Deleterious AGXT Missense Variant Associated with Type 1 Primary Hyperoxaluria (PH1) in Zwartbles Sheep

 , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animals and DNA Samples
2.3. Histopathology
2.4. Whole-Genome Sequencing
2.5. Candidate Variant Validation
2.6. Protein Predictions
2.7. Availability of Data and Material
3. Results
3.1. Clinical Findings
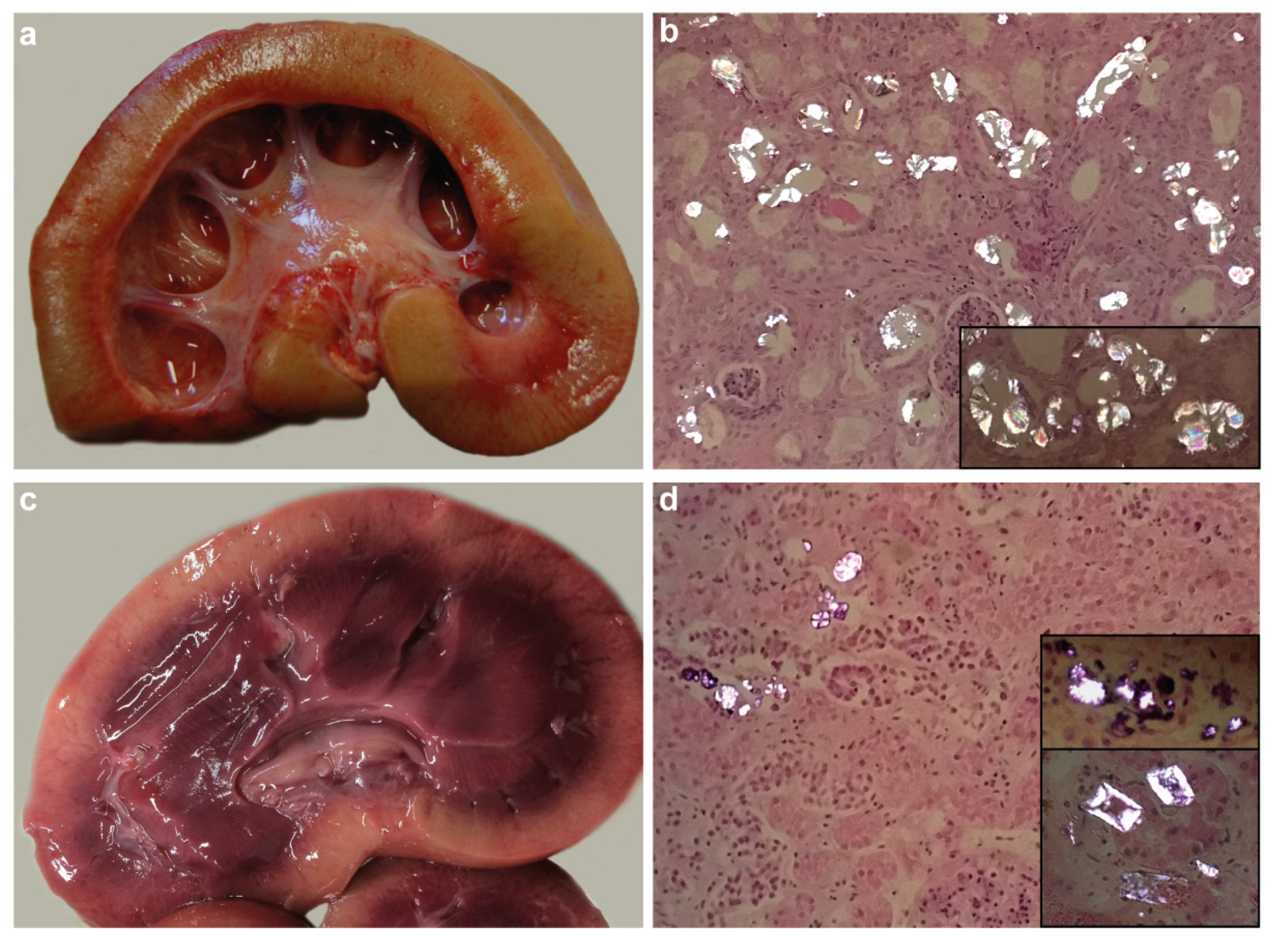
3.2. Macroscopic and Histopathological Findings
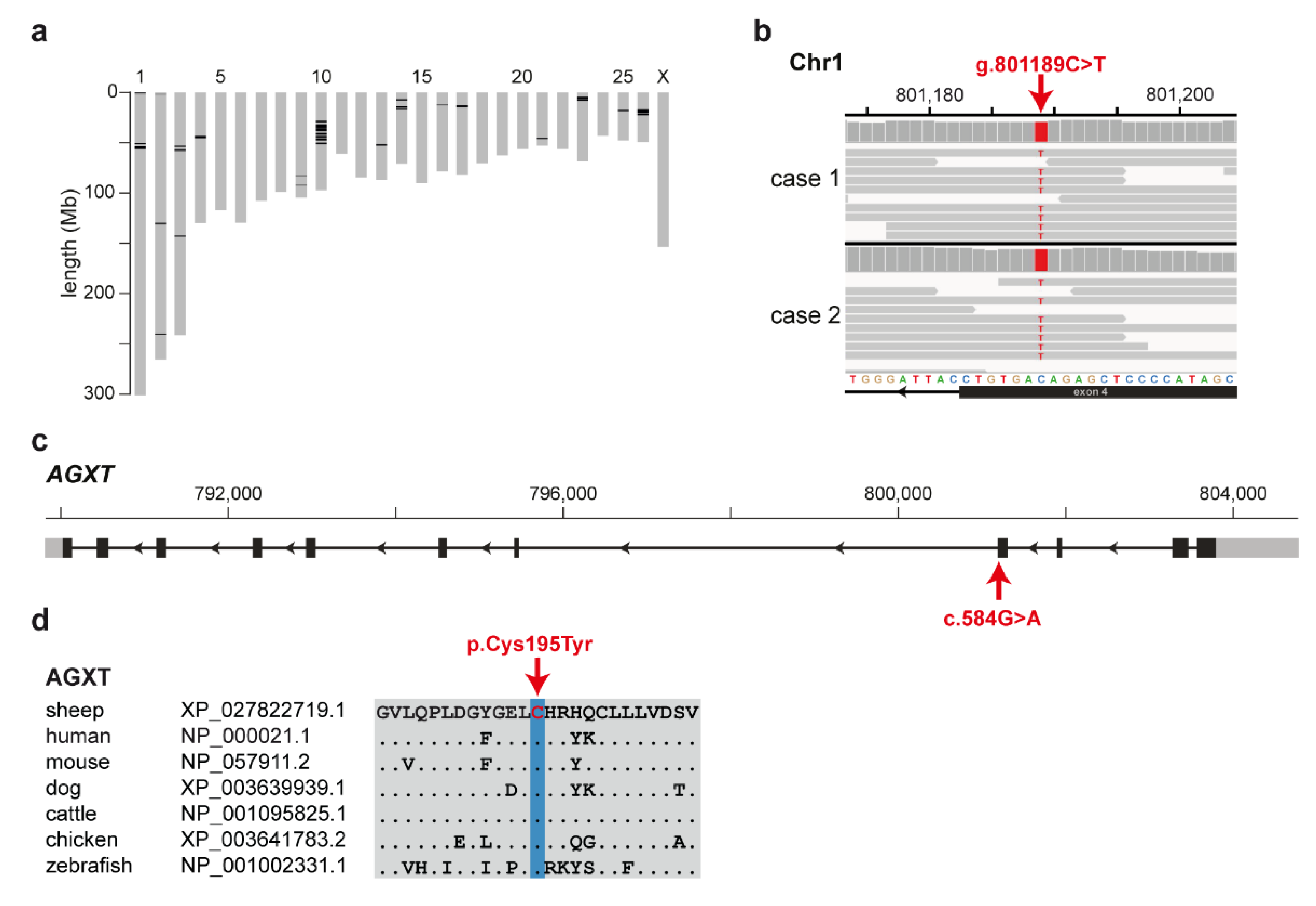
3.3. Identification of the Causative Variant
3.4. Targeted Genotyping
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cochat, P.; Rumsby, G. Primary hyperoxaluria. N. Engl. J. Med. 2013, 369, 649–658. [Google Scholar] [CrossRef]
- Vidgren, G.; Vainio-Siukola, K.; Honkasalo, S.; Dillard, K.; Anttila, M.; Vauhkonen, H. Primary hyperoxaluria in Coton de Tulear. Anim. Genet. 2012, 43, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, R.E.; Narala, S.; Sabet, N.; Goldstein, O.; McDonough, S.P. Primary hyperoxaluria in cats is caused by a mutation in the feline GRHPR gene. J. Hered. 2009, 100, S2–S7. [Google Scholar] [CrossRef]
- Rhyan, J.C.; Sartin, E.A.; Powers, R.D.; Wolfe, D.F.; Dowling, P.M.; Spano, J.S. Severe renal oxalosis in five young Beefmaster calves. J. Am. Vet. Med. Assoc. 1992, 201, 1907–1910. [Google Scholar]
- Yavuz Gülbahar, M.; Kaya, A.; Gölen, Ý. Renal oxalosis in a calf. Turkish J. Vet. Anim. Sci. 2002, 26, 1197–1200. [Google Scholar]
- Strugnell, B.W.; Gaudie, C.M.; Wessels, M.; Schock, A.; Davies, I. Sheep: Severe oxalate nephropathy in zwartbles sheep. Vet. Rec. 2011, 169, 81. [Google Scholar] [CrossRef] [PubMed]
- Barley, J.; Hanna, R.; McConnell, S. Oxalate nephrosis in Zwartble sheep. Vet. Irel. J. 2015, 5, 46–48. [Google Scholar]
- Paris, J.M.; Letko, A.; Häfliger, I.M.; Ammann, P.; Flury, C.; Drögemüller, C. Identification of two TYRP1 loss-of-function alleles in Valais Red sheep. Anim. Genet. 2019, 50, 778–782. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.A.M.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 1–16. [Google Scholar] [CrossRef]
- Ceballos, F.C.; Joshi, P.K.; Clark, D.W.; Ramsay, M.; Wilson, J.F. Runs of homozygosity: Windows into population history and trait architecture. Nat. Rev. Genet. 2018, 19, 220–234. [Google Scholar] [CrossRef]
- Naval-Sanchez, M.; Nguyen, Q.; McWilliam, S.; Porto-Neto, L.R.; Tellam, R.; Vuocolo, T.; Reverter, A.; Perez-Enciso, M.; Brauning, R.; Clarke, S.; et al. Sheep genome functional annotation reveals proximal regulatory elements contributed to the evolution of modern breeds. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [PubMed]
- Pejaver, V.; Urresti, J.; Lugo-Martinez, J.; Pagel, K.A.; Lin, G.N.; Nam, H.-J.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M.; et al. MutPred2: Inferring the molecular and phenotypic impact of amino acid variants. BioRxiv 2017, 134981. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. BioRxiv 2019. [Google Scholar] [CrossRef]
- McIntosh, G.H.; Belling, G.B. An isotopic study of oxalate excretion in sheep. Aust. J. Exp. Biol. Med. Sci. 1975, 53, 479–487. [Google Scholar] [CrossRef]
- Williams, E.L.; Acquaviva, C.; Amoroso, A.; Chevalier, F.; Coulter-Mackie, M.; Monico, C.G.; Giachino, D.; Owen, T.; Robbiano, A.; Salido, E.; et al. Primary hyperoxaluria type 1: Update and additional mutation analysis of the AGXT gene. Hum. Mutat. 2009, 30, 910–917. [Google Scholar] [CrossRef]
- Williams, E.; Rumsby, G. Selected exonic sequencing of the AGXT gene provides a genetic diagnosis in 50% of patients with primary hyperoxaluria type I. Clin. Chem. 2007, 53, 1216–1221. [Google Scholar] [CrossRef]
- Lu, X.; Chen, W.; Li, L.; Zhu, X.; Huang, C.; Liu, S.; Yang, Y.; Zhao, Y. Two novel AGXT mutations cause the infantile form of primary hyperoxaluria type I in a Chinese family: Research on missed mutation. Front. Pharmacol. 2019, 10, 1–9. [Google Scholar] [CrossRef]
- Cochat, P.; Groothoff, J. Primary hyperoxaluria type 1: Practical and ethical issues. Pediatr. Nephrol. 2013, 28, 2273–2281. [Google Scholar] [CrossRef] [PubMed]
- Salido, E.C.; Li, X.M.; Lu, Y.; Wang, X.; Santana, A.; Roy-Chowdhury, N.; Torres, A.; Shapiro, L.J.; Roy-Chowdhury, J. Alanine-glyoxylate aminotransferase-deficient mice, a model for primary hyperoxaluria that responds to adenoviral gene transfer. Proc. Natl. Acad. Sci. USA 2006, 103, 18249–18254. [Google Scholar] [CrossRef] [PubMed]
- Salido, E.; Pey, A.L.; Rodriguez, R.; Lorenzo, V. Primary hyperoxalurias: Disorders of glyoxylate detoxification. Biochim. Biophys. Acta Mol. Basis Dis. 2012, 1822, 1453–1464. [Google Scholar] [CrossRef] [PubMed]
- Van Woerden, C.S.; Groothoff, J.W.; Wijburg, F.A.; Annink, C.; Wanders, R.J.A.; Waterham, H.R. Clinical implications of mutation analysis in primary hyperoxaluria type 1. Kidney Int. 2004, 66, 746–752. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Variant Position 1 | Gene | Protein Change | Allele Frequency 2 | PROVEAN Score 3 | MutPred2 Score 4 |
|---|---|---|---|---|---|
| chr1:652874 | SNED1 | p.Glu747Lys | 0 | −1.279 | 0.628 |
| chr1:801189 | AGXT | p.Cys195Tyr | 0 | −9.768 | 0.891 |
| chr1:54671486 | ERICH3 | p.Gly23Glu | 0.0099 | −4.526 | 0.852 |
| chr10:36256345 | SPATA13 | p.Asp1073= | 0.0036 | NA | NA |
| chr10:38336210 | MPHOSPH8 | p.Arg426Gln | 0 | −0.638 | 0.085 |
| chr14:14488283 | ZNF469 | p.Glu2351Lys | 0 | −1.756 | 0.271 |
| chr17:13778922 | ZNF827 | p.Asn694Ser | 0 | −0.462 | 0.098 |
| chr26:16667329 | CCDC110 | p.Asn649= | 0 | NA | NA |
| chr26:16820162 | SORBS2 | p.Pro394Leu | 0 | −0.301 | 0.121 |
| chr26:17153454 | TLR3 | p.Leu244= | 0 | NA | NA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Letko, A.; Dijkman, R.; Strugnell, B.; Häfliger, I.M.; Paris, J.M.; Henderson, K.; Geraghty, T.; Orr, H.; Scholes, S.; Drögemüller, C. Deleterious AGXT Missense Variant Associated with Type 1 Primary Hyperoxaluria (PH1) in Zwartbles Sheep. Genes 2020, 11, 1147. https://doi.org/10.3390/genes11101147
Letko A, Dijkman R, Strugnell B, Häfliger IM, Paris JM, Henderson K, Geraghty T, Orr H, Scholes S, Drögemüller C. Deleterious AGXT Missense Variant Associated with Type 1 Primary Hyperoxaluria (PH1) in Zwartbles Sheep. Genes. 2020; 11(10):1147. https://doi.org/10.3390/genes11101147
Chicago/Turabian StyleLetko, Anna, Reinie Dijkman, Ben Strugnell, Irene M. Häfliger, Julia M. Paris, Katrina Henderson, Tim Geraghty, Hannah Orr, Sandra Scholes, and Cord Drögemüller. 2020. "Deleterious AGXT Missense Variant Associated with Type 1 Primary Hyperoxaluria (PH1) in Zwartbles Sheep" Genes 11, no. 10: 1147. https://doi.org/10.3390/genes11101147
APA StyleLetko, A., Dijkman, R., Strugnell, B., Häfliger, I. M., Paris, J. M., Henderson, K., Geraghty, T., Orr, H., Scholes, S., & Drögemüller, C. (2020). Deleterious AGXT Missense Variant Associated with Type 1 Primary Hyperoxaluria (PH1) in Zwartbles Sheep. Genes, 11(10), 1147. https://doi.org/10.3390/genes11101147