Dynamics of a Protein Interaction Network Associated to the Aggregation of polyQ-Expanded Ataxin-1

 ,
,  ,
,  and
and
Abstract
1. Introduction
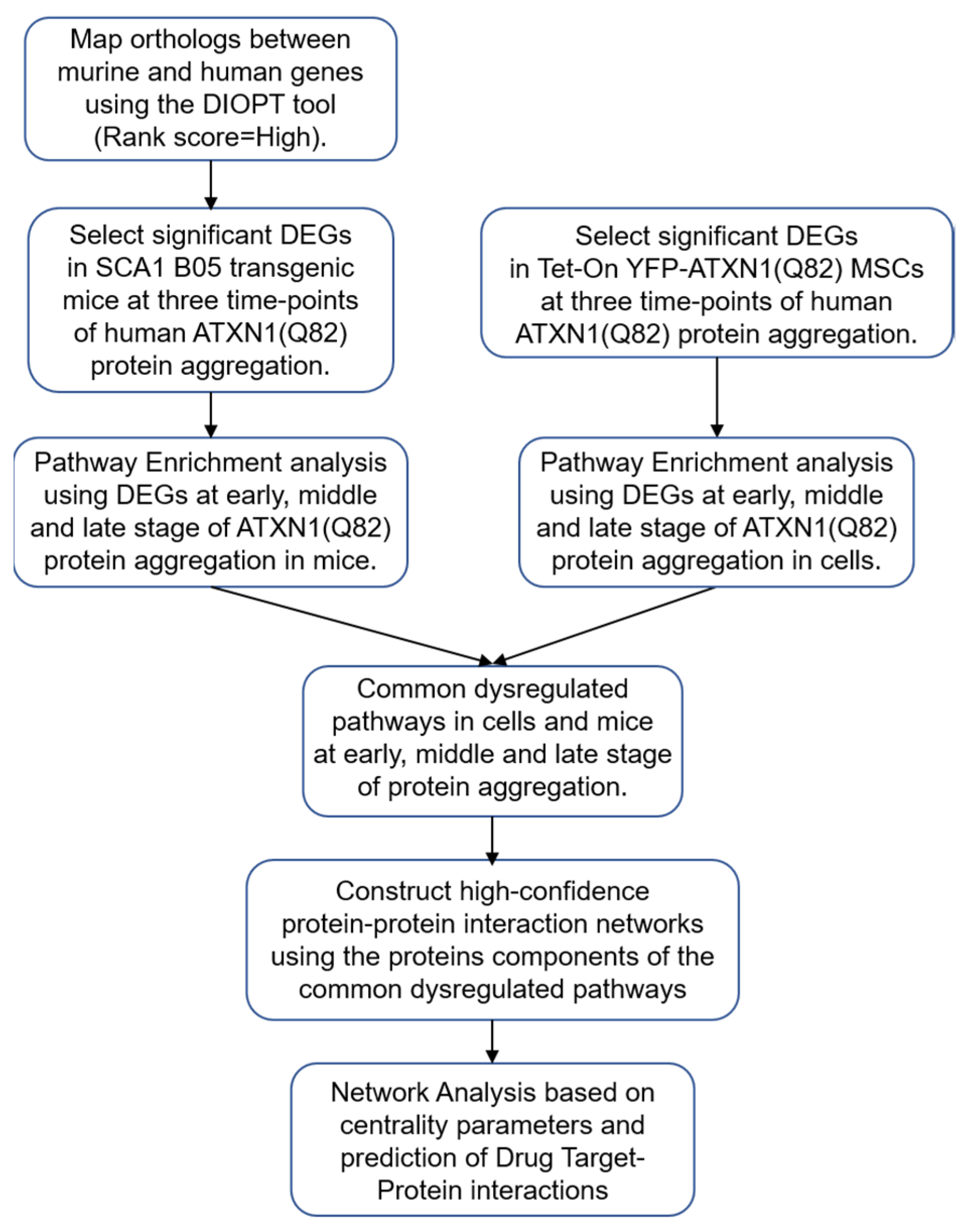
2. Materials and Methods
2.1. Datasets of SCA1 B05 Transgenic Mice and Tet-On YFP-ATXN1(Q82) MSCs
2.2. Differential Gene Expression Analysis
2.3. Pathway Enrichment Analysis
2.4. Construction of PPI Networks
2.5. Selection of Genes Analysis
2.6. Network Analysis
2.7. Drug-Protein Interaction Network
3. Results
3.1. Identification of Dysregulated Pathways in Tet-On YFP-ATXN1(Q82) MSCs and SCA1 B05 Transgenic Mice
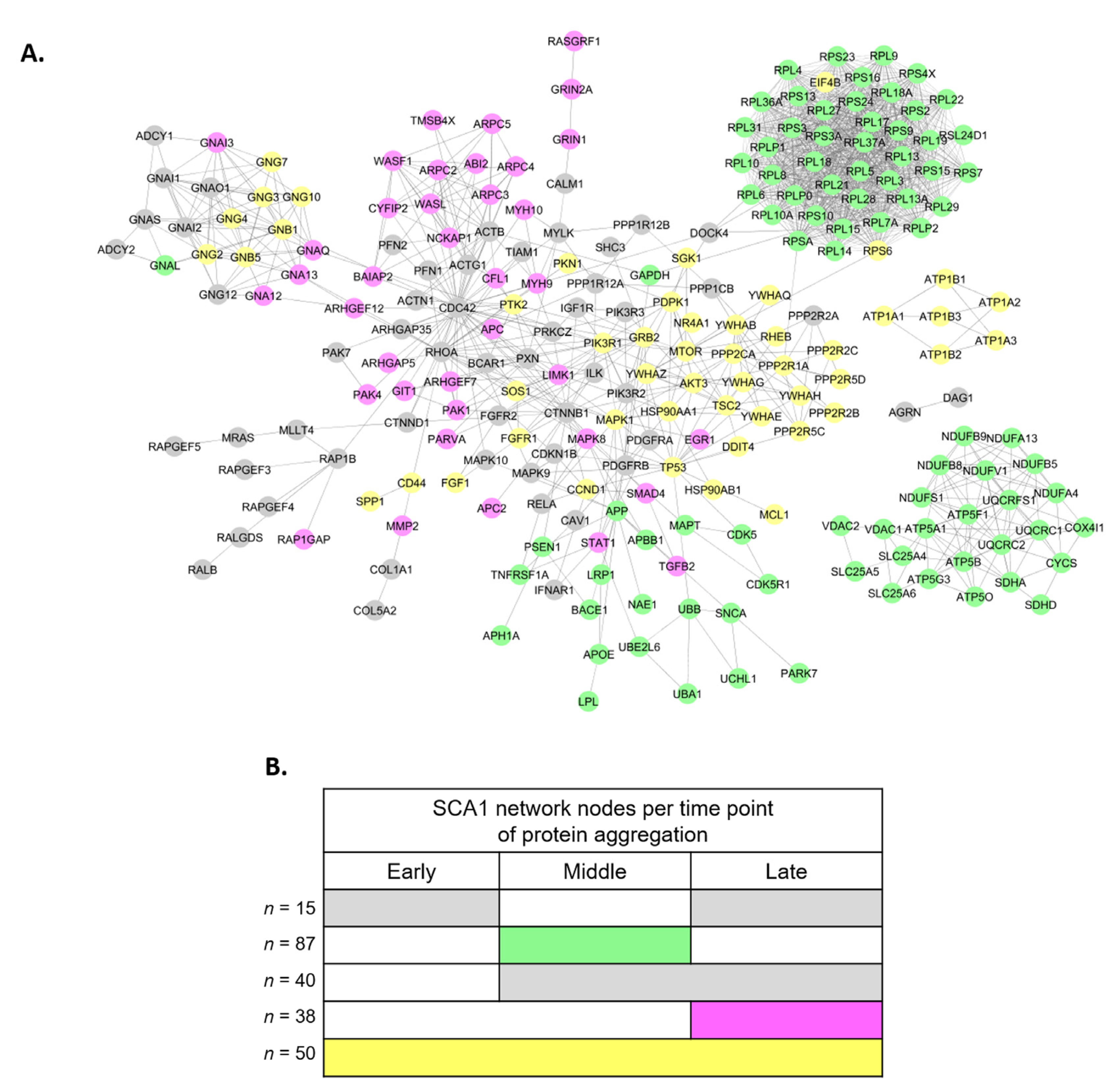
3.2. Perturbed PPI Networks in SCA1 Models
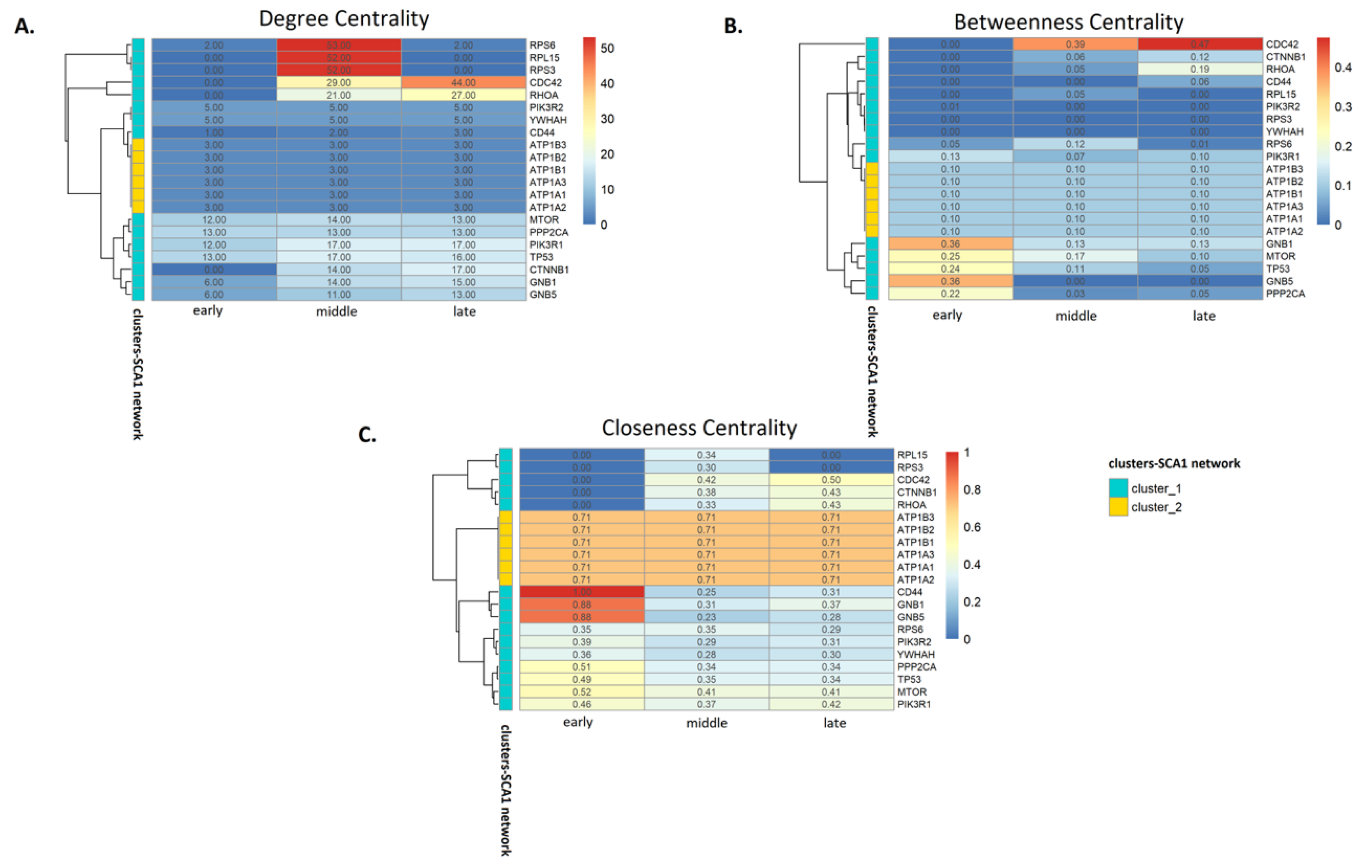
3.3. Analysis of SCA1 PPI Networks
3.4. Drug-Protein Interaction Network in SCA1
4. Discussion
4.1. Dysregulated Pathways Associated to polyQ-Expanded ATXN1 Aggregation
4.2. Network Analysis Indicates Critical Protein Nodes for SCA1 Pathogenesis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Banfi, S.; Chung, M.Y.; Kwiatkowski, T.J., Jr.; Ranum, L.P.; McCall, A.E.; Chinault, A.C.; Orr, H.T.; Zoghbi, H.Y. Mapping and cloning of the critical region for the spinocerebellar ataxia type 1 gene (SCA1) in a yeast artificial chromosome contig spanning 1.2 Mb. Genomics 1993, 18, 627–635. [Google Scholar] [CrossRef]
- Orr, H.T. Cell biology of spinocerebellar ataxia. J. Cell Biol. 2012, 197, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Klockgether, T. Update on degenerative ataxias. Curr. Opin. Neurol. 2011, 24, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, A.H. The pathogenesis of spinocerebellar ataxia. Cerebellum 2005, 4, 62–73. [Google Scholar] [CrossRef]
- Lam, Y.C.; Bowman, A.B.; Jafar-Nejad, P.; Lim, J.; Richman, R.; Fryer, J.D.; Hyun, E.D.; Duvick, L.A.; Orr, H.T.; Botas, J.; et al. ATAXIN-1 interacts with the repressor Capicua in its native complex to cause SCA1 neuropathology. Cell 2006, 127, 1335–1347. [Google Scholar] [CrossRef]
- Lim, J.; Crespo-Barreto, J.; Jafar-Nejad, P.; Bowman, A.B.; Richman, R.; Hill, D.E.; Orr, H.T.; Zoghbi, H.Y. Opposing effects of polyglutamine expansion on native protein complexes contribute to SCA1. Nature 2008, 452, 713–718. [Google Scholar] [CrossRef]
- Serra, H.G.; Duvick, L.; Zu, T.; Carlson, K.; Stevens, S.; Jorgensen, N.; Lysholm, A.; Burright, E.; Zoghbi, H.Y.; Clark, H.B.; et al. RORalpha-mediated Purkinje cell development determines disease severity in adult SCA1 mice. Cell 2006, 127, 697–708. [Google Scholar] [CrossRef]
- Tsuda, H.; Jafar-Nejad, H.; Patel, A.J.; Sun, Y.; Chen, H.K.; Rose, M.F.; Venken, K.J.; Botas, J.; Orr, H.T.; Bellen, H.J.; et al. The AXH domain of Ataxin-1 mediates neurodegeneration through its interaction with Gfi-1/Senseless proteins. Cell 2005, 122, 633–644. [Google Scholar] [CrossRef]
- Rousseaux, M.W.C.; Tschumperlin, T.; Lu, H.C.; Lackey, E.P.; Bondar, V.V.; Wan, Y.W.; Tan, Q.; Adamski, C.J.; Friedrich, J.; Twaroski, K.; et al. ATXN1-CIC Complex Is the Primary Driver of Cerebellar Pathology in Spinocerebellar Ataxia Type 1 through a Gain-of-Function Mechanism. Neuron 2018, 97, 1235–1243. [Google Scholar] [CrossRef]
- Yeger-Lotem, E.; Sharan, R. Human protein interaction networks across tissues and diseases. Front. Genet. 2015, 6, 257. [Google Scholar] [CrossRef]
- Taylor, I.W.; Wrana, J.L. Protein interaction networks in medicine and disease. Proteomics 2012, 12, 1706–1716. [Google Scholar] [CrossRef] [PubMed]
- Petrakis, S.; Andrade-Navarro, M.A. Editorial: Protein Interaction Networks in Health and Disease. Front. Genet. 2016, 7, 111. [Google Scholar] [CrossRef] [PubMed]
- Jucker, M.; Walker, L.C. Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1341–1349. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, R.I. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev. 2008, 22, 1427–1438. [Google Scholar] [CrossRef]
- Labbadia, J.; Morimoto, R.I. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef]
- Buijsen, R.A.M.; Gardiner, S.L.; Bouma, M.J.; van der Graaf, L.M.; Boogaard, M.W.; Pepers, B.A.; Eussen, B.; de Klein, A.; Freund, C.; van Roon-Mom, W.M.C. Generation of 3 spinocerebellar ataxia type 1 (SCA1) patient-derived induced pluripotent stem cell lines LUMCi002-A, B, and C and 2 unaffected sibling control induced pluripotent stem cell lines LUMCi003-A and B. Stem Cell Res. 2018, 29, 125–128. [Google Scholar] [CrossRef]
- Burright, E.N.; Clark, H.B.; Servadio, A.; Matilla, T.; Feddersen, R.M.; Yunis, W.S.; Duvick, L.A.; Zoghbi, H.Y.; Orr, H.T. SCA1 transgenic mice: A model for neurodegeneration caused by an expanded CAG trinucleotide repeat. Cell 1995, 82, 937–948. [Google Scholar] [CrossRef]
- Watase, K.; Weeber, E.J.; Xu, B.; Antalffy, B.; Yuva-Paylor, L.; Hashimoto, K.; Kano, M.; Atkinson, R.; Sun, Y.; Armstrong, D.L.; et al. A long CAG repeat in the mouse Sca1 locus replicates SCA1 features and reveals the impact of protein solubility on selective neurodegeneration. Neuron 2002, 34, 905–919. [Google Scholar] [CrossRef]
- Nitschke, L.; Tewari, A.; Coffin, S.L.; Xhako, E.; Pang, K.; Gennarino, V.A.; Johnson, J.L.; Blanco, F.A.; Liu, Z.; Zoghbi, H.Y. miR760 regulates ATXN1 levels via interaction with its 5’ untranslated region. Genes Dev. 2020, 34, 1147–1160. [Google Scholar] [CrossRef]
- Laidou, S.; Alanis-Lobato, G.; Pribyl, J.; Rasko, T.; Tichy, B.; Mikulasek, K.; Tsagiopoulou, M.; Oppelt, J.; Kastrinaki, G.; Lefaki, M.; et al. Nuclear inclusions of pathogenic ataxin-1 induce oxidative stress and perturb the protein synthesis machinery. Redox Biol. 2020, 32, 101458. [Google Scholar] [CrossRef]
- Ingram, M.; Wozniak, E.A.L.; Duvick, L.; Yang, R.; Bergmann, P.; Carson, R.; O’Callaghan, B.; Zoghbi, H.Y.; Henzler, C.; Orr, H.T. Cerebellar Transcriptome Profiles of ATXN1 Transgenic Mice Reveal SCA1 Disease Progression and Protection Pathways. Neuron 2016, 89, 1194–1207. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Flockhart, I.; Vinayagam, A.; Bergwitz, C.; Berger, B.; Perrimon, N.; Mohr, S.E. An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC Bioinform. 2011, 12, 357. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Santos, A.; Tsafou, K.; Stolte, C.; Pletscher-Frankild, S.; O’Donoghue, S.I.; Jensen, L.J. Comprehensive comparison of large-scale tissue expression datasets. PeerJ 2015, 3, e1054. [Google Scholar] [CrossRef]
- Assenov, Y.; Ramirez, F.; Schelhorn, S.E.; Lengauer, T.; Albrecht, M. Computing topological parameters of biological networks. Bioinformatics 2008, 24, 282–284. [Google Scholar] [CrossRef]
- Fontaine, J.F.; Priller, F.; Barbosa-Silva, A.; Andrade-Navarro, M.A. Genie: Literature-based gene prioritization at multi genomic scale. Nucleic Acids Res. 2011, 39, W455–W461. [Google Scholar] [CrossRef]
- Kutmon, M.; Kelder, T.; Mandaviya, P.; Evelo, C.T.; Coort, S.L. CyTargetLinker: A cytoscape app to integrate regulatory interactions in network analysis. PLoS ONE 2013, 8, e82160. [Google Scholar] [CrossRef]
- Law, V.; Knox, C.; Djoumbou, Y.; Jewison, T.; Guo, A.C.; Liu, Y.; Maciejewski, A.; Arndt, D.; Wilson, M.; Neveu, V.; et al. DrugBank 4.0: Shedding new light on drug metabolism. Nucleic Acids Res. 2014, 42, D1091–D1097. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, L.; Lv, M.; Pei, R.; Li, P.; Pei, Z.; Wang, Y.; Su, W.; Xie, X.Q. AlzPlatform: An Alzheimer’s disease domain-specific chemogenomics knowledgebase for polypharmacology and target identification research. J. Chem. Inf. Model. 2014, 54, 1050–1060. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Fryer, J.D.; Kang, H.; Crespo-Barreto, J.; Bowman, A.B.; Gao, Y.; Kahle, J.J.; Hong, J.S.; Kheradmand, F.; Orr, H.T.; et al. ATXN1 protein family and CIC regulate extracellular matrix remodeling and lung alveolarization. Dev. Cell 2011, 21, 746–757. [Google Scholar] [CrossRef]
- Ozgur, A.; Vu, T.; Erkan, G.; Radev, D.R. Identifying gene-disease associations using centrality on a literature mined gene-interaction network. Bioinformatics 2008, 24, i277–i285. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Kim, P.M.; Sprecher, E.; Trifonov, V.; Gerstein, M. The importance of bottlenecks in protein networks: Correlation with gene essentiality and expression dynamics. PLoS Comput. Biol. 2007, 3, e59. [Google Scholar] [CrossRef]
- Koschutzki, D.; Schreiber, F. Centrality analysis methods for biological networks and their application to gene regulatory networks. Gene Regul. Syst. Biol. 2008, 2, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Nobes, C.D.; Hall, A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 1995, 81, 53–62. [Google Scholar] [CrossRef]
- Huang, Y.; Thathiah, A. Regulation of neuronal communication by G protein-coupled receptors. FEBS Lett. 2015, 589, 1607–1619. [Google Scholar] [CrossRef]
- Roszkowska, M.; Skupien, A.; Wojtowicz, T.; Konopka, A.; Gorlewicz, A.; Kisiel, M.; Bekisz, M.; Ruszczycki, B.; Dolezyczek, H.; Rejmak, E.; et al. CD44: A novel synaptic cell adhesion molecule regulating structural and functional plasticity of dendritic spines. Mol. Biol. Cell 2016, 27, 4055–4066. [Google Scholar] [CrossRef]
- Kotlyar, M.; Fortney, K.; Jurisica, I. Network-based characterization of drug-regulated genes, drug targets, and toxicity. Methods 2012, 57, 499–507. [Google Scholar] [CrossRef]
- Tong, Z.; Zhou, Y.; Wang, J. Identifying potential drug targets in hepatocellular carcinoma based on network analysis and one-class support vector machine. Sci. Rep. 2019, 9, 10442. [Google Scholar] [CrossRef] [PubMed]
- Bonneh-Barkay, D.; Wiley, C.A. Brain extracellular matrix in neurodegeneration. Brain Pathol. 2009, 19, 573–585. [Google Scholar] [CrossRef] [PubMed]
- Anzilotti, S.; Giampa, C.; Laurenti, D.; Perrone, L.; Bernardi, G.; Melone, M.A.; Fusco, F.R. Immunohistochemical localization of receptor for advanced glycation end (RAGE) products in the R6/2 mouse model of Huntington’s disease. Brain Res. Bull. 2012, 87, 350–358. [Google Scholar] [CrossRef]
- Hosp, F.; Vossfeldt, H.; Heinig, M.; Vasiljevic, D.; Arumughan, A.; Wyler, E.; Landthaler, M.; Hubner, N.; Wanker, E.E.; Lannfelt, L.; et al. Quantitative interaction proteomics of neurodegenerative disease proteins. Cell Rep. 2015, 11, 1134–1146. [Google Scholar] [CrossRef] [PubMed]
- Barabasi, A.L.; Oltvai, Z.N. Network biology: Understanding the cell’s functional organization. Nat. Rev. Genet. 2004, 5, 101–113. [Google Scholar] [CrossRef]
- Milo, R.; Shen-Orr, S.; Itzkovitz, S.; Kashtan, N.; Chklovskii, D.; Alon, U. Network motifs: Simple building blocks of complex networks. Science 2002, 298, 824–827. [Google Scholar] [CrossRef]
- Raman, K.; Damaraju, N.; Joshi, G.K. The organisational structure of protein networks: Revisiting the centrality-lethality hypothesis. Syst. Synth. Biol. 2014, 8, 73–81. [Google Scholar] [CrossRef]
- Ashtiani, M.; Salehzadeh-Yazdi, A.; Razaghi-Moghadam, Z.; Hennig, H.; Wolkenhauer, O.; Mirzaie, M.; Jafari, M. A systematic survey of centrality measures for protein-protein interaction networks. BMC Syst. Biol. 2018, 12, 80. [Google Scholar] [CrossRef]
- DeGeer, J.; Lamarche-Vane, N. Rho GTPases in neurodegeneration diseases. Exp. Cell Res. 2013, 319, 2384–2394. [Google Scholar] [CrossRef]
- Tourette, C.; Li, B.; Bell, R.; O’Hare, S.; Kaltenbach, L.S.; Mooney, S.D.; Hughes, R.E. A large scale Huntingtin protein interaction network implicates Rho GTPase signaling pathways in Huntington disease. J. Biol. Chem. 2014, 289, 6709–6726. [Google Scholar] [CrossRef]
- Shirasaki, D.I.; Greiner, E.R.; Al-Ramahi, I.; Gray, M.; Boontheung, P.; Geschwind, D.H.; Botas, J.; Coppola, G.; Horvath, S.; Loo, J.A.; et al. Network organization of the huntingtin proteomic interactome in mammalian brain. Neuron 2012, 75, 41–57. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Cui, X.; Al-Ramahi, I.; Sun, X.; Li, B.; Hou, J.; Difiglia, M.; Palacino, J.; Wu, Z.Y.; Ma, L.; et al. A striatal-enriched intronic GPCR modulates huntingtin levels and toxicity. eLife 2015, 4, e05449. [Google Scholar] [CrossRef] [PubMed]
- Bae, B.I.; Xu, H.; Igarashi, S.; Fujimuro, M.; Agrawal, N.; Taya, Y.; Hayward, S.D.; Moran, T.H.; Montell, C.; Ross, C.A.; et al. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington’s disease. Neuron 2005, 47, 29–41. [Google Scholar] [CrossRef]
- Krauss, S.; Griesche, N.; Jastrzebska, E.; Chen, C.; Rutschow, D.; Achmuller, C.; Dorn, S.; Boesch, S.M.; Lalowski, M.; Wanker, E.; et al. Translation of HTT mRNA with expanded CAG repeats is regulated by the MID1-PP2A protein complex. Nat. Commun. 2013, 4, 1511. [Google Scholar] [CrossRef] [PubMed]
- Deyts, C.; Galan-Rodriguez, B.; Martin, E.; Bouveyron, N.; Roze, E.; Charvin, D.; Caboche, J.; Betuing, S. Dopamine D2 receptor stimulation potentiates PolyQ-Huntingtin-induced mouse striatal neuron dysfunctions via Rho/ROCK-II activation. PLoS ONE 2009, 4, e8287. [Google Scholar] [CrossRef]
- Roscic, A.; Baldo, B.; Crochemore, C.; Marcellin, D.; Paganetti, P. Induction of autophagy with catalytic mTOR inhibitors reduces huntingtin aggregates in a neuronal cell model. J. Neurochem. 2011, 119, 398–407. [Google Scholar] [CrossRef]
- Hong, H.; Koon, A.C.; Chen, Z.S.; Wei, Y.; An, Y.; Li, W.; Lau, M.H.Y.; Lau, K.F.; Ngo, J.C.K.; Wong, C.H.; et al. AQAMAN, a bisamidine-based inhibitor of toxic protein inclusions in neurons, ameliorates cytotoxicity in polyglutamine disease models. J. Biol. Chem. 2019, 294, 2757–2770. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Cells | Mice | |||
|---|---|---|---|---|
| Enrichment Term | Overlap | p-Value | Overlap | p-Value |
| A. Day 2 (D2) Cells vs. Week 5 (W5) Mice | ||||
| Protein digestion and absorption | 8/90 | 0.012 | 6/90 | 0.005 |
| ECM-receptor interaction | 14/82 | >0.001 | 5/82 | 0.016 |
| PI3K-Akt signaling pathway | 26/341 | >0.001 | 12/341 | 0.002 |
| B. Day 5 (D5) Cells vs. Week 12 (W12) Mice | ||||
| Ribosome | 38/137 | >0.001 | 17/137 | 0.003 |
| ECM-receptor interaction | 18/82 | >0.001 | 12/82 | 0.003 |
| Focal adhesion | 24/202 | >0.001 | 21/202 | 0.009 |
| PI3K-Akt signaling pathway | 29/341 | >0.001 | 30/341 | 0.022 |
| Protein digestion and absorption | 12/90 | 0.001 | 11/90 | 0.018 |
| Alzheimer’s disease | 15/168 | 0.002 | 18/168 | 0.011 |
| Rap1 signaling pathway | 16/211 | 0.001 | 21/211 | 0.015 |
| Parkinson’s disease | 11/142 | 0.024 | 14/142 | 0.045 |
| C. Day 10 (D10) Cells vs. Week 28 (W28) Mice | ||||
| AGE-RAGE signaling pathway | 9/101 | 0.019 | 11/101 | 0.012 |
| ECM-receptor interaction | 13/82 | >0.001 | 9/82 | 0.03 |
| Focal adhesion | 21/202 | >0.001 | 17/202 | 0.042 |
| PI3K-Akt signaling pathway | 26/341 | 0.001 | 27/341 | 0.026 |
| Protein digestion and absorption | 8/90 | 0.027 | 9/90 | 0.05 |
| Rap1 signaling pathway | 16/211 | 0.01 | 22/211 | 0.002 |
| Regulation of actin cytoskeleton | 18/214 | 0.002 | 22/214 | 0.002 |
| Target | Drug | Algorithm | Fingerprint | BBB Permeability Prediction |
|---|---|---|---|---|
| PPP2AC | Vitamin E | ADABoost | MACCS | BBB+ |
| Openbabel | BBB+ | |||
| Molprint | BBB+ | |||
| PubChem | BBB+ | |||
| SVM | MACCS | BBB+ | ||
| Openbabel | BBB+ | |||
| Molprint | BBB+ | |||
| PubChem | BBB+ | |||
| TP53 | PhiKan 083 | ADABoost | MACCS | BBB+ |
| Openbabel | BBB+ | |||
| Molprint | BBB+ | |||
| PubChem | BBB+ | |||
| SVM | MACCS | BBB+ | ||
| Openbabel | BBB+ | |||
| Molprint | BBB+ | |||
| PubChem | BBB+ | |||
| AZD 3355 | ADABoost | MACCS | BBB+ | |
| Openbabel | BBB+ | |||
| Molprint | BBB+ | |||
| PubChem | BBB+ | |||
| SVM | MACCS | BBB+ | ||
| Openbabel | BBB+ | |||
| Molprint | BBB+ | |||
| PubChem | BBB+ | |||
| GNB1 | FARNESYL | ADABoost | MACCS | BBB+ |
| Openbabel | BBB+ | |||
| Molprint | BBB+ | |||
| PubChem | BBB+ | |||
| SVM | MACCS | BBB+ | ||
| Openbabel | BBB+ | |||
| Molprint | BBB+ | |||
| PubChem | BBB+ | |||
| ATP1A1 | Bretylium | ADABoost | MACCS | BBB+ |
| Openbabel | BBB+ | |||
| Molprint | BBB+ | |||
| PubChem | BBB+ | |||
| SVM | MACCS | BBB+ | ||
| Openbabel | BBB+ | |||
| Molprint | BBB+ | |||
| PubChem | BBB+ | |||
| Ciclopirox | ADABoost | MACCS | BBB+ | |
| Openbabel | BBB+ | |||
| Molprint | BBB+ | |||
| PubChem | BBB+ | |||
| SVM | MACCS | BBB+ | ||
| Openbabel | BBB+ | |||
| Molprint | BBB+ | |||
| PubChem | BBB+ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vagiona, A.-C.; Andrade-Navarro, M.A.; Psomopoulos, F.; Petrakis, S. Dynamics of a Protein Interaction Network Associated to the Aggregation of polyQ-Expanded Ataxin-1. Genes 2020, 11, 1129. https://doi.org/10.3390/genes11101129
Vagiona A-C, Andrade-Navarro MA, Psomopoulos F, Petrakis S. Dynamics of a Protein Interaction Network Associated to the Aggregation of polyQ-Expanded Ataxin-1. Genes. 2020; 11(10):1129. https://doi.org/10.3390/genes11101129
Chicago/Turabian StyleVagiona, Aimilia-Christina, Miguel A. Andrade-Navarro, Fotis Psomopoulos, and Spyros Petrakis. 2020. "Dynamics of a Protein Interaction Network Associated to the Aggregation of polyQ-Expanded Ataxin-1" Genes 11, no. 10: 1129. https://doi.org/10.3390/genes11101129
APA StyleVagiona, A.-C., Andrade-Navarro, M. A., Psomopoulos, F., & Petrakis, S. (2020). Dynamics of a Protein Interaction Network Associated to the Aggregation of polyQ-Expanded Ataxin-1. Genes, 11(10), 1129. https://doi.org/10.3390/genes11101129