acids Res. 2020, 48, D606–D612. [Google Scholar] [CrossRef]
- Davis, J.J.; Gerdes, S.; Olsen, G.J.; Olson, R.; Pusch, G.D.; Shukla, M.; Vonstein, V.; Wattam, A.R.; Yoo, H. PATtyFams: Protein families for the microbial genomes in the PATRIC database. Front. Microbiol. 2016, 7, 118. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A.; Hoover, P.; Rougemont, J. A rapid bootstrap algorithm for the RAxML web servers. Syst. Biol. 2008, 57, 758–771. [Google Scholar] [CrossRef]
- Castillo-Ramírez, S.; Corander, J.; Marttinen, P.; Aldeljawi, M.; Hanage, W.P.; Westh, H.; Boye, K.; Gulay, Z.; Bentley, S.D.; Parkhill, J.; et al. Phylogeographic variation in recombination rates within a global clone of methicillin-resistant Staphylococcus aureus. Genome Biol. 2012, 13, R126. [Google Scholar] [CrossRef]
- Alkharsah, K.; Rehman, S.; Alkhamis, F.; AlNimr, A.M.; Diab, A.; Al-Ali, A.K. Comparative and molecular analysis of MRSA isolates from infection sites and carrier colonization sites. Ann. Clin. Microbiol. Antimicrob. 2018, 17, 7. [Google Scholar] [CrossRef]
- Lee, C.-Y.; Fang, Y.-P.; Chang, Y.-F.; Wu, T.-H.; Yang, Y.-Y.; Huang, Y.-C. Comparison of molecular epidemiology of bloodstream methicillin-resistant Staphylococcus aureus isolates between a new and an old hospital in central Taiwan. Int. J. Infect. Dis. 2019, 79, 162–168. [Google Scholar] [CrossRef]
- Durand, G.; Javerliat, F.; Bes, M.; Veyrieras, J.-B.; Guigon, G.; Mugnier, N.; Schicklin, S.; Kaneko, G.; Santiago-Allexant, E.; Bouchiat, C.; et al. Routine whole-genome sequencing for outbreak investigations of Staphylococcus aureus in a national reference center. Front. Microbiol. 2018, 9, 511. [Google Scholar] [CrossRef]
- Shady, H.M.A.; Bakr, A.E.A.; Hashad, M.E.; Alzohairy, M.A. Staphylococcus aureus nasal carriage among outpatients attending primary health care centers: A comparative study of two cities in Saudi Arabia and Egypt. Braz. J. Infect. Dis. 2015, 19, 68–76. [Google Scholar] [CrossRef]
- Hadyeh, E.; Azmi, K.; Abu Seir, R.; Abdellatief, I.; Abdeen, Z. Molecular characterization of methicillin resistant Staphylococcus aureus in West Bank-Palestine. Front. Public Health 2019, 7, 130. [Google Scholar] [CrossRef] [PubMed]
- Elshabrawy, W.O.; Zaki, M.E.; Kamel, M.F. Genetic and phenotypic study of methicillin-resistant Staphylococcus aureus among patients and health care workers in Mansoura University Hospital, Egypt. Iran. J. Microbiol. 2017, 9, 82–88. [Google Scholar] [PubMed]
- Öksüz, L.; Dupieux, C.; Tristan, A.; Bes, M.; Etienne, J.; Gürler, N. The high diversity of MRSA clones detected in a University Hospital in Istanbul. Int. J. Med. Sci. 2013, 10, 1740–1745. [Google Scholar] [CrossRef]
- O’Hara, F.P.; Amrine-Madsen, H.; Mera, R.M.; Brown, M.L.; Close, N.M.; Suaya, J.A.; Acosta, C.J. Molecular characterization of Staphylococcus aureus in the United States 2004–2008 reveals the rapid expansion of USA300 among inpatients and Outpatients. Microb. Drug Resist. 2012, 18, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Hassan, R.; Barwa, R.; EL-Sokkary, M.M.; Ashraf, D. Virulence characteristics of methicillin resistant Staphylococcus aureus isolated from different clinical sources. N. Egypt J. Microbiol. 2017, 48, 14–29. [Google Scholar]
- Zarfel, G.; Krziwanek, K.; Johler, S.; Hoenigl, M.; Leitner, E.; Kittinger, C.; Masoud, L.; Feierl, G.; Grisold, A.J. Virulence and antimicrobial resistance genes in human MRSA ST398 isolates in Austria. Epidemiol. Infect. 2012, 141, 888–892. [Google Scholar] [CrossRef]
- Enany, S.; Yaoita, E.; Yoshida, Y.; Enany, M.; Yamamoto, T. Molecular characterization of Panton-Valentine leukocidin-positive community-acquired methicillin-resistant Staphylococcus aureus isolates in Egypt. Microbiol. Res. 2010, 165, 152–162. [Google Scholar] [CrossRef]
- Maltezou, H.C.; Giamarellou, H. Community-acquired methicillin-resistant Staphylococcus aureus infections. Int. J. Antimicrob. Agents 2006, 27, 87–96. [Google Scholar] [CrossRef]
- Bastidas, C.; Villacrés-Granda, I.; Navarrete, D.; Monsalve, M.; Coral, M.; Cifuentes, S.G. Antibiotic susceptibility profile and prevalence of mecA and lukS-PV/lukF-PV genes in Staphylococcus aureus isolated from nasal and pharyngeal sources of medical students in Ecuador. Infect. Drug Resist. 2019, 12, 2553–2560. [Google Scholar] [CrossRef]
- Ahmad, N.I.; Yean, C.Y.; Foo, P.C.; Safiee, A.W.M.; Hassan, S.A. Prevalence and association of Panton-Valentine Leukocidin gene with the risk of sepsis in patients infected with Methicillin Resistant Staphylococcus aureus. J. Infect. Public Health 2020, 13, 1508–1512. [Google Scholar] [CrossRef]
- Elshimy, R.; Khattab, R.A.; Zedan, H.; Hosny, A.S.; Elmorsy, T.H. Study on prevalence and genetic discrimination of methicillin-resistant Staphylococcus aureus (MRSA) in Egyptian hospitals. Afr. J. Microbiol. Res. 2018, 12, 629–646. [Google Scholar]
- Senok, A.; Ehricht, R.; Monecke, S.; Al-Saedan, R.; Somily, A. Molecular characterization of methicillin-resistant Staphylococcus aureus in nosocomial infections in a tertiary-care facility: Emergence of new clonal complexes in Saudi Arabia. New Microbes New Infect. 2016, 14, 13–18. [Google Scholar] [CrossRef]
- Monecke, S.; Slickers, P.; Gawlik, D.; Müller, E.; Reissig, A.; Ruppelt-Lorz, A.; Akpaka, P.E.; Bandt, D.; Bes, M.; Boswihi, S.S.; et al. Molecular typing of ST239-MRSA-III from diverse geographic locations and the evolution of the SCCmec III element during its intercontinental spread. Front. Microbiol. 2018, 9, 1436. [Google Scholar] [CrossRef]
- Abdulgader, S.M.; Shittu, A.O.; Nicol, M.P.; Ekaba, M. Molecular epidemiology of Methicillin-resistant Staphylococcus aureus in Africa: A systematic review. Front. Microbiol. 2015, 6, 348. [Google Scholar] [CrossRef]
- Boswihi, S.S.; Udo, E.E.; Al-Sweih, N. Shifts in the clonal distribution of methicillin-resistant Staphylococcus aureus in Kuwait Hospitals: 1992–2010. PLoS ONE 2016, 11, e0162744. [Google Scholar] [CrossRef] [PubMed]
- Junie, L.M.; Jeican, I.I.; Matroș, L.; Pandrea, S.L. Molecular epidemiology of the community-associated methicillin-resistant Staphylococcus aureus clones: A synthetic review. Clujul Med. 2018, 91, 7–11. [Google Scholar] [CrossRef]
- De Carvalho, S.P.; de Almeida, J.B.; de Freitas, L.M.; Guimarães, A.M.S.; Nascimento, N.C.D.; dos Santos, A.P.; Campos, G.B.; Messick, J.B.; Timenetsky, J.; Marques, L.M. Genomic profile of Brazilian methicillin-resistant Staphylococcus aureus resembles clones dispersed worldwide. J. Med. Microbiol. 2019, 68, 693–702. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Sequence Types | SCCmec Element Type * (ccr Complex Type) | Count and Frequency (%) | Isolate No. (Date of Isolation) | Diagnosis | Sample | Ward/Unit | Acquisition | Outcome |
|---|---|---|---|---|---|---|---|---|
| ST8 | V(5C2) | 1 (5.5%) | i01 (3/6/2018) | Sepsis | Blood | SICU2 | HA | Death |
| STu | I(1B), VI(4B) | 2 (11.1%) | i02 (5/5/2018) | CAUTI | ETA | MICU | CO | DC |
| STu | VI(4b) | i06 (11/4/2018) | SSI | Tissue | MSU1 | CA | DC | |
| ST1-like | V(5C2 and 5) | 1 (5.5%) | i08 (6/4/2018) | Osteomyelitis | Wound | MSU5 | CA | DC |
| ST121 | V(5C2) | 1 (5.5%) | i10 (3/4/2018) | Infected prosthesis | Infected prothesis | MSU6 | HA | DC |
| ST5 | VI(4B) | 2 (11.1%) | i11 (13/3/2018) | LTX recipient | ETA | SICU | CO | DC |
| ST5 | VI(4B) | i29 (15/4/2017) | Acute kidney injury | Blood | MSU1 | CA | DC | |
| ST239 | III(3A) | 2 (11.1%) | i13 (14/1/2018) | Urosepsis | Blood | MICU | CA | Death |
| ST239 | III(3A) | i23 (10/6/2018) | Diabetic foot | Tissue | MSU2 | CA | DC | |
| ST1535 | V(5C2) | 3 (16.6%) | i14 (7/1/2018) | Fever with Osteosarcoma | Blood | MSU3 | CA | Death |
| ST1535 | V(5C2) | i26 (7/9/2018) | Sepsis | Blood | MSU6 | CA | DC | |
| ST1535 | V(5C2) | i27 (9/6/2017) | Febrile neutropenia | Blood | BMTU | HA | DC | |
| ST80 | IVc(2B) | 1 (5.5%) | i15 (10/12/2017) | IAI | Wound | MSU4 | CO | DC |
| ST22 | IVa(2B) | 1 (5.5%) | i17 (11/10/2017) | Pneumonia in breast cancer | ETA | MICU | HA | death |
| ST1 | V(5C2 and 5), | 2 (11.1%) | i18 (9/10/2017) | SSTI | Wound | MSU6 | HA | DC |
| ST1 | V(5C2 and 5) | i19 (16/8/2017) | Pneumonia | ETA | MSU6 | CA | DC | |
| ST8-like | No SCC detected | 1 (5.5%) | i21 (19/3/2018) | Pneumonia | ETA | SICU | CO | Death |
| ST913 | V(5C2) | 1 (5.5%) | i32 (15/12/2017) | Diabetic foot | Tissue | MSU2 | CA | DC |
| Sample ID | Specimen | Genome Accession |
|---|---|---|
| i01 | Blood | JACVZG000000000 |
| i02 | ETA | JACYHC000000000 |
| i06 | Tissue | JACVZH000000000 |
| i08 | Wound | JACVZI000000000 |
| i10 | Infected prothesis | JACVZJ000000000 |
| i11 | ETA | JACVZK000000000 |
| i13 | Blood | JACVZL000000000 |
| i14 | Blood | JACVZM000000000 |
| i15 | Wound | JACVZN000000000 |
| i17 | ETA | JACVZO000000000 |
| i18 | Wound | JACYVY000000000 |
| i19 | ETA | JACVZP000000000 |
| i21 | ETA | JACVZQ000000000 |
| i23 | Tissue | JACVZR000000000 |
| i26 | Blood | JACYHD000000000 |
| i27 | Blood | JACVZS000000000 |
| i29 | Blood | JACVZT000000000 |
| i32 | Tissue | JACVZU000000000 |
| Sequence Type/ SCCmec | Isolate No. | Aureolysin | Serine Proteases | Leucocidins | Enterotoxins | TSST | Exfoliative A | Haemolysins | Staphylokinase | SCIN | Epidermal Cell Diff Inhibitor |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ST8-V | i01 | aur | splA, splB, splE | lukE, lukD | seb | – | – | hlb, hlgA, hlgB, hlgC | sak | scn | – |
| Stu-VI | i02 | – | – | – | – | – | – | – | – | – | – |
| Stu-VI | i06 | aur | splA, splB, splE | lukE, lukD | – | – | – | hlb, hlgA, hlgB, hlgC | – | scn | – |
| ST1-like-V | i08 | aur | splA, splB, splE | lukE, lukD, lukS-PV, lukF-PV | seq, sek | – | – | hlb, hlgA, hlgB, hlgC | sak | scn | – |
| ST121-V | i10 | aur | splB | lukE, lukD, lukS-PV, lukF-PV | seg, seo, seb, sem, sei, seu | – | – | hlb, hlgA, hlgB, hlgC | sak | scn | – |
| ST5-VI | i11 | – | splB | lukE, lukD | seg, seo, seb, sem, sei, seu, ser, sej, sed | – | – | hlb, hlgA, hlgB, hlgC | sak | scn | – |
| ST5-VI | i29 | aur | splA, splB | lukE, lukD | seg, seo, seb, sen, sei, seu | – | – | hlb, hlgA, hlgB, hlgC | sak | scn | – |
| ST239-III | i13 | aur | splA, splB, splE | lukE, lukD | seq, sek | – | – | hlb, hlgA, hlgB, hlgC | sak | – | – |
| ST239-III | i23 | aur | splA, splB, splE | lukE, lukD | seq, sek | hlb, hlgA, hlgB, hlgC | |||||
| ST1535-V | i14 | aur | splA, splB, splE | lukE, lukD | – | – | – | hlb, hlgA, hlgB, hlgC | – | scn | – |
| ST1535-V | i26 | aur | splA, splB, splE | lukD | – | – | – | hlb, hlgA, hlgB, hlgC | – | scn | – |
| ST1535-V | i27 | aur | splA, splB,, splE | lukE, lukD | – | – | – | hlb, hlgA, hlgB, hlgC | – | scn | – |
| ST80-IV | i15 | aur | splA, splB | lukE, lukD, LukS-PV, lukF-PV | sen | – | – | hlb, hlgA, hlgB, hlgC | sak | – | edinB |
| ST22-IV | i17 | aur | – | – | seo, sei, sen, seg | tst | – | hlb, hlgA, hlgB, hlgC | – | – | |
| ST1-V | i18 | aur | splA, splB, splE | LukE, LukD, LukS-PV, LukF-PV | seh, seq, sek | – | – | hlb, hlgB | sak | scn | – |
| ST1-V | i19 | aur | splA, splB, splE | LukE, LukD | seh | – | – | hlb, hlgC | sak | scn | – |
| ST8-like | i21 | aur | splA, splB, splE | LukE, LukD | seb, sen | – | – | hlb, hlgA, hlgB | sak | scn | – |
| ST913-V | i32 | aur | splA, splB, splE | LukE, LukD | – | – | eta | hlb, hlgA hlgB, hlgC | sak | scn | edinB |
| Sequence Type/ SCCmec Element | Isolate Serial No. | Resistome and Susceptibility Profile (Genotypic/Phenotypic Data Represented) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fox, β -Lactams 1 | Penicillin | VA 3 | CN | E | CIP, LEV | CD | Phenicol 2 | PSL 2 | Fuscidic Acid 2 | DO | MLS | SXT | ||
| ST8-V | i01 | mecA/R | blaZ/R | –/S | –/S | –/R | norA/S,S | –/R | – | – | – | –/R | – | –/S |
| STu-VI | i02 | mecA/R | blaZ/R | –/S | aac(6′)-aph(2″)/R | –/S | norA/S,S | –/S | Fex A | – | – | tet M/R | – | –/S |
| STu-VI | i06 | mecA/R | blaZ/R | –/S | aac(6′)-aph(2″), ant 6-Ia, aph(3′)-III, aadD/R | mph C/S | –/S,S | InuA/S | – | cfr | – | –/R | msrA | –/S |
| ST1-like-V | i08 | mecA/R | blaZ/R | –/S | aac(6′)-aph(2″), ant 6-Ia, aph(3′)-III/R | –/R | norA/R,R | –/R | – | – | – | tet L/R | – | –/R |
| ST121-V | i10 | mecA/R | blaZ/R | –/S | aac(6′)-aph(2″)/R | –/S | norA/R,R | –/S | – | – | – | –/R | – | –/R |
| ST5-VI | i11 | mecA/R | blaZ/R | –/S | –/S | –/R | norA/R,R | –/S | Fex A | – | – | tet M/R | – | –/R |
| ST5-VI | i29 | mecA/R | blaZ/R | –/S | –/S | erm C/R | norA/R,R | –/R | – | – | – | –/R | – | dfrG/R |
| ST239-III | i13 | mecA/R | blaZ/R | –/S | aac(6′)-aph(2″), aph(3′)-III/R | erm C/R | norA/R,R | –/R | cat (pc221) | – | – | tet K, tet M/R | – | –/R |
| ST239-III | i23 | mecA/R | blaZ/R | –/S | aac(6′)-aph(2″), aph(3′)-III/R | erm C/R | norA/R,R | –/R | – | – | – | tet K, tet M/R | – | – |
| ST1535-V | i14 | mecA/R | blaZ/R | –/S | aac(6′)-aph(2″), aadD/R | –/S | norA/S,S | –/R | – | – | – | tet K/R | – | –/S |
| ST1535-V | i26 | mecA/R | blaZ/R | –/S | aac(6′)-aph(2″), aadD/S | –/S | norA/S,S | InuA/S | – | – | – | tet K/R | – | –/R |
| ST1535-V | i27 | mecA/R | blaZ/R | –/S | aac(6′)-aph(2″), aadD/S | –/S | norA/S,S | InuA/S | – | – | – | tet K/R | – | –/S |
| ST80-IV | i15 | mecA/R | blaZ/R | –/S | ant 6-Ia, aph(3′)-III/S | –/S | norA/S,S | –/S | – | – | fus B | tet K/R | – | –/S |
| ST22-IV | i17 | mecA/R | blaZ/R | –/S | –/S | erm C/S | norA/S,S | –/S | – | – | – | tet L/R | – | –/S |
| ST1-V | i18 | mecA/R | blaZ/R | –/S | aac(6′)-aph(2″), ant 6-Ia, aph(3′)-III/S | erm C/S | norA/S,S | –/S | – | – | – | tet K/S | – | –/S |
| ST1-V | i19 | mecA/R | blaZ/R | –/S | aac(6′)-aph(2″)/R | –/S | norA/R,R | –/S | – | – | – | –/R | – | –/S |
| ST8-like | i21 | mecA/R | blaZ/R | –/S | –/S | –/S | norA/S,S | –/S | – | – | – | –/S | – | –/R |
| ST913-V | i32 | mecA/R | blaZ/R | –/S | aac(6′)-aph(2″)/R | –/S | norA/S,S | –/S | – | – | – | –/R | – | –/R |
| Variable 1 | Variable 2 | Fisher’s Test p-Value | Correlation Coefficient (If Applicable) | Type of Association |
|---|---|---|---|---|
| lukS-PV, lukF PV | Severity | 0.0237 | N/A | Genes absent in severe cases |
| aph(3″)-III | Severity | 0.0128 | N/A | Gene rare in severe cases, enriched in non-severe ones |
| ant.6.Ia | Severity | 0.011 | N/A | Gene absent in severe cases |
| ant.6.Ia | Sample | 0.003 | N/A | Gene enriched in wound isolates |
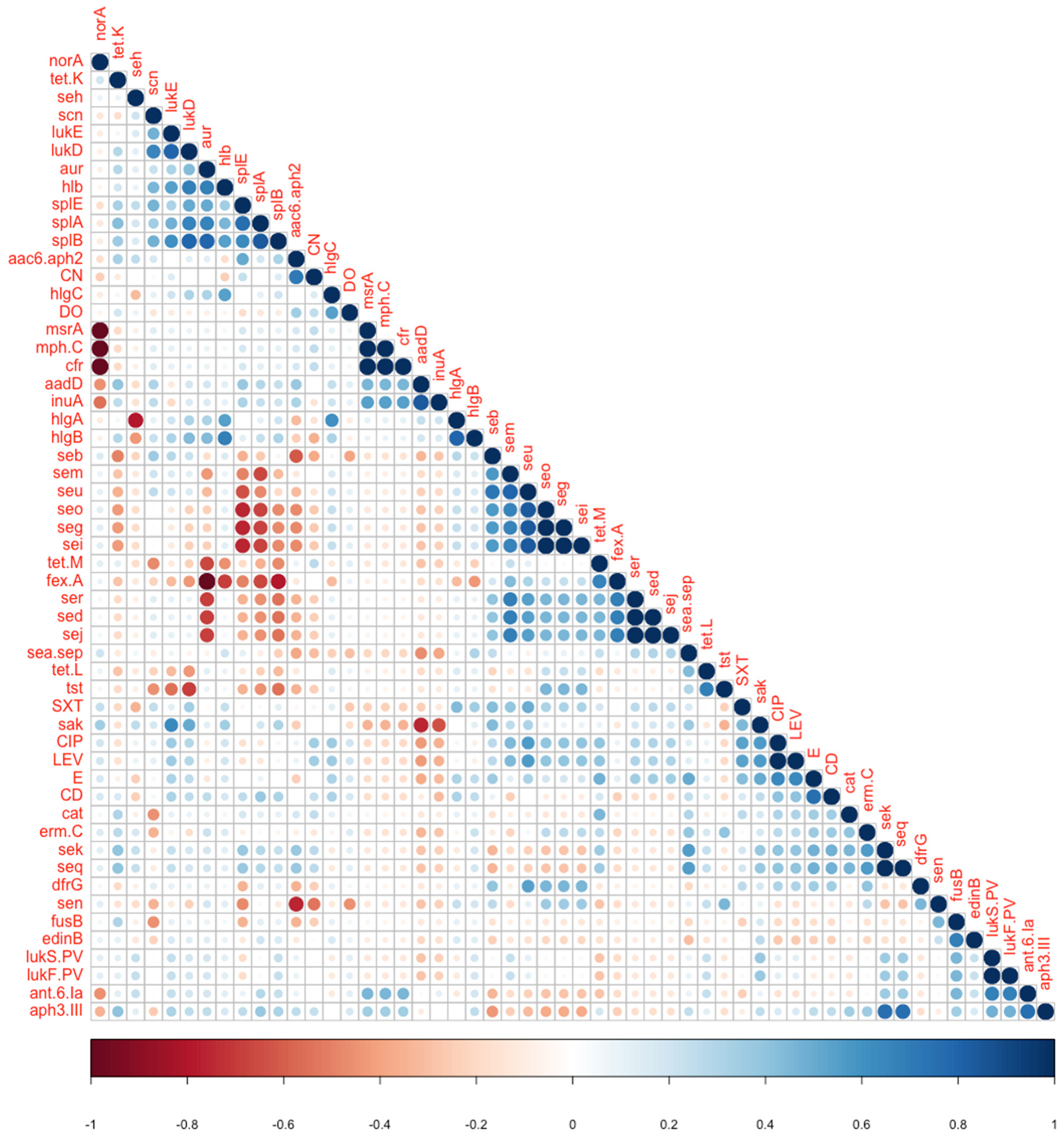
| aur | fexA | 0.007 | −1 | Negative association |
| norA | msrA, cfr | >0.05 | −1 | Negative association |
| seg, seo | splA | 0.019 | −0.679 | Negative association |
| seg, seo | splE | 0.005 | −0.756 | Negative association |
| aur | splB | 0.0196 | 0.79 | Positive association |
| lukS-PV lukF PV | ant.6.Ia | 0.019 | 0.679 | Positive association |
| lukD | splA | 0.0392 | 0.661 | Positive association |
| lukD | splB | 0.0196 | 0.791 | Positive association |
| lukE | sak | 0.0245 | 0.632 | Positive association |
| aadD | sak | 0.005 | −0.756 | Reverse association |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soliman, M.S.; Soliman, N.S.; El-Manakhly, A.R.; ElBanna, S.A.; Aziz, R.K.; El-Kholy, A.A. Genomic Characterization of Methicillin-Resistant Staphylococcus aureus (MRSA) by High-Throughput Sequencing in a Tertiary Care Hospital. Genes 2020, 11, 1219. https://doi.org/10.3390/genes11101219
Soliman MS, Soliman NS, El-Manakhly AR, ElBanna SA, Aziz RK, El-Kholy AA. Genomic Characterization of Methicillin-Resistant Staphylococcus aureus (MRSA) by High-Throughput Sequencing in a Tertiary Care Hospital. Genes. 2020; 11(10):1219. https://doi.org/10.3390/genes11101219
Chicago/Turabian StyleSoliman, May Sherif, Noha Salah Soliman, Arwa Ramadan El-Manakhly, Shahira AbdelSalam ElBanna, Ramy Karam Aziz, and Amani Ali El-Kholy. 2020. "Genomic Characterization of Methicillin-Resistant Staphylococcus aureus (MRSA) by High-Throughput Sequencing in a Tertiary Care Hospital" Genes 11, no. 10: 1219. https://doi.org/10.3390/genes11101219
APA StyleSoliman, M. S., Soliman, N. S., El-Manakhly, A. R., ElBanna, S. A., Aziz, R. K., & El-Kholy, A. A. (2020). Genomic Characterization of Methicillin-Resistant Staphylococcus aureus (MRSA) by High-Throughput Sequencing in a Tertiary Care Hospital. Genes, 11(10), 1219. https://doi.org/10.3390/genes11101219