Application of CRISPR/Cas9-Based Reverse Genetics in Leishmania braziliensis: Conserved Roles for HSP100 and HSP23

 ,
,  and
and
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Leishmania Strains and Culture
2.2. Promastigote Cultivation
2.3. Transfections, Selection, and Cell Cloning
2.4. Construction and Preparation of Recombinant DNA
2.5. PCR-Amplification of Targeting Constructs
2.6. Analytical PCR
2.7. RNA Extraction, cDNA Synthesis, and Quantitative Real-Time PCR (qRT-PCR)
2.8. Next Generation Sequencing
2.9. Western Blotting
2.10. Immunofluorescence Assays
2.11. Flow Cytometry Cell Analysis
2.12. In Vitro Infection of Murine Bone Marrow-Derived Macrophages
2.13. In Silico Procedures
3. Results
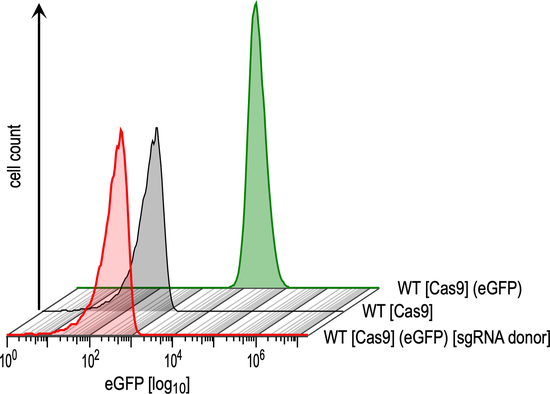
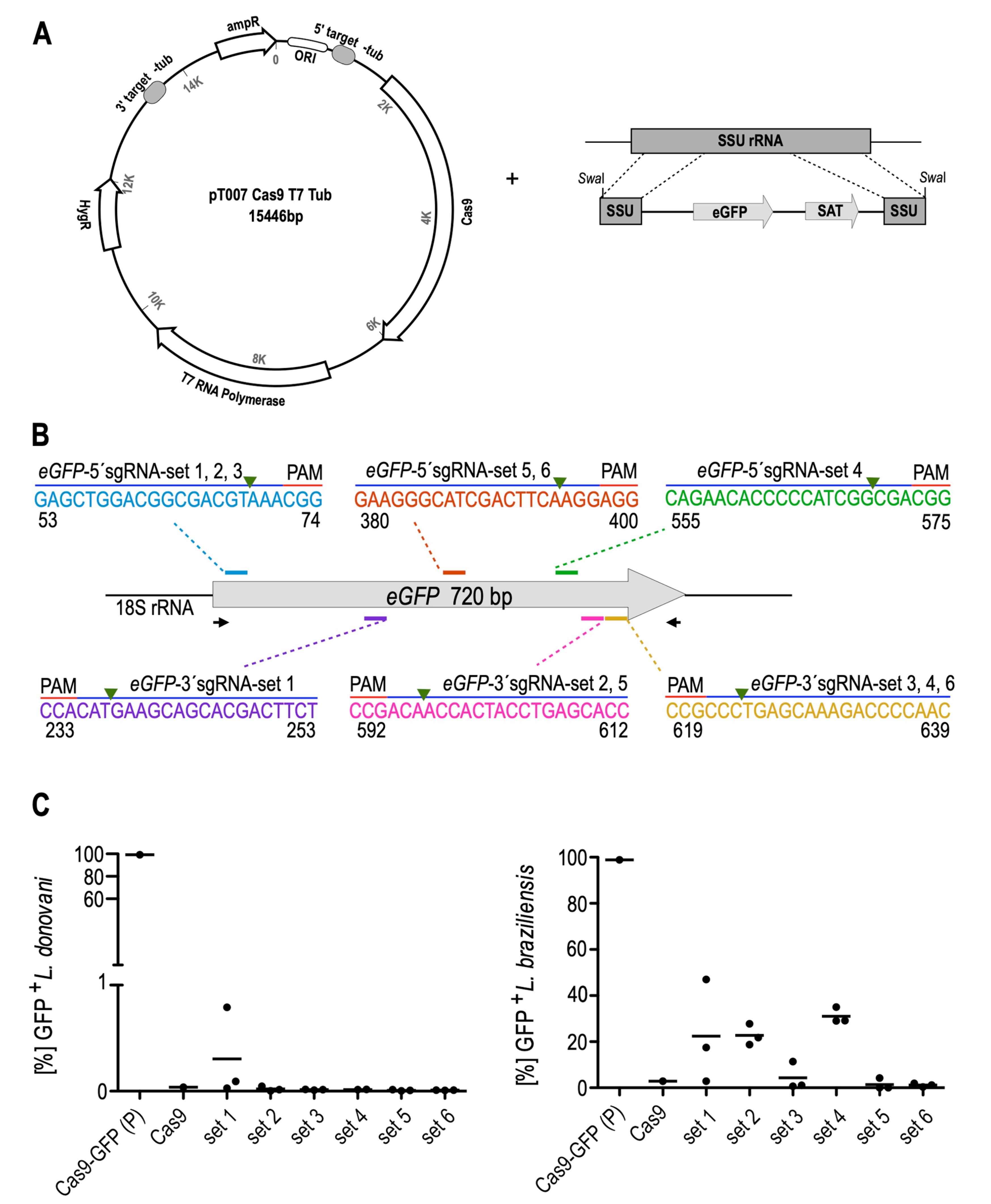
3.1. Optimisation and Validation of the CRISPR-Cas9 System in L. braziliensis
3.2. CRISPR–Cas9-Mediated Disruption of Endogenous HSP23 and HSP100 Genes in L. braziliensis
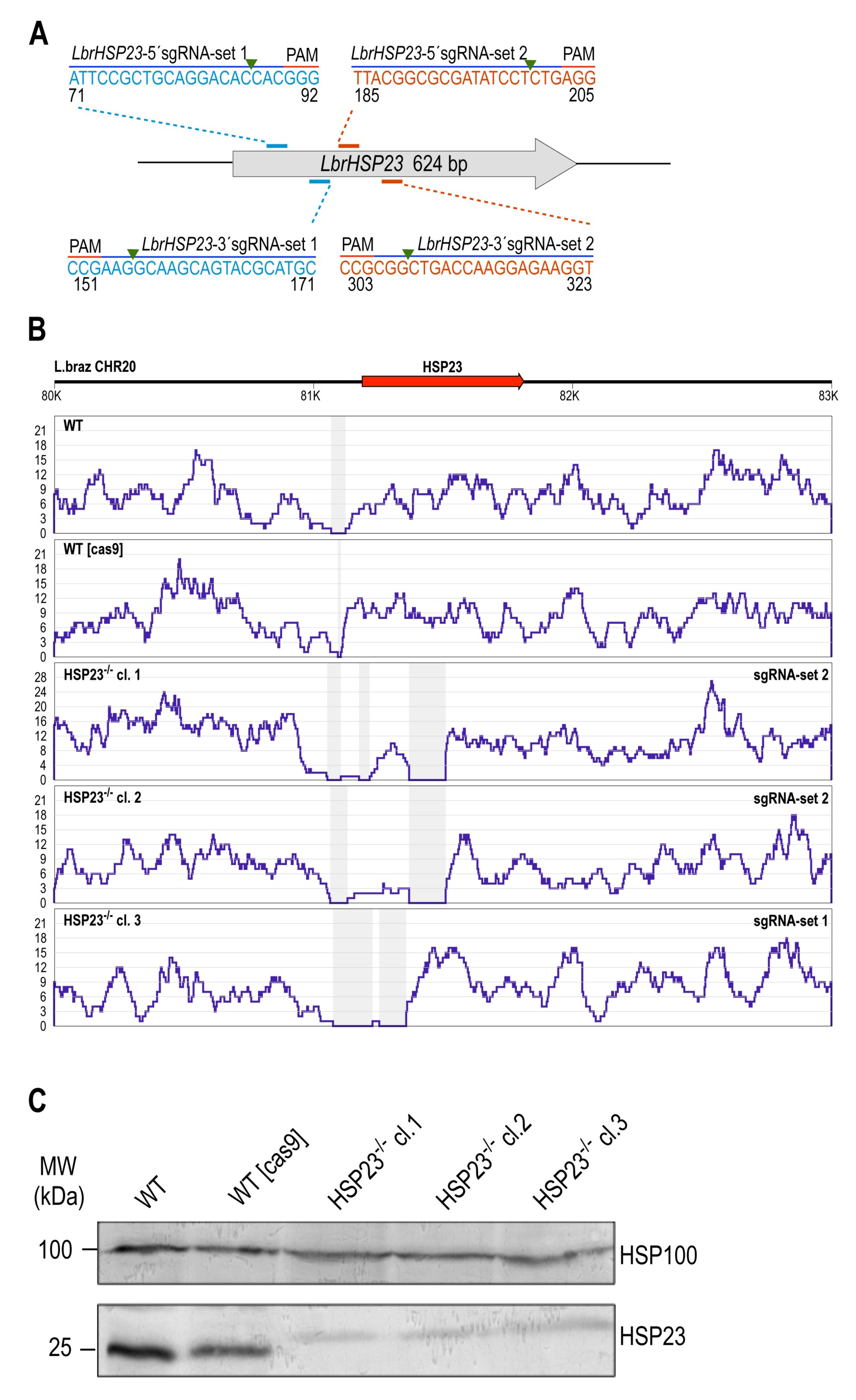
3.2.1. LbrHSP23 Gene Replacement
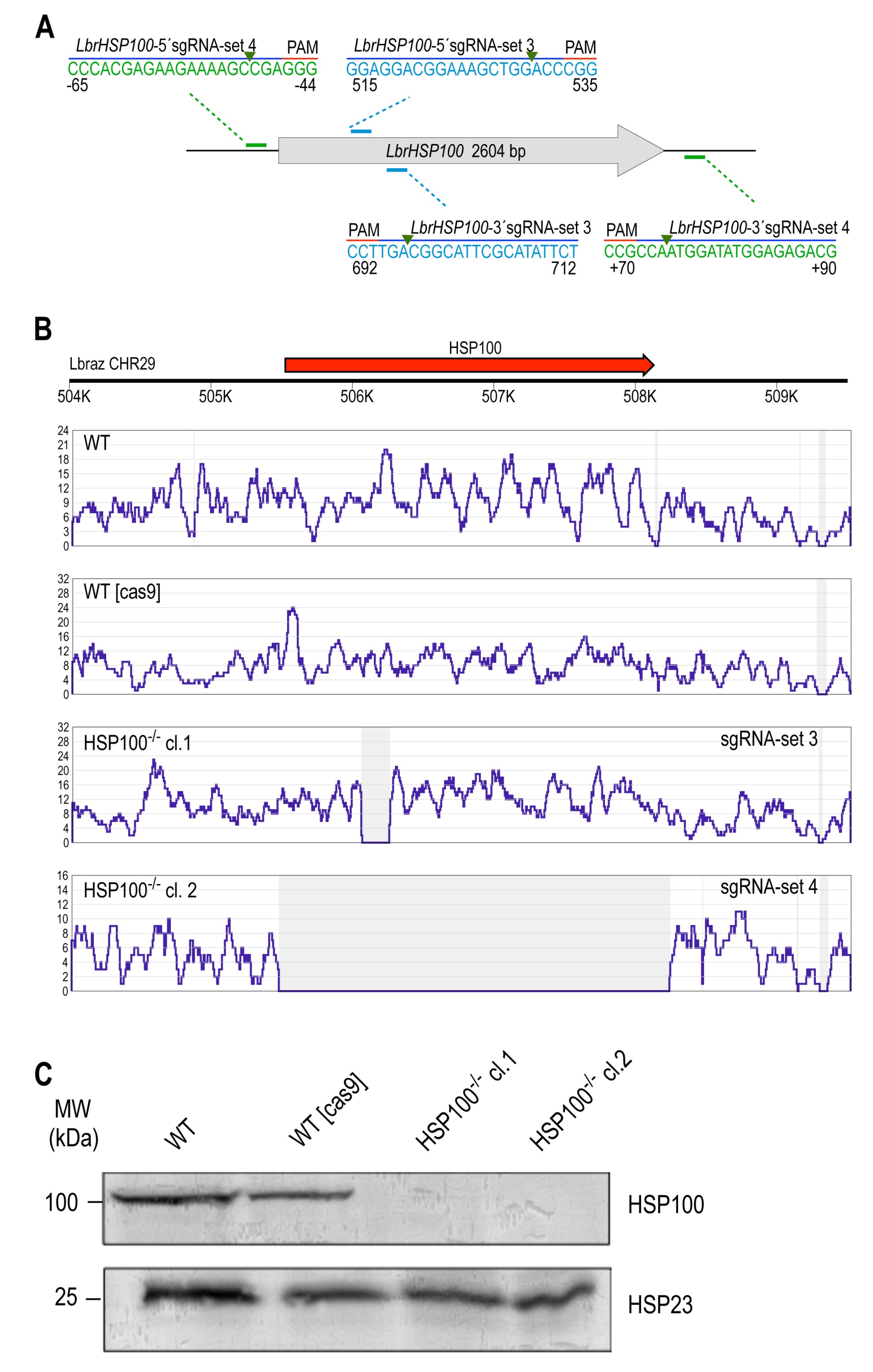
3.2.2. LbrHSP100 Gene Replacement
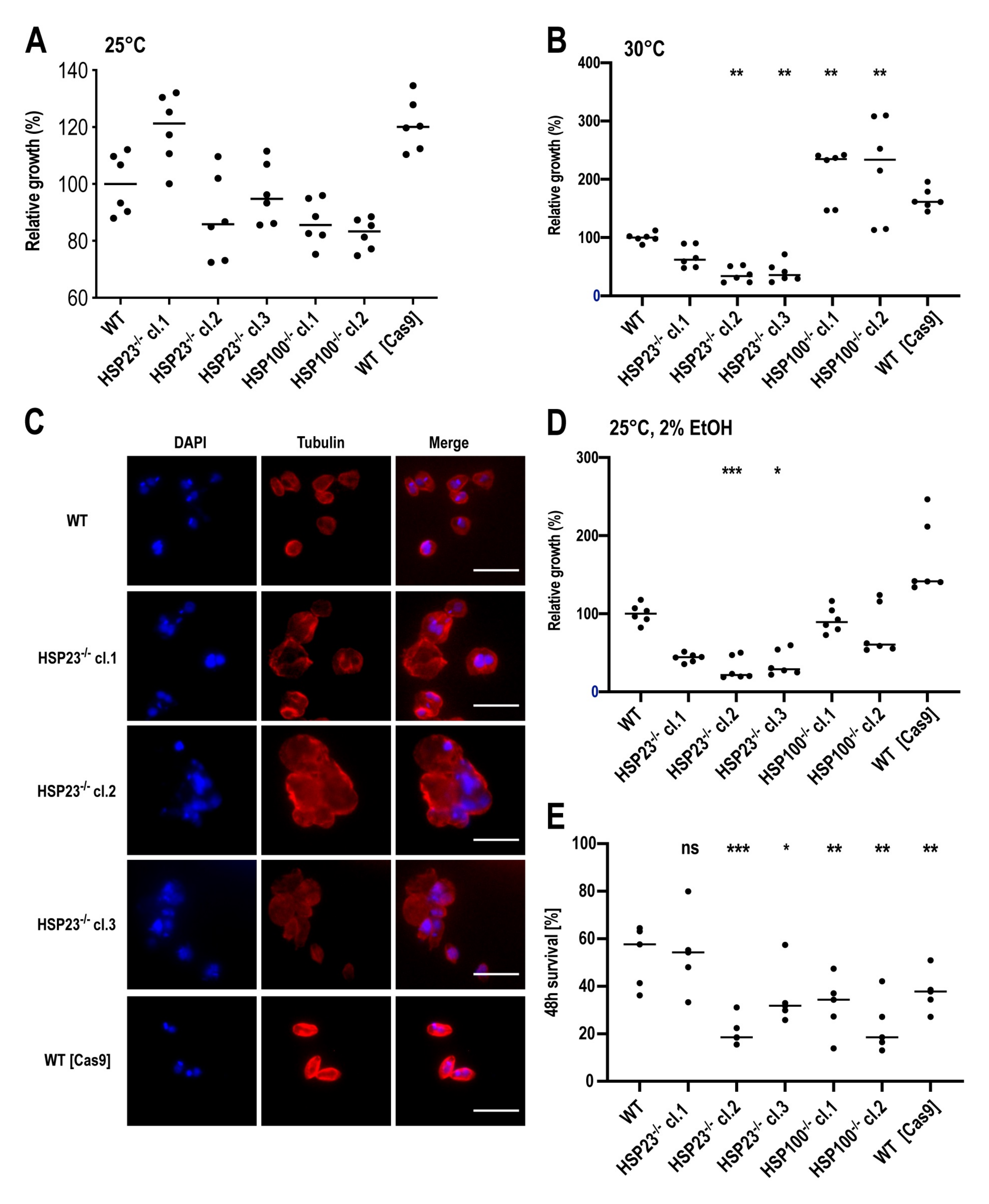
3.3. L. braziliensis HSP23- and HSP100-Null Mutant Phenotypes Resemble Those Described for Old World Leishmania
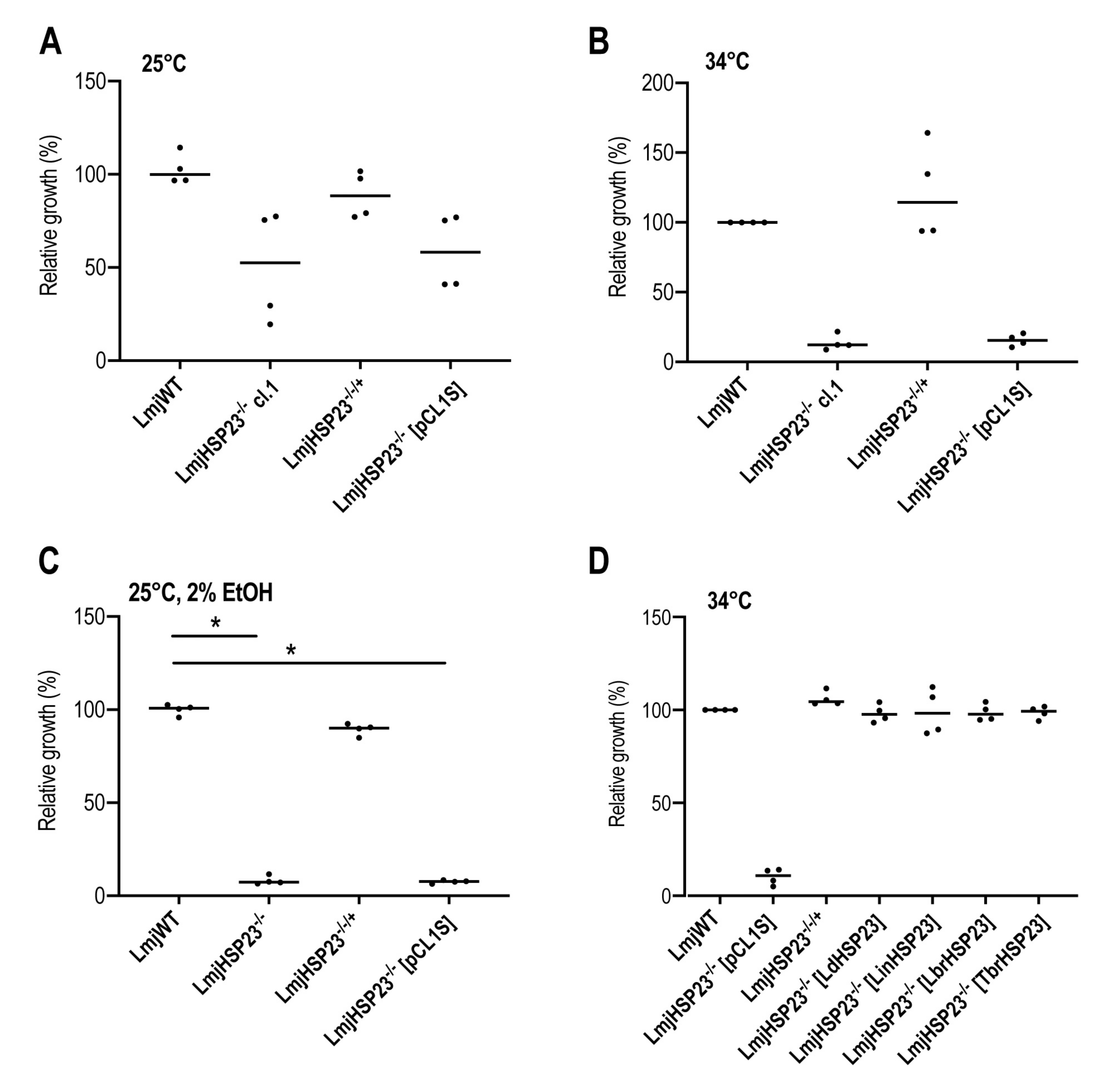
3.4. Complementation Studies in L. major HSP23-Null Mutants Indicate a Conserved Function in Thermotolerance for Trypanosomatid HSP23
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Marsden, P.D. Mucosal leishmaniasis ("espundia" Escomel, 1911). Trans R Soc. Trop. Med. Hyg. 1986, 80, 859–876. [Google Scholar] [CrossRef]
- Amato, V.S.; Tuon, F.F.; Siqueira, A.M.; Nicodemo, A.C.; Neto, V.A. Treatment of mucosal leishmaniasis in Latin America: systematic review. Am. J. Trop. Med. Hyg. 2007, 77, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Arevalo, J.; Ramirez, L.; Adaui, V.; Zimic, M.; Tulliano, G.; Miranda-Verastegui, C.; Lazo, M.; Loayza-Muro, R.; De Doncker, S.; Maurer, A.; et al. Influence of Leishmania (Viannia) species on the response to antimonial treatment in patients with American tegumentary leishmaniasis. J. Infect. Dis. 2007, 195, 1846–1851. [Google Scholar] [CrossRef] [PubMed]
- Reithinger, R.; Dujardin, J.C.; Louzir, H.; Pirmez, C.; Alexander, B.; Brooker, S. Cutaneous leishmaniasis. Lancet Infect. Dis. 2007, 7, 581–596. [Google Scholar] [CrossRef]
- Cruz, A.; Beverley, S.M. Gene replacement in parasitic protozoa. Nature 1990, 348, 171–173. [Google Scholar] [CrossRef]
- Cruz, A.; Coburn, C.M.; Beverley, S.M. Double targeted gene replacement for creating null mutants. Proc. Natl. Acad. Sci. USA 1991, 88, 7170–7174. [Google Scholar] [CrossRef]
- Zirpel, H.; Clos, J. Gene Replacement by Homologous Recombination. Methods Mol. Biol. 2019, 1971, 169–188. [Google Scholar]
- Peacock, C.S.; Seeger, K.; Harris, D.; Murphy, L.; Ruiz, J.C.; Quail, M.A.; Peters, N.; Adlem, E.; Tivey, A.; Aslett, M.; et al. Comparative genomic analysis of three Leishmania species that cause diverse human disease. Nat. Genet. 2007, 39, 839–847. [Google Scholar] [CrossRef]
- Lye, L.F.; Owens, K.; Shi, H.; Murta, S.M.; Vieira, A.C.; Turco, S.J.; Tschudi, C.; Ullu, E.; Beverley, S.M. Retention and loss of RNA interference pathways in trypanosomatid protozoans. PLoS Pathog. 2010, 6, e1001161. [Google Scholar] [CrossRef]
- De Paiva, R.M.; Grazielle-Silva, V.; Cardoso, M.S.; Nakagaki, B.N.; Mendonca-Neto, R.P.; Canavaci, A.M.; Souza Melo, N.; Martinelli, P.M.; Fernandes, A.P.; daRocha, W.D.; et al. Amastin Knockdown in Leishmania braziliensis Affects Parasite–Macrophage Interaction and Results in Impaired Viability of Intracellular Amastigotes. PLoS Pathog. 2015, 11, e1005296. [Google Scholar] [CrossRef]
- Jackson, A.L.; Bartz, S.R.; Schelter, J.; Kobayashi, S.V.; Burchard, J.; Mao, M.; Li, B.; Cavet, G.; Linsley, P.S. Expression profiling reveals off–target gene regulation by RNAi. Nat Biotechnol. 2003, 21, 635–637. [Google Scholar] [CrossRef] [PubMed]
- Knott, G.J.; Doudna, J.A. CRISPR–Cas guides the future of genetic engineering. Science 2018, 361, 866–869. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual–RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double–Strand Break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef]
- Bibikova, M.; Beumer, K.; Trautman, J.K.; Carroll, D. Enhancing gene targeting with designed zinc finger nucleases. Science 2003, 300, 764. [Google Scholar] [CrossRef]
- Peng, D.; Kurup, S.P.; Yao, P.Y.; Minning, T.A.; Tarleton, R.L. CRISPR–Cas9–mediated single–gene and gene family disruption in Trypanosoma cruzi. mBio 2014, 6, e02097-14. [Google Scholar] [CrossRef]
- Beneke, T.; Madden, R.; Makin, L.; Valli, J.; Sunter, J.; Gluenz, E. A CRISPR Cas9 high–throughput genome editing toolkit for kinetoplastids. R Soc. Open Sci. 2017, 4, 170095. [Google Scholar] [CrossRef]
- Vasquez, J.J.; Wedel, C.; Cosentino, R.O.; Siegel, T.N. Exploiting CRISPR–Cas9 technology to investigate individual histone modifications. Nucleic Acids Res. 2018, 46, e106. [Google Scholar] [CrossRef]
- Sollelis, L.; Ghorbal, M.; MacPherson, C.R.; Martins, R.M.; Kuk, N.; Crobu, L.; Bastien, P.; Scherf, A.; Lopez-Rubio, J.-J.; Sterkers, Y. First efficient CRISPR–Cas9–mediated genome editing in Leishmania parasites. Cell. Microbiol. 2015, 17, 1405–1412. [Google Scholar] [CrossRef]
- Zhang, W.W.; Matlashewski, G. CRISPR–Cas9–Mediated Genome Editing in Leishmania donovani. MBio 2015, 6, e00861. [Google Scholar] [CrossRef]
- Martel, D.; Beneke, T.; Gluenz, E.; Spath, G.F.; Rachidi, N. Characterisation of Casein Kinase 1.1 in Leishmania donovani Using the CRISPR Cas9 Toolkit. Biomed. Res. Int. 2017, 2017, 4635605. [Google Scholar] [CrossRef] [PubMed]
- Soares Medeiros, L.C.; South, L.; Peng, D.; Bustamante, J.M.; Wang, W.; Bunkofske, M.; Perumal, N.; Sanchez-Valdez, F.; Tarleton, R.L. Rapid, Selection–Free, High–Efficiency Genome Editing in Protozoan Parasites Using CRISPR–Cas9 Ribonucleoproteins. mBio 2017, 8. [Google Scholar] [CrossRef]
- Fernandez-Prada, C.; Sharma, M.; Plourde, M.; Bresson, E.; Roy, G.; Leprohon, P.; Ouellette, M. High–throughput Cos–Seq screen with intracellular Leishmania infantum for the discovery of novel drug–resistance mechanisms. Int. J. Parasitol. Drugs Drug Resist. 2018, 8, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Ishemgulova, A.; Hlavacova, J.; Majerova, K.; Butenko, A.; Lukes, J.; Votypka, J.; Volf, P.; Yurchenko, V. CRISPR/Cas9 in Leishmania mexicana: A case study of LmxBTN1. PloS ONE 2018, 13, e0192723. [Google Scholar] [CrossRef] [PubMed]
- Bryant, J.M.; Baumgarten, S.; Glover, L.; Hutchinson, S.; Rachidi, N. CRISPR in Parasitology: Not Exactly Cut and Dried! Trends Parasitol 2019, 35, 409–422. [Google Scholar] [CrossRef]
- Cruz, A.K.; Titus, R.; Beverley, S.M. Plasticity in chromosome number and testing of essential genes in Leishmania by targeting. Proc. Natl. Acad. Sci. USA 1993, 90, 1599–1603. [Google Scholar] [CrossRef]
- Sterkers, Y.; Crobu, L.; Lachaud, L.; Pages, M.; Bastien, P. Parasexuality and mosaic aneuploidy in Leishmania: alternative genetics. Trends Parasitol. 2014, 30, 429–435. [Google Scholar] [CrossRef]
- Dumetz, F.; Imamura, H.; Sanders, M.; Seblova, V.; Myskova, J.; Pescher, P.; Vanaerschot, M.; Meehan, C.J.; Cuypers, B.; De Muylder, G.; et al. Modulation of Aneuploidy in Leishmania donovani during Adaptation to Different In Vitro and In Vivo Environments and Its Impact on Gene Expression. MBio 2017, 8. [Google Scholar] [CrossRef]
- Duncan, S.M.; Jones, N.G.; Mottram, J.C. Recent advances in Leishmania reverse genetics: Manipulating a manipulative parasite. Mol. Biochem. Parasitol. 2017, 216, 30–38. [Google Scholar] [CrossRef]
- Zhang, W.W.; Lypaczewski, P.; Matlashewski, G. Optimized CRISPR–Cas9 Genome Editing for Leishmania and Its Use To Target a Multigene Family, Induce Chromosomal Translocation, and Study DNA Break Repair Mechanisms. mSphere 2017, 2. [Google Scholar] [CrossRef]
- Zhang, W.W.; Matlashewski, G. Single–Strand Annealing Plays a Major Role in Double–Strand DNA Break Repair following CRISPR–Cas9 Cleavage in Leishmania. mSphere 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Damasceno, J.D.; Reis-Cunha, J.; Crouch, K.; Beraldi, D.; Lapsley, C.; Tosi, L.R.O.; Bartholomeu, D.; McCulloch, R. Conditional knockout of RAD51–related genes in Leishmania major reveals a critical role for homologous recombination during genome replication. PLoS Genet. 2020, 16, e1008828. [Google Scholar] [CrossRef] [PubMed]
- Yagoubat, A.; Crobu, L.; Berry, L.; Kuk, N.; Lefebvre, M.; Sarrazin, A.; Bastien, P.; Sterkers, Y. Universal highly efficient conditional knockout system in Leishmania, with a focus on untranscribed region preservation. Cell. Microbiol. 2020, 22, e13159. [Google Scholar] [CrossRef] [PubMed]
- Yardley, V.; Ortuno, N.; Llanos-Cuentas, A.; Chappuis, F.; Doncker, S.D.; Ramirez, L.; Croft, S.; Arevalo, J.; Adaui, V.; Bermudez, H.; et al. American tegumentary leishmaniasis: Is antimonial treatment outcome related to parasite drug susceptibility? J. Infect. Dis. 2006, 194, 1168–1175. [Google Scholar] [CrossRef]
- Rosenzweig, D.; Smith, D.; Opperdoes, F.; Stern, S.; Olafson, R.W.; Zilberstein, D. Retooling Leishmania metabolism: from sand fly gut to human macrophage. FASEB J 2008, 22, 590–602. [Google Scholar] [CrossRef] [PubMed]
- Al-Jawabreh, A.; Diezmann, S.; Muller, M.; Wirth, T.; Schnur, L.F.; Strelkova, M.V.; Kovalenko, D.A.; Razakov, S.A.; Schwenkenbecher, J.; Kuhls, K.; et al. Identification of geographically distributed sub–populations of Leishmania (Leishmania) major by microsatellite analysis. BMC Evol. Biol. 2008, 8, 183. [Google Scholar] [CrossRef]
- Kapler, G.M.; Coburn, C.M.; Beverley, S.M. Stable transfection of the human parasite Leishmania major delineates a 30–kilobase region sufficient for extrachromosomal replication and expression. Mol. Cell. Biol. 1990, 10, 1084–1094. [Google Scholar] [CrossRef] [PubMed]
- Krobitsch, S.; Brandau, S.; Hoyer, C.; Schmetz, C.; Hübel, A.; Clos, J. Leishmania donovani heat shock protein 100: characterization and function in amastigote stage differentiation. J. Biol. Chem. 1998, 273, 6488–6494. [Google Scholar] [CrossRef]
- Ommen, G.; Lorenz, S.; Clos, J. One–step generation of double–allele gene replacement mutants in Leishmania donovani. Int. J. Parasitol. 2009, 39, 541–546. [Google Scholar] [CrossRef]
- Beneke, T.; Gluenz, E. LeishGEdit: A Method for Rapid Gene Knockout and Tagging Using CRISPR–Cas9. Methods Mol. Biol. 2019, 1971, 189–210. [Google Scholar]
- Schumann Burkard, G.; Jutzi, P.; Roditi, I. Genome–wide RNAi screens in bloodstream form trypanosomes identify drug transporters. Mol. Biochem. Parasitol. 2011, 175, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, K.; Hombach-Barrigah, A.; Clos, J. Hsp90 inhibitors radicicol and geldanamycin have opposing effects on Leishmania Aha1–dependent proliferation. Cell Stress Chaperones 2017, 22, 729–742. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, K.; Zander, D.; Kube, M.; Reinhardt, R.; Clos, J. Identification of a Leishmania infantum gene mediating resistance to miltefosine and SbIII. Int. J. Parasitol. 2008, 38, 1411–1423. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real–time quantitative PCR and the 2(–Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Kröber-Boncardo, C.; Lorenzen, S.; Brinker, C.; Clos, J. Casein Kinase 1.2 Over Expression Restores Stress Resistance toLeishmania donovaniHSP23 Null Mutants. Sci. Rep. 2020, 10. in press. [Google Scholar]
- Van den Broeck, F.; Savill, N.J.; Imamura, H.; Sanders, M.; Maes, I.; Cooper, S.; Mateus, D.; Jara, M.; Adaui, V.; Arevalo, J.; et al. Ecological divergence and hybridization of Neotropical Leishmania parasites. Proc. Natl. Acad. Sci. USA 2020, 10, 210. [Google Scholar] [CrossRef]
- Hombach, A.; Ommen, G.; MacDonald, A.; Clos, J. A small heat shock protein is essential for thermotolerance and intracellular survival of Leishmania donovani. J. Cell Sci. 2014, 127, 4762–4773. [Google Scholar] [CrossRef]
- Hombach-Barrigah, A.; Bartsch, K.; Smirlis, D.; Rosenqvist, H.; MacDonald, A.; Dingli, F.; Loew, D.; Spath, G.F.; Rachidi, N.; Wiese, M.; et al. Leishmania donovani 90 kD Heat Shock Protein – Impact of Phosphosites on Parasite Fitness, Infectivity and Casein Kinase Affinity. Sci. Rep. 2019, 9, 5074. [Google Scholar] [CrossRef]
- Bifeld, E.; Tejera Nevado, P.; Bartsch, J.; Eick, J.; Clos, J. A versatile qPCR assay to quantify trypanosomatidic infections of host cells and tissues. Med. Microbiol. Immunol. 2016, 205, 449–458. [Google Scholar] [CrossRef]
- Bifeld, E. Quantification of Intracellular Leishmania spp. Using Real–Time Quantitative PCR (qPCR). Methods Mol. Biol. 2019, 1971, 249–263. [Google Scholar]
- Bifeld, E. Generation of Bone Marrow–Derived Macrophages for In Vitro Infection Experiments. Methods Mol. Biol. 2019, 1971, 237–247. [Google Scholar] [PubMed]
- Peng, D.; Tarleton, R. EuPaGDT: a web tool tailored to design CRISPR guide RNAs for eukaryotic pathogens. Microb. Genom. 2015, 1, e000033. [Google Scholar] [CrossRef]
- Hoyer, C.; Zander, D.; Fleischer, S.; Schilhabel, M.; Kroener, M.; Platzer, M.; Clos, J. A Leishmania donovani gene that confers accelerated recovery from stationary phase growth arrest. Int. J. Parasitol. 2004, 34, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, D.J.; Ward, J.D.; Reiner, D.J.; Goldstein, B. Engineering the Caenorhabditis elegans genome using Cas9–triggered homologous recombination. Nat. Methods 2013, 10, 1028–1034. [Google Scholar] [CrossRef] [PubMed]
- Bottcher, R.; Hollmann, M.; Merk, K.; Nitschko, V.; Obermaier, C.; Philippou-Massier, J.; Wieland, I.; Gaul, U.; Forstemann, K. Efficient chromosomal gene modification with CRISPR/cas9 and PCR–based homologous recombination donors in cultured Drosophila cells. Nucleic Acids Res. 2014, 42, e89. [Google Scholar] [CrossRef]
- Hübel, A.; Krobitsch, S.; Horauf, A.; Clos, J. Leishmania major Hsp100 is required chiefly in the mammalian stage of the parasite. Mol. Cell Biol. 1997, 17, 5987–5995. [Google Scholar] [CrossRef]
- Krobitsch, S.; Clos, J. A novel role for 100 kD heat shock proteins in the parasite Leishmania donovani. Cell Stress Chaperones 1999, 4, 191–198. [Google Scholar] [CrossRef]
- Van Montfort, R.L.; Basha, E.; Friedrich, K.L.; Slingsby, C.; Vierling, E. Crystal structure and assembly of a eukaryotic small heat shock protein. Nat. Struct. Biol. 2001, 8, 1025–1030. [Google Scholar] [CrossRef]
- Nuhs, A.; Schafer, C.; Zander, D.; Trube, L.; Tejera Nevado, P.; Schmidt, S.; Arevalo, J.; Adaui, V.; Maes, L.; Dujardin, J.C.; et al. A novel marker, ARM58, confers antimony resistance to Leishmania spp. Int. J. Parasitol. Drugs Drug Resist. 2014, 4, 37–47. [Google Scholar] [CrossRef]
- Callahan, H.L.; Portal, I.F.; Bensinger, S.J.; Grogl, M. Leishmania spp: temperature sensitivity of promastigotes in vitro as a model for tropism in vivo. Exp. Parasitol. 1996, 84, 400–409. [Google Scholar] [CrossRef]
- Piper, P.W. The heat shock and ethanol stress responses of yeast exhibit extensive similarity and functional overlap. FEMS Microbiol. Lett. 1995, 134, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Barak, E.; Amin-Spector, S.; Gerliak, E.; Goyard, S.; Holland, N.; Zilberstein, D. Differentiation of Leishmania donovani in host–free system: analysis of signal perception and response. Mol. Biochem. Parasitol. 2005, 141, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Cupolillo, E.; Brahim, L.R.; Toaldo, C.B.; de Oliveira-Neto, M.P.; de Brito, M.E.; Falqueto, A.; de Farias Naiff, M.; Grimaldi, G., Jr. Genetic polymorphism and molecular epidemiology of Leishmania (Viannia) braziliensis from different hosts and geographic areas in Brazil. J. Clin. Microbiol. 2003, 41, 3126–3132. [Google Scholar] [CrossRef] [PubMed]
- Tobin, J.F.; Laban, A.; Wirth, D.F. Homologous recombination in Leishmania enriettii. Proc. Natl. Acad. Sci. USA 1991, 88, 864–868. [Google Scholar] [CrossRef]
- Beverley, S.M. Protozomics: trypanosomatid parasite genetics comes of age. Nat. Rev. Genet. 2003, 4, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Wiese, M. A mitogen–activated protein (MAP) kinase homologue of Leishmania mexicana is essential for parasite survival in the infected host. Embo. J. 1998, 17, 2619–2628. [Google Scholar] [CrossRef]
- Coelho, A.C.; Oliveira, J.C.; Espada, C.R.; Reimao, J.Q.; Trinconi, C.T.; Uliana, S.R. A Luciferase–Expressing Leishmania braziliensis Line That Leads to Sustained Skin Lesions in BALB/c Mice and Allows Monitoring of Miltefosine Treatment Outcome. PLoS Negl. Trop. Dis. 2016, 10, e0004660. [Google Scholar] [CrossRef]
- Bastos, M.S.; Souza, L.A.; Onofre, T.S.; Silva, A.J.; Almeida, M.R.; Bressan, G.C.; Fietto, J.L. Achievement of constitutive fluorescent pLEXSY–egfp Leishmania braziliensis and its application as an alternative method for drug screening in vitro. Mem. Inst. Oswaldo. Cruz. 2017, 112, 155–159. [Google Scholar] [CrossRef]
- Sharma, R.; Silveira-Mattos, P.S.; Ferreira, V.C.; Rangel, F.A.; Oliveira, L.B.; Celes, F.S.; Viana, S.M.; Wilson, M.E.; de Oliveira, C.I. Generation and Characterization of a Dual–Reporter Transgenic Leishmania braziliensis Line Expressing eGFP and Luciferase. Front. Cell. Infect. Microbiol. 2019, 9, 468. [Google Scholar] [CrossRef]
- Andrade, J.M.; Murta, S.M. Functional analysis of cytosolic tryparedoxin peroxidase in antimony–resistant and –susceptible Leishmania braziliensis and Leishmania infantum lines. Parasites Vectors 2014, 7, 406. [Google Scholar] [CrossRef]
- Andrade, J.M.; Baba, E.H.; Machado-de-Avila, R.A.; Chavez-Olortegui, C.; Demicheli, C.P.; Frezard, F.; Monte-Neto, R.L.; Murta, S.M. Silver and Nitrate Oppositely Modulate Antimony Susceptibility through Aquaglyceroporin 1 in Leishmania (Viannia) Species. Antimicrob. Agents Chemother 2016, 60, 4482–4489. [Google Scholar] [CrossRef] [PubMed]
- Moreira, D.S.; Xavier, M.V.; Murta, S.M.F. Ascorbate peroxidase overexpression protects Leishmania braziliensis against trivalent antimony effects. Mem. Inst. Oswaldo Cruz 2018, 113, e180377. [Google Scholar] [CrossRef] [PubMed]
- De Toledo, J.S.; Junqueira dos Santos, A.F.; Rodrigues de Moura, T.; Antoniazi, S.A.; Brodskyn, C.; Indiani de Oliveira, C.; Barral, A.; Cruz, A.K. Leishmania (Viannia) braziliensis transfectants overexpressing the miniexon gene lose virulence in vivo. Parasitol. Int. 2009, 58, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Smith, I.; Greenside, P.G.; Natoli, T.; Lahr, D.L.; Wadden, D.; Tirosh, I.; Narayan, R.; Root, D.E.; Golub, T.R.; Subramanian, A.; et al. Evaluation of RNAi and CRISPR technologies by large–scale gene expression profiling in the Connectivity Map. PLoS Biol. 2017, 15, e2003213. [Google Scholar] [CrossRef] [PubMed]
- Adaui, V.; Lye, L.F.; Akopyants, N.S.; Zimic, M.; Llanos-Cuentas, A.; Garcia, L.; Maes, I.; De Doncker, S.; Dobson, D.E.; Arevalo, J.; et al. Association of the Endobiont Double–Stranded RNA Virus LRV1 With Treatment Failure for Human Leishmaniasis Caused by Leishmania braziliensis in Peru and Bolivia. J. Infect. Dis. 2016, 213, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Bourreau, E.; Ginouves, M.; Prevot, G.; Hartley, M.A.; Gangneux, J.P.; Robert-Gangneux, F.; Dufour, J.; Sainte-Marie, D.; Bertolotti, A.; Pratlong, F.; et al. Presence of Leishmania RNA Virus 1 in Leishmania guyanensis Increases the Risk of First–Line Treatment Failure and Symptomatic Relapse. J. Infect. Dis. 2016, 213, 105–111. [Google Scholar] [CrossRef]
- Cantanhede, L.M.; Fernandes, F.G.; Ferreira, G.E.M.; Porrozzi, R.; Ferreira, R.G.M.; Cupolillo, E. New insights into the genetic diversity of Leishmania RNA Virus 1 and its species–specific relationship with Leishmania parasites. PloS ONE 2018, 13, e0198727. [Google Scholar] [CrossRef]
- Ives, A.; Ronet, C.; Prevel, F.; Ruzzante, G.; Fuertes-Marraco, S.; Schutz, F.; Zangger, H.; Revaz-Breton, M.; Lye, L.F.; Hickerson, S.M.; et al. Leishmania RNA virus controls the severity of mucocutaneous leishmaniasis. Science 2011, 331, 775–778. [Google Scholar] [CrossRef]
- Eren, R.O.; Reverte, M.; Rossi, M.; Hartley, M.A.; Castiglioni, P.; Prevel, F.; Martin, R.; Desponds, C.; Lye, L.F.; Drexler, S.K.; et al. Mammalian Innate Immune Response to a Leishmania–Resident RNA Virus Increases Macrophage Survival to Promote Parasite Persistence. Cell. Host Microbe. 2016, 20, 318–328. [Google Scholar] [CrossRef]
- Doench, J.G.; Hartenian, E.; Graham, D.B.; Tothova, Z.; Hegde, M.; Smith, I.; Sullender, M.; Ebert, B.L.; Xavier, R.J.; Root, D.E. Rational design of highly active sgRNAs for CRISPR–Cas9–mediated gene inactivation. Nat. Biotechnol. 2014, 32, 1262–1267. [Google Scholar] [CrossRef]
- Wong, N.; Liu, W.; Wang, X. WU–CRISPR: characteristics of functional guide RNAs for the CRISPR/Cas9 system. Genome Biol. 2015, 16, 218. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Xiao, T.; Chen, C.H.; Li, W.; Meyer, C.A.; Wu, Q.; Wu, D.; Cong, L.; Zhang, F.; Liu, J.S.; et al. Sequence determinants of improved CRISPR sgRNA design. Genome Res. 2015, 25, 1147–1157. [Google Scholar] [CrossRef] [PubMed]
- Labuhn, M.; Adams, F.F.; Ng, M.; Knoess, S.; Schambach, A.; Charpentier, E.M.; Schwarzer, A.; Mateo, J.L.; Klusmann, J.H.; Heckl, D. Refined sgRNA efficacy prediction improves large– and small–scale CRISPR–Cas9 applications. Nucleic Acids Res. 2018, 46, 1375–1385. [Google Scholar] [CrossRef] [PubMed]
- Graf, R.; Li, X.; Chu, V.T.; Rajewsky, K. sgRNA Sequence Motifs Blocking Efficient CRISPR/Cas9–Mediated Gene Editing. Cell Rep. 2019, 26, 1098–1103.e3. [Google Scholar] [CrossRef]
- Yuen, G.; Khan, F.J.; Gao, S.; Stommel, J.M.; Batchelor, E.; Wu, X.; Luo, J. CRISPR/Cas9–mediated gene knockout is insensitive to target copy number but is dependent on guide RNA potency and Cas9/sgRNA threshold expression level. Nucleic Acids Res. 2017, 45, 12039–12053. [Google Scholar] [CrossRef]
- Ng, H.; Dean, N. Dramatic Improvement of CRISPR/Cas9 Editing in Candida albicans by Increased Single Guide RNA Expression. mSphere 2017, 2. [Google Scholar] [CrossRef]
- Jara, M.; Maes, I.; Imamura, H.; Domagalska, M.A.; Dujardin, J.C.; Arevalo, J. Tracking of quiescence in Leishmania by quantifying the expression of GFP in the ribosomal DNA locus. Sci. Rep. 2019, 9, 18951. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adaui, V.; Kröber-Boncardo, C.; Brinker, C.; Zirpel, H.; Sellau, J.; Arévalo, J.; Dujardin, J.-C.; Clos, J. Application of CRISPR/Cas9-Based Reverse Genetics in Leishmania braziliensis: Conserved Roles for HSP100 and HSP23. Genes 2020, 11, 1159. https://doi.org/10.3390/genes11101159
Adaui V, Kröber-Boncardo C, Brinker C, Zirpel H, Sellau J, Arévalo J, Dujardin J-C, Clos J. Application of CRISPR/Cas9-Based Reverse Genetics in Leishmania braziliensis: Conserved Roles for HSP100 and HSP23. Genes. 2020; 11(10):1159. https://doi.org/10.3390/genes11101159
Chicago/Turabian StyleAdaui, Vanessa, Constanze Kröber-Boncardo, Christine Brinker, Henner Zirpel, Julie Sellau, Jorge Arévalo, Jean-Claude Dujardin, and Joachim Clos. 2020. "Application of CRISPR/Cas9-Based Reverse Genetics in Leishmania braziliensis: Conserved Roles for HSP100 and HSP23" Genes 11, no. 10: 1159. https://doi.org/10.3390/genes11101159
APA StyleAdaui, V., Kröber-Boncardo, C., Brinker, C., Zirpel, H., Sellau, J., Arévalo, J., Dujardin, J.-C., & Clos, J. (2020). Application of CRISPR/Cas9-Based Reverse Genetics in Leishmania braziliensis: Conserved Roles for HSP100 and HSP23. Genes, 11(10), 1159. https://doi.org/10.3390/genes11101159