Multiple Isolated Transcription Factors Act as Switches and Contribute to Species Uniqueness

Abstract
1. Introduction
2. Materials and Methods
2.1. Mammalian TF Database Construction
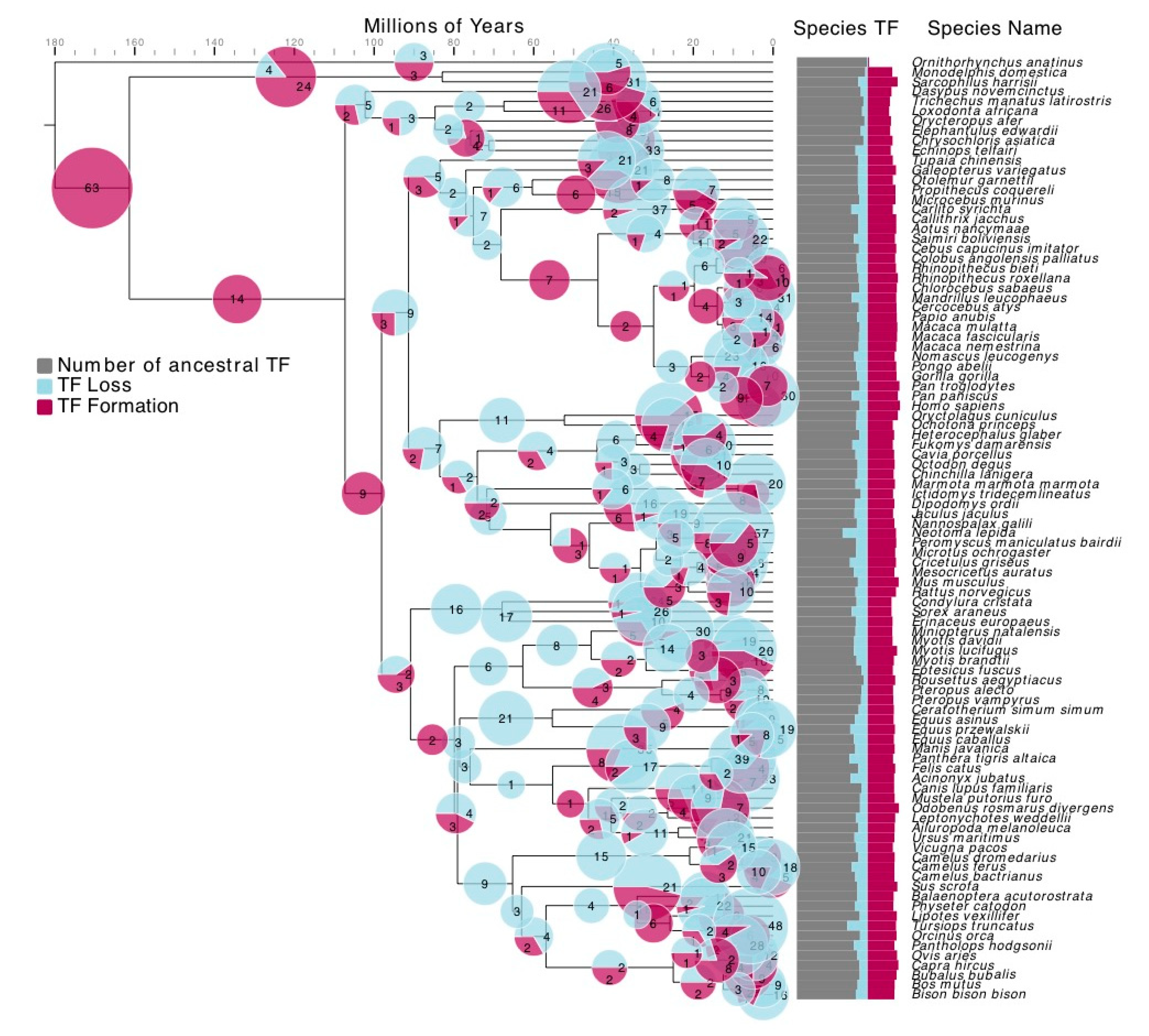
2.2. TF Formation and Loss Detection
2.3. Construction of Protein–Protein Interaction (PPI) Networks
2.4. Functional Cartography of the Human PPI Network
2.5. Negative Binomial Regression Analysis of the Effect of TF Membership Variation on Gene Expression
2.6. TF-GO Bipartite Graphs for Humans and Mice
2.7. Data Availability
3. Results
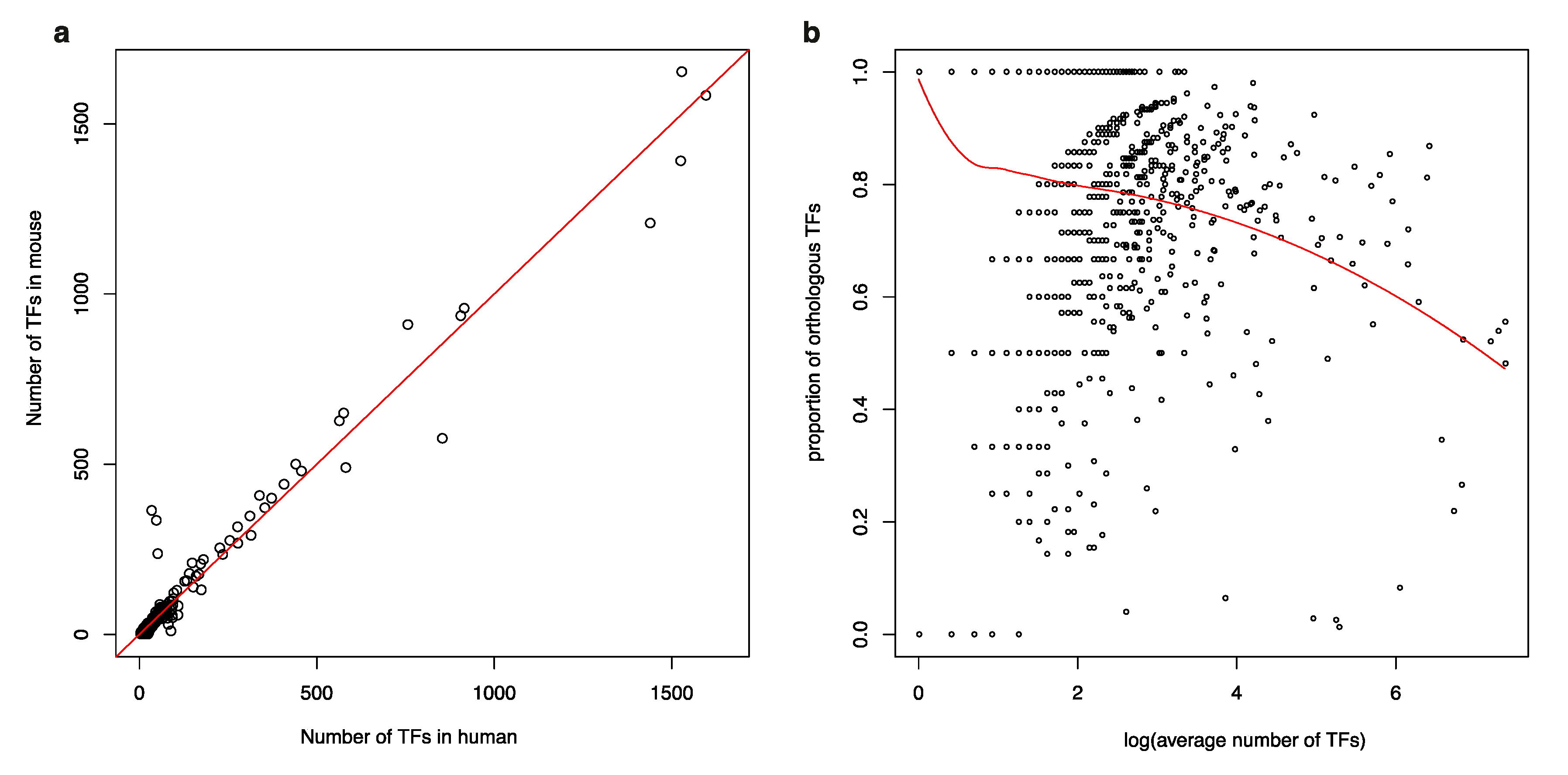
3.1. Isolated TFs in a Human TF-to-TF Network Often Have No Orthologs in Mouse
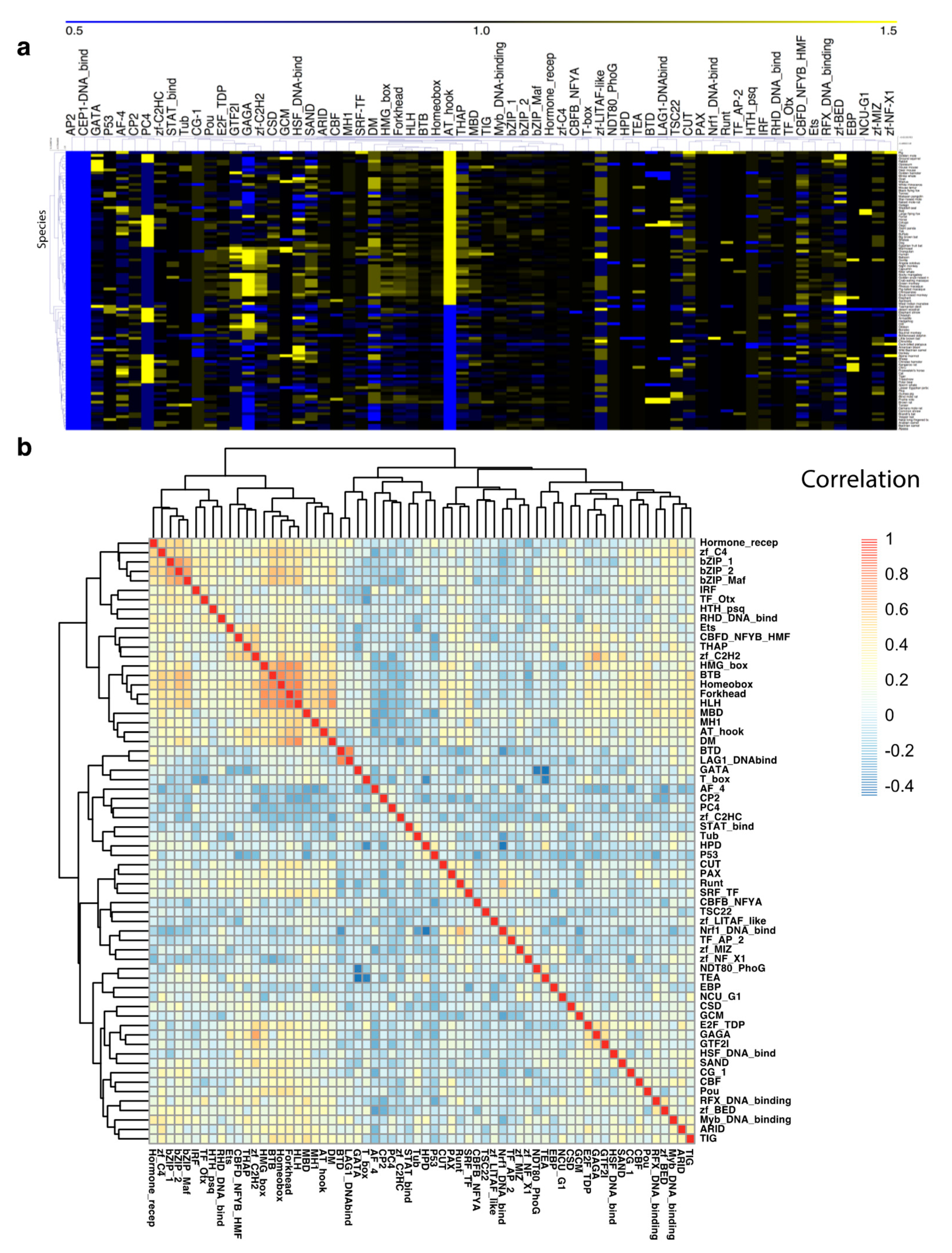
3.2. TF Families Vary Greatly in Scale among Mammalian Species
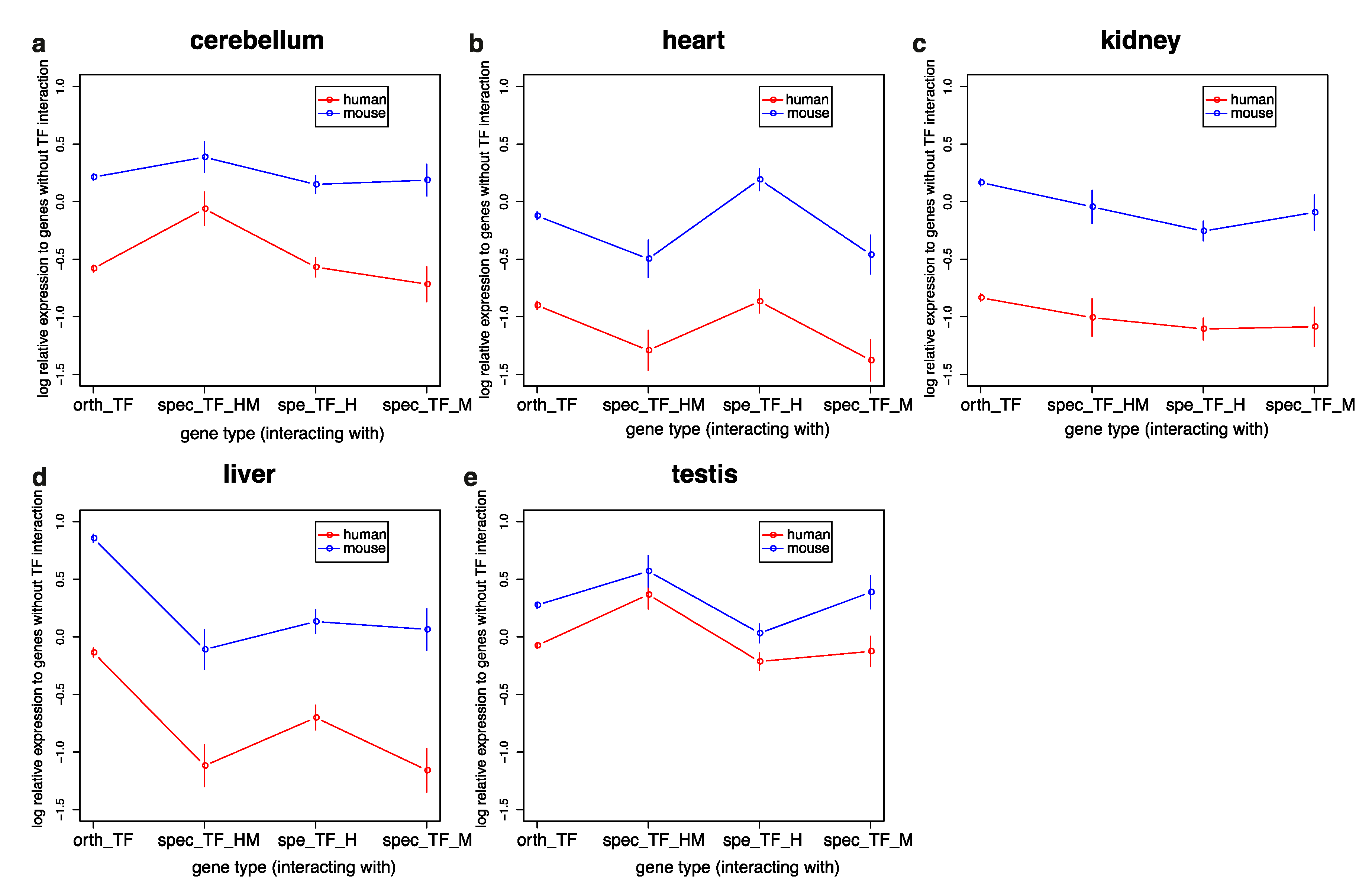
3.3. Genes Interacting with TFs in Humans and Mice Have Similar Expression Profiles but Are More Highly Expressed in Mice
3.4. Loss of Human TFs in Mice Reveals Knockout-Phenotypes of Their Targets in Humans
3.5. Human and Mouse Biological Functions Are Regulated by Similar Numbers of TFs but Different TF Family Members
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Erwin, D.H.; Davidson, E.H. The evolution of hierarchical gene regulatory networks. Nat. Rev. Genet. 2009, 10, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Britten, R.J.; Davidson, E.H. Gene regulation for higher cells: A theory. Science 1969, 165, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Stern, D.L. Evolutionary developmental biology and the problem of variation. Evolution 2000, 54, 1079–1091. [Google Scholar] [CrossRef] [PubMed]
- Wray, G.A. The evolutionary significance of cis-regulatory mutations. Nat. Rev. Genet. 2007, 8, 206–216. [Google Scholar] [CrossRef]
- Stergachis, A.B.; Neph, S.; Sandstrom, R.; Haugen, E.; Reynolds, A.P.; Zhang, M.H.; Byron, R.; Canfield, T.; Stelhing-Sun, S.; Lee, K.; et al. Conservation of trans-acting circuitry during mammalian regulatory evolution. Nature 2014, 515, 365. [Google Scholar] [CrossRef]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The human transcription factors. Cell 2018, 175, 598–599. [Google Scholar] [CrossRef]
- Krumlauf, R. Hox Genes in Vertebrate Development. Cell 1994, 78, 191–201. [Google Scholar] [CrossRef]
- Jager, R.J.; Anvret, M.; Hall, K.; Scherer, G. A Human Xy Female with a Frame Shift Mutation in the Candidate Testis-Determining Gene Sry. Nature 1990, 348, 452–454. [Google Scholar] [CrossRef]
- Shannon, M.; Hamilton, A.T.; Gordon, L.; Branscomb, E.; Stubbs, L. Differential expansion of zinc-finger transcription factor loci in homologous human and mouse gene clusters. Genome Res. 2003, 13, 1097–1110. [Google Scholar] [CrossRef]
- Liu, H.; Chang, L.H.; Sun, Y.; Lu, X.; Stubbs, L. Deep vertebrate roots for mammalian zinc finger transcription factor subfamilies. Genome Biol. Evol. 2014, 6, 510–525. [Google Scholar] [CrossRef]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef] [PubMed]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2018. [Google Scholar] [CrossRef]
- Zhang, H.M.; Liu, T.; Liu, C.J.; Song, S.Y.; Zhang, X.T.; Liu, W.; Jia, H.B.; Xue, Y.; Guo, A.Y. AnimalTFDB 2.0: A resource for expression, prediction and functional study of animal transcription factors. Nucleic Acids Res. 2015, 43, D76–D81. [Google Scholar] [CrossRef] [PubMed]
- Kanamori, M.; Konno, H.; Osato, N.; Kawai, J.; Hayashizaki, Y.; Suzuki, H. A genome-wide and nonredundant mouse transcription factor database. Biochem. Biophys. Res. Commun. 2004, 322, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Pfreundt, U.; James, D.P.; Tweedie, S.; Wilson, D.; Teichmann, S.A.; Adryan, B. FlyTF: Improved annotation and enhanced functionality of the Drosophila transcription factor database. Nucleic Acids Res. 2010, 38, D443–D447. [Google Scholar] [CrossRef]
- Fulton, D.L.; Sundararajan, S.; Badis, G.; Hughes, T.R.; Wasserman, W.W.; Roach, J.C.; Sladek, R. TFCat: The curated catalog of mouse and human transcription factors. Genome Biol. 2009, 10, R29. [Google Scholar] [CrossRef]
- Lee, A.P.; Yang, Y.; Brenner, S.; Venkatesh, B. TFCONES: A database of vertebrate transcription factor-encoding genes and their associated conserved noncoding elements. BMC Genom. 2007, 8, 441. [Google Scholar] [CrossRef]
- Zheng, G.Y.; Tu, K.; Yang, Q.; Xiong, Y.; Wei, C.C.; Xie, L.; Zhu, Y.Y.; Li, Y.X. ITFP: An integrated platform of mammalian transcription factors. Bioinformatics 2008, 24, 2416–2417. [Google Scholar] [CrossRef]
- Bork, P.; Jensen, L.J.; von Mering, C.; Ramani, A.K.; Lee, I.; Marcotte, E.M. Protein interaction networks from yeast to human. Curr. Opin. Struct. Biol. 2004, 14, 292–299. [Google Scholar] [CrossRef]
- Rual, J.F.; Venkatesan, K.; Hao, T.; Hirozane-Kishikawa, T.; Dricot, A.; Li, N.; Berriz, G.F.; Gibbons, F.D.; Dreze, M.; Ayivi-Guedehoussou, N.; et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature 2005, 437, 1173–1178. [Google Scholar] [CrossRef]
- Nowick, K.; Stubbs, L. Lineage-specific transcription factors and the evolution of gene regulatory networks. Brief. Funct. Genom. 2010, 9, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Miao, Y.R.; Jia, L.H.; Yu, Q.Y.; Zhang, Q.; Guo, A.Y. AnimalTFDB 3.0: A comprehensive resource for annotation and prediction of animal transcription factors. Nucleic Acids Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Agarwala, R.; Barrett, T.; Beck, J.; Benson, D.A.; Bollin, C.; Bolton, E.; Bourexis, D.; Brister, J.R.; Bryant, S.H.; Lanese, K.; et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2016, 44, D7–D19. [Google Scholar]
- Yamada, K.D.; Tomii, K.; Katoh, K. Application of the MAFFT sequence alignment program to large data-reexamination of the usefulness of chained guide trees. Bioinformatics 2016, 32, 3246–3251. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.H. DAMBE6: New tools for microbial genomics, phylogenetics, and molecular evolution. J. Hered. 2017, 108, 431–437. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. TimeTree: A resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef]
- Darby, C.A.; Stolzer, M.; Ropp, P.J.; Barker, D.; Durand, D. Xenolog classification. Bioinformatics 2017, 33, 640–649. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef]
- Guimera, R.; Nunes Amaral, L.A. Functional cartography of complex metabolic networks. Nature 2005, 433, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Brawand, D.; Soumillon, M.; Necsulea, A.; Julien, P.; Csardi, G.; Harrigan, P.; Weier, M.; Liechti, A.; Aximu-Petri, A.; Kircher, M.; et al. The evolution of gene expression levels in mammalian organs. Nature 2011, 478, 343–348. [Google Scholar] [CrossRef]
- Petryszak, R.; Keays, M.; Tang, Y.A.; Fonseca, N.A.; Barrera, E.; Burdett, T.; Fullgrabe, A.; Fuentes, A.M.; Jupp, S.; Koskinen, S.; et al. Expression Atlas update--an integrated database of gene and protein expression in humans, animals and plants. Nucleic Acids Res. 2016, 44, D746–752. [Google Scholar] [CrossRef] [PubMed]
- Venables, W.N.; Ripley, B.D.; Venables, W.N. Modern Applied Statistics with S, 4th ed.; Springer: New York, NY, USA, 2002; p. xi. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2014. [Google Scholar]
- Carbon, S.; Dietze, H.; Lewis, S.E.; Mungall, C.J.; Munoz-Torres, M.C.; Basu, S.; Chisholm, R.L.; Dodson, R.J.; Fey, P.; Thomas, P.D.; et al. Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res. 2017, 45, D331–D338. [Google Scholar]
- Reimand, J.; Arak, T.; Adler, P.; Kolberg, L.; Reisberg, S.; Peterson, H.; Vilo, J. g:Profiler-a web server for functional interpretation of gene lists (2016 update). Nucleic Acids Res. 2016, 44, W83–W89. [Google Scholar] [CrossRef] [PubMed]
- Cleveland, W.S. Robust locally weighted regression and smoothing scatterplots. J. Am. Stat. Assoc. 1979, 74, 829–836. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Smith, C.L.; Blake, J.A.; Kadin, J.A.; Richardson, J.E.; Bult, C.J. Mouse Genome Database Group Mouse Genome Database (MGD)-2018: Knowledgebase for the laboratory mouse. Nucleic Acids Res. 2018, 46, D836–D842. [Google Scholar] [CrossRef]
- Shu, J.; Zhang, K.; Zhang, M.; Yao, A.; Shao, S.; Du, F.; Yang, C.; Chen, W.; Wu, C.; Yang, W.; et al. GATA family members as inducers for cellular reprogramming to pluripotency. Cell Res. 2015, 25, 169. [Google Scholar] [CrossRef]
- Golson, M.L.; Kaestner, K.H. Fox transcription factors: From development to disease. Development 2016, 143, 4558–4570. [Google Scholar] [CrossRef]
- Lupo, A.; Cesaro, E.; Montano, G.; Zurlo, D.; Izzo, P.; Costanzo, P. KRAB-zinc finger proteins: A repressor family displaying multiple biological functions. Curr. Genom. 2013, 14, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Cho, J.W.; Lee, S.; Yun, A.; Kim, H.; Bae, D.; Yang, S.; Kim, C.Y.; Lee, M.; Kim, E.; et al. TRRUST v2: An expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018, 46, D380–D386. [Google Scholar] [CrossRef] [PubMed]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards suite: From gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinform. 2016, 54, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Huang, X.; Muruganujan, A.; Tang, H.; Mills, C.; Kang, D.; Thomas, P.D. PANTHER version 11: Expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017, 45, D183–D189. [Google Scholar] [CrossRef] [PubMed]
- Barrera, L.A.; Vedenko, A.; Kurland, J.V.; Rogers, J.M.; Gisselbrecht, S.S.; Rossin, E.J.; Woodard, J.; Mariani, L.; Kock, K.H.; Inukai, S.; et al. Survey of variation in human transcription factors reveals prevalent DNA binding changes. Science 2016, 351, 1450–1454. [Google Scholar] [CrossRef]
- Lambert, S.A.; Yang, A.W.H.; Sasse, A.; Cowley, G.; Albu, M.; Caddick, M.X.; Morris, Q.D.; Weirauch, M.T.; Hughes, T.R. Similarity regression predicts evolution of transcription factor sequence specificity. Nat. Genet. 2019. [Google Scholar] [CrossRef]
- Wagner, G.P.; Lynch, V.J. The gene regulatory logic of transcription factor evolution. Trends Ecol. Evol. 2008, 23, 377–385. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TF Type | TF Number | TFs with Lethal Phenotype | TFs Absent in Mouse | Degrees |
|---|---|---|---|---|
| large component TFs | 1040 | 507 (48.8%) | 40 (3.8%) | 77.9 ± 127.8 |
| Isolated TFs | 515 | 26 (5.0%) | 189 (36.7%) | 10.6 ± 8.8 |
| Organ | Mouse Normal Phenotype | Gene with KRAB-C2H2 Interactions | Mammalian Phenotype | Mouse Knock-Out Phenotype | Human (Monkey) Normal Phenotype |
|---|---|---|---|---|---|
| Tail | Horizontal tail | CACNA1B | MP:0003382 | Straub tail | (Vertical tail) |
| Long tail | RIPK4 | MP:0000592 | Short tail | Without tail | |
| Tooth | Continually growing teeth | OTUD7A | MP:0003053 | Delayed tooth eruption | Permanent tooth |
| Hair | Fur-covered | CTSL2 | MP:0000414 | Alopecia | Hairless |
| CTSL2 | MP:0000417 | Short hair | |||
| Skin | Epidermis < 25 μm | CTSL2 | MP:0001219 | Thick epidermis | Epidermis > 50 μm |
| Normal dermis | CTSL2 | MP:0001245 | Thick dermal layer | Thicker dermis than mouse |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, X.-W.; Kishino, H. Multiple Isolated Transcription Factors Act as Switches and Contribute to Species Uniqueness. Genes 2020, 11, 1148. https://doi.org/10.3390/genes11101148
Zhao X-W, Kishino H. Multiple Isolated Transcription Factors Act as Switches and Contribute to Species Uniqueness. Genes. 2020; 11(10):1148. https://doi.org/10.3390/genes11101148
Chicago/Turabian StyleZhao, Xin-Wei, and Hirohisa Kishino. 2020. "Multiple Isolated Transcription Factors Act as Switches and Contribute to Species Uniqueness" Genes 11, no. 10: 1148. https://doi.org/10.3390/genes11101148
APA StyleZhao, X.-W., & Kishino, H. (2020). Multiple Isolated Transcription Factors Act as Switches and Contribute to Species Uniqueness. Genes, 11(10), 1148. https://doi.org/10.3390/genes11101148