Comparative Transcriptomics and Co-Expression Networks Reveal Tissue- and Genotype-Specific Responses of qDTYs to Reproductive-Stage Drought Stress in Rice (Oryza sativa L.)

Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Experimental Treatments
2.2. RNA Extraction and Sequencing
2.3. Transcriptome Assembly and Expression Level Quantification
2.4. Differential Expression Analysis
2.5. Gene Co-Expression Network Analysis
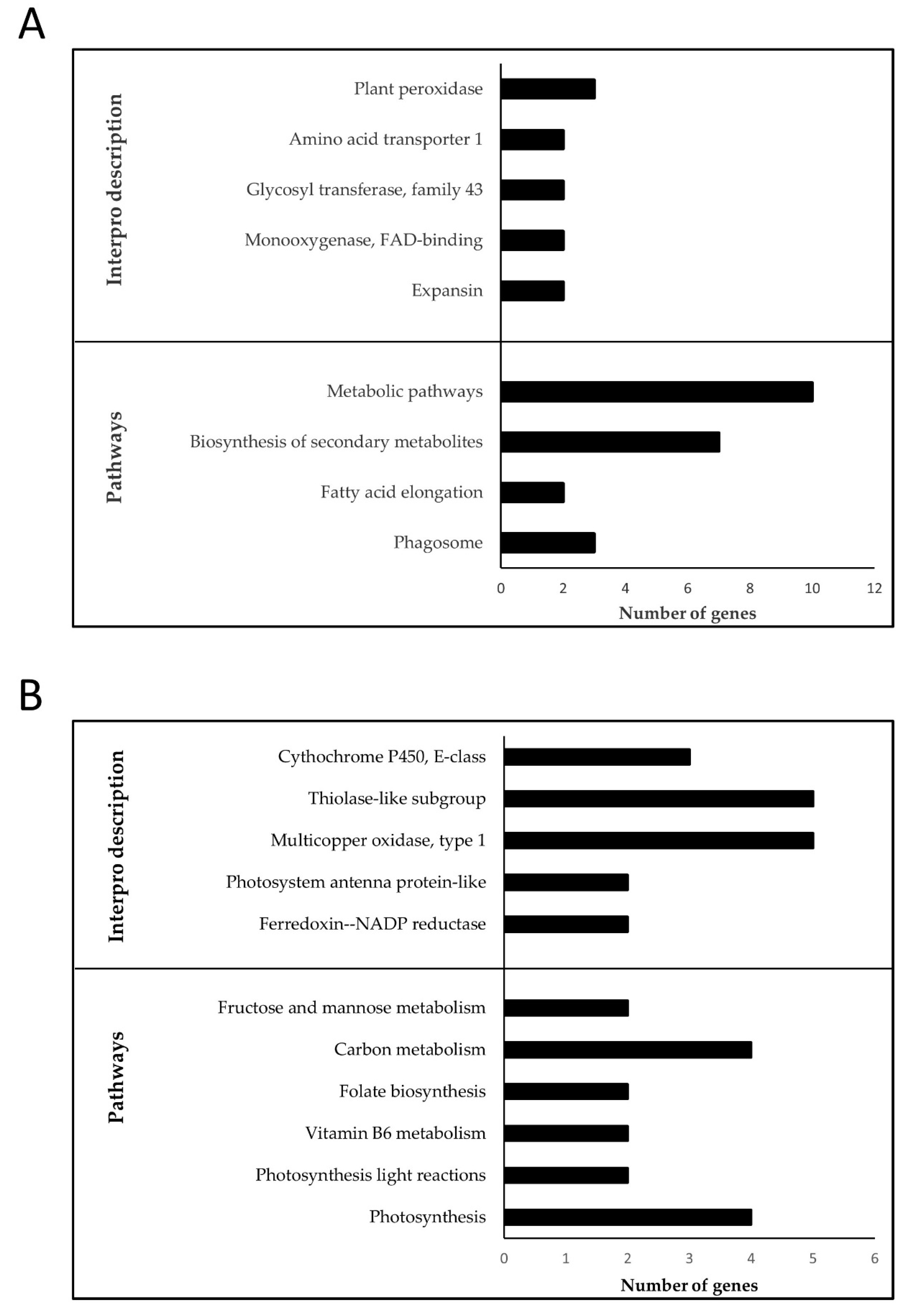
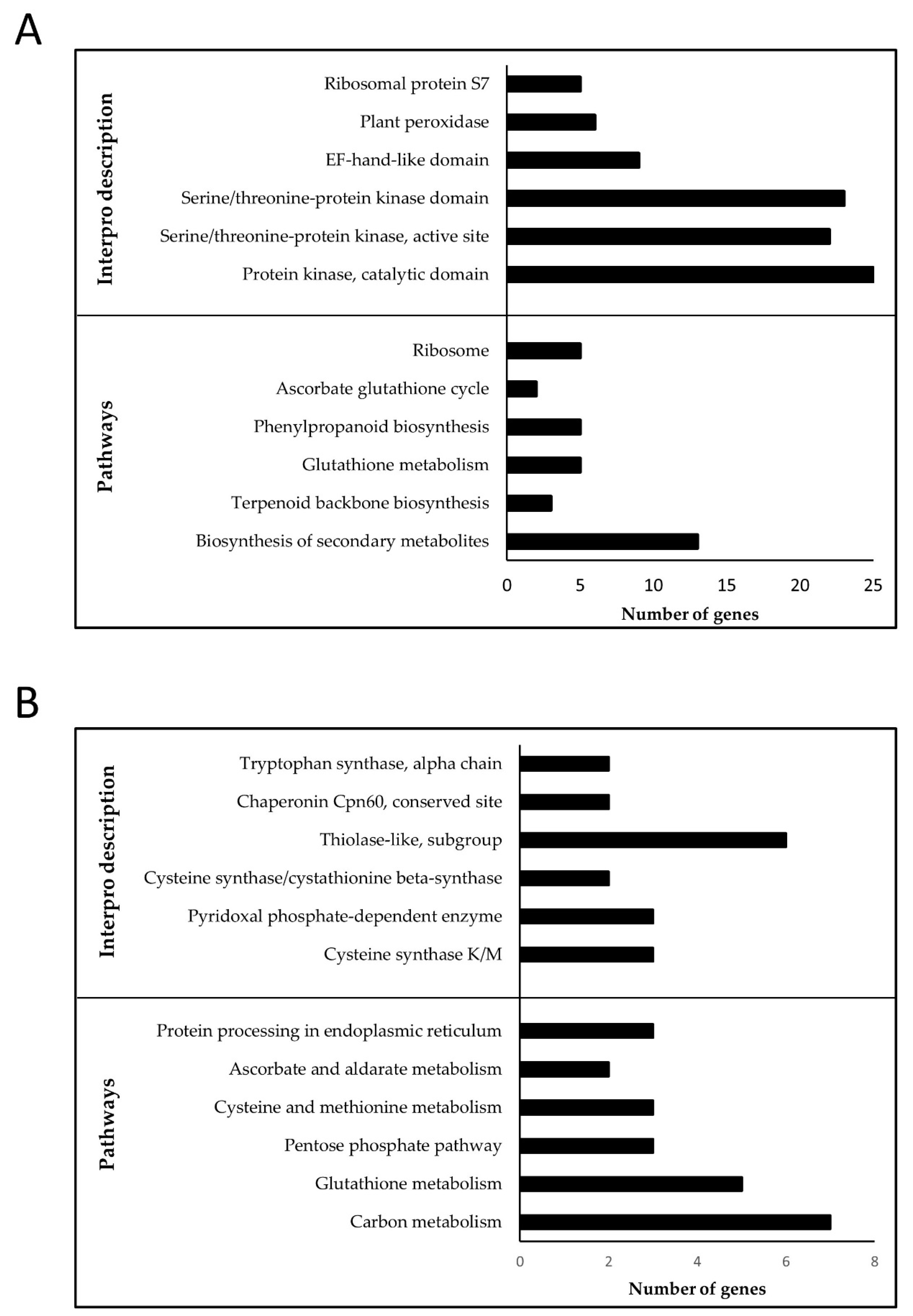
2.6. Over-Representation Analysis (ORA)
2.7. Quantitative PCR Analysis
2.8. Genotyping
2.9. DNA Extraction and Sanger Sequencing
3. Results
3.1. Phenotypes of Swarna and DTY-IL under Reproductive Stage Drought Stress
3.2. Generating a Transcriptional Map of the Moderate RDS Response in Rice
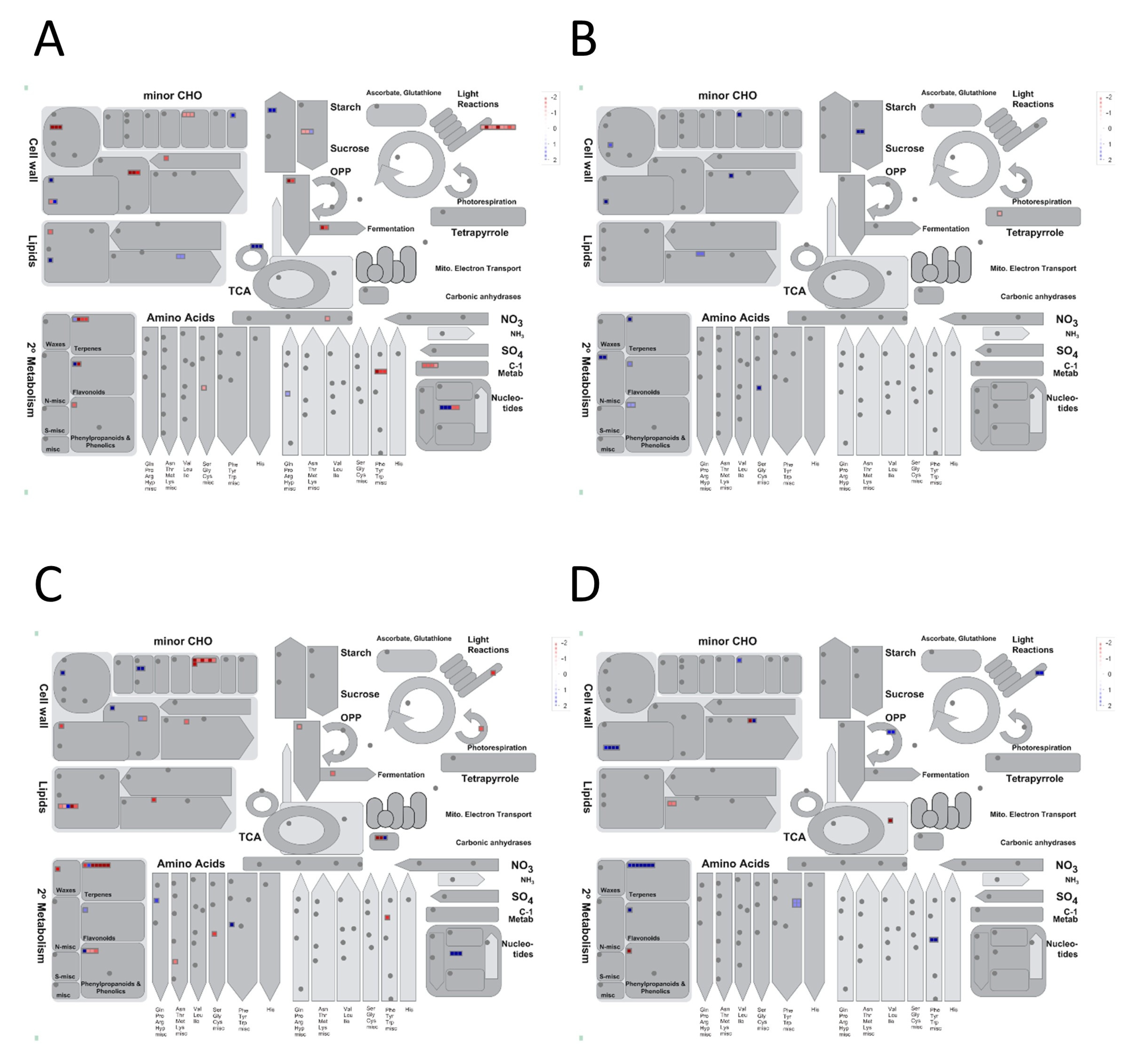
3.2.1. Expression Profiles of Drought-Responsive Genes in the Flag-Leaf Tissue under RDS
3.2.2. Expression Profiles of Drought-Responsive Genes in the Panicle Tissue under RDS
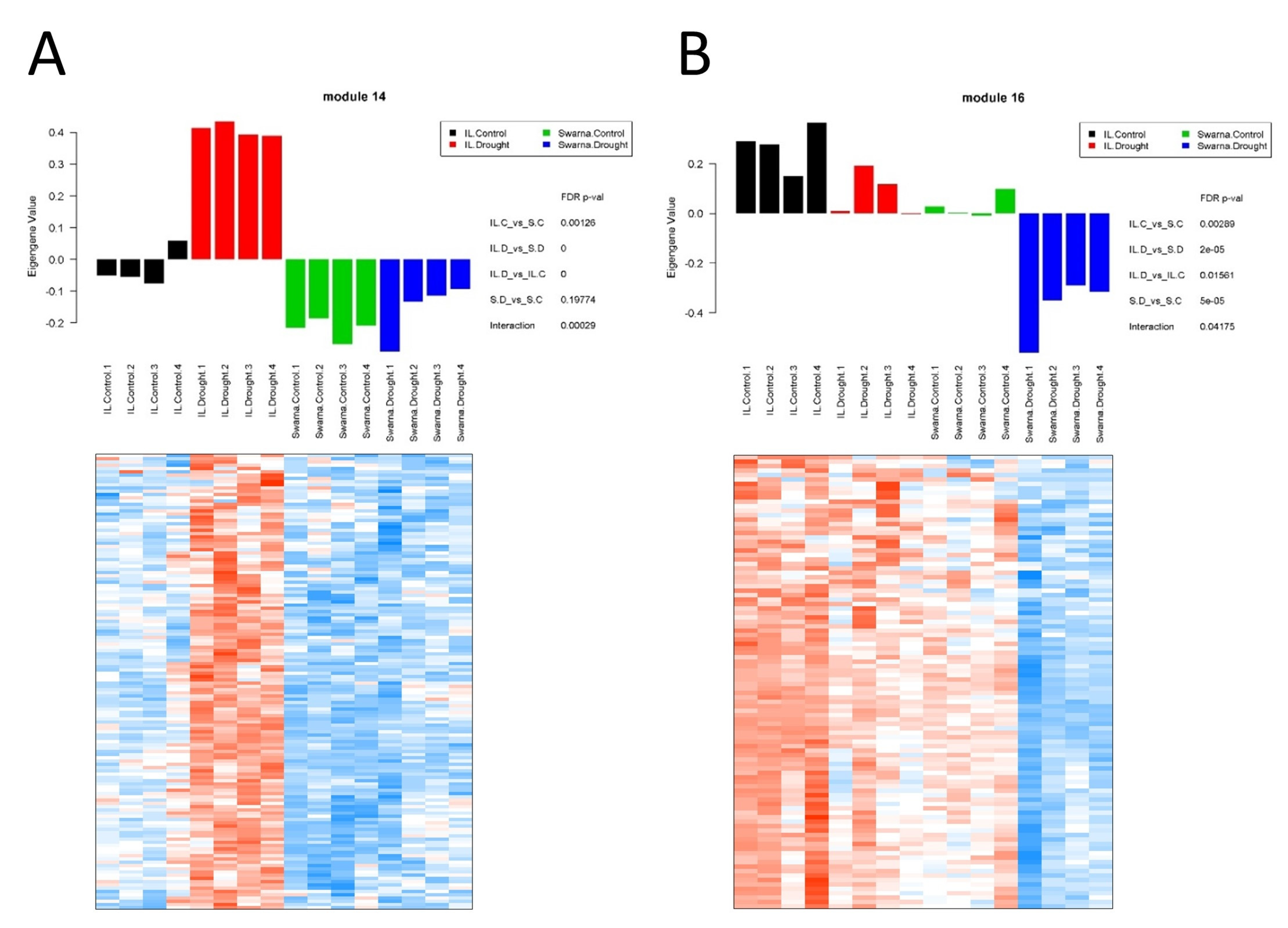
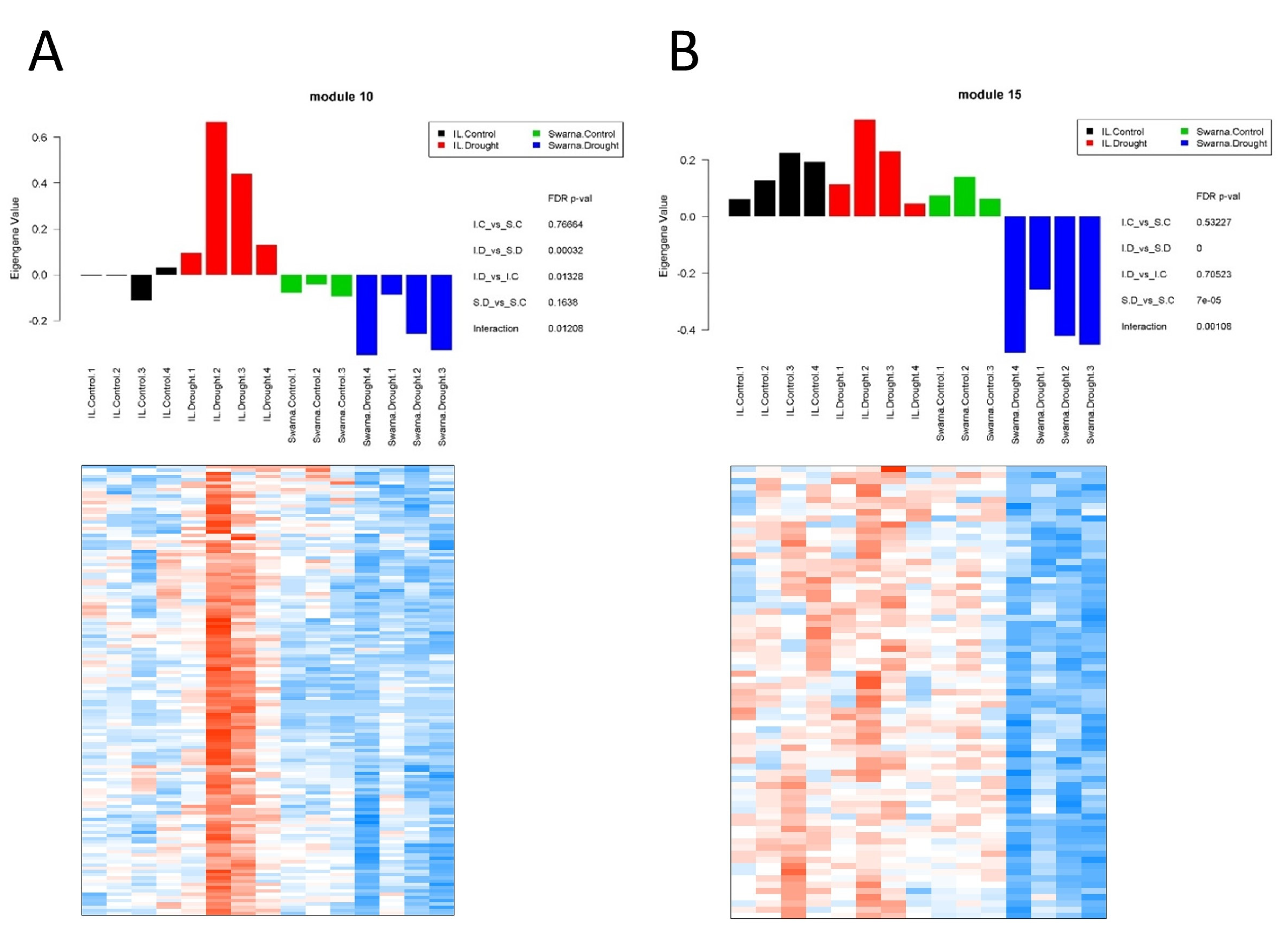
3.3. Drought-Stress-Related Gene Modules within the Transcriptional Map
3.3.1. Flag-Leaf Specific Modules
3.3.2. Panicle Specific Modules
3.4. Validation of Differential Gene Expression
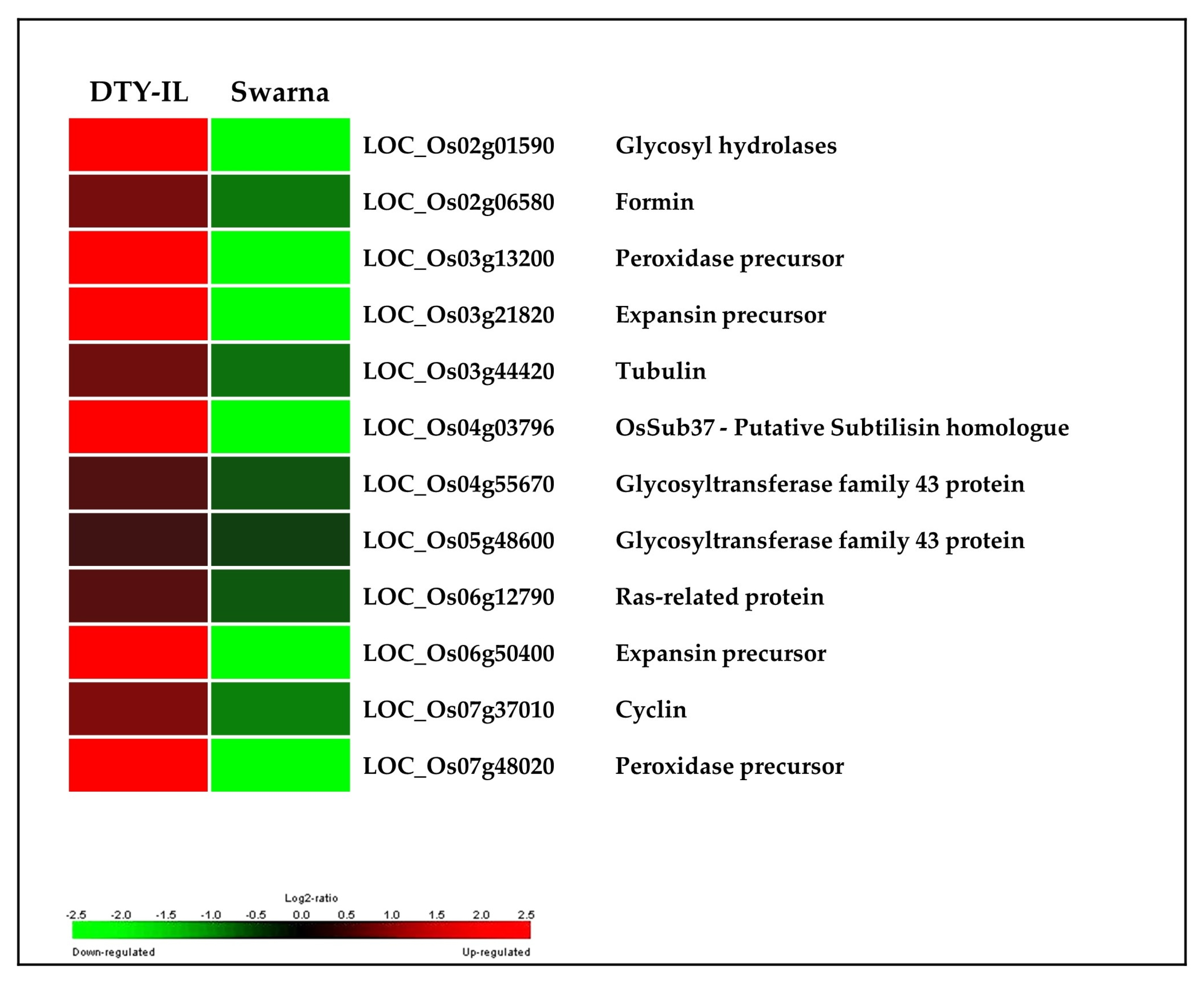
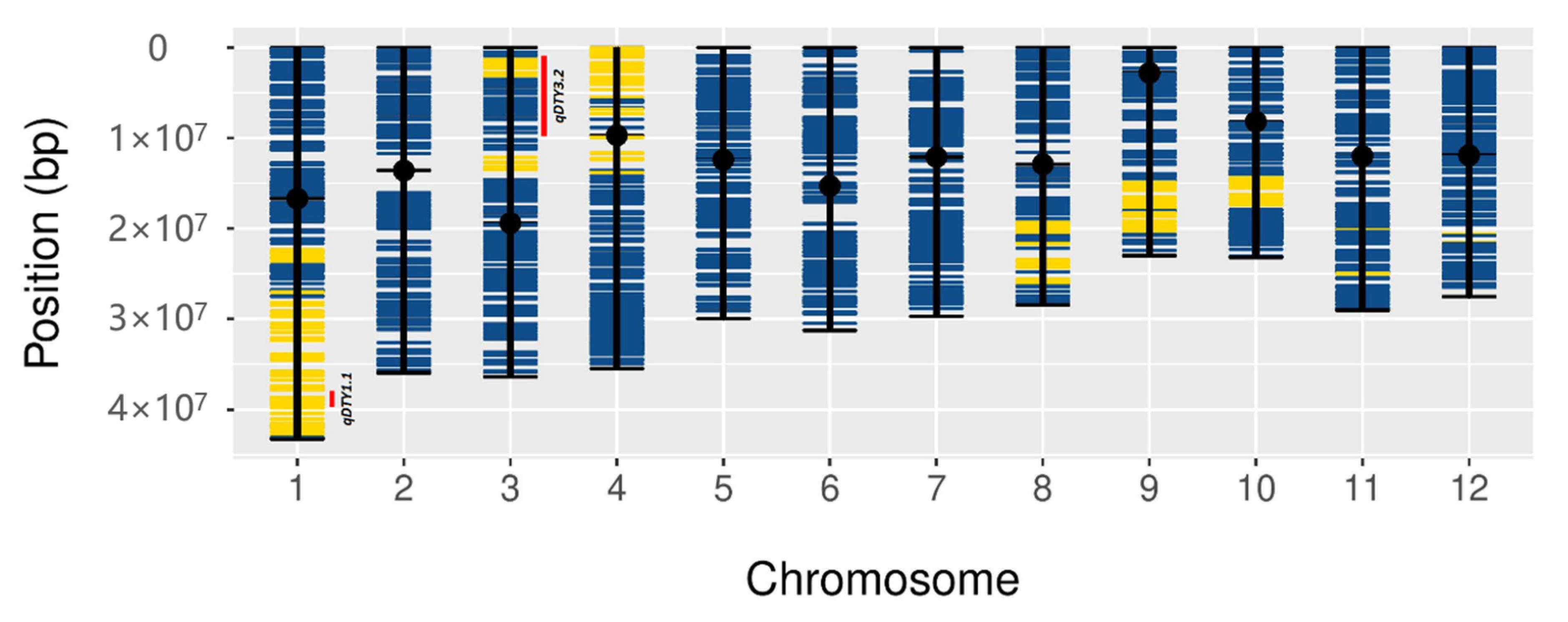
3.5. Colocalization of DEGs in the Introgression Fragments
4. Discussion
4.1. Source Effects—Flag-Leaf-Specific Differences between DTY-IL and Swarna
4.2. Sink Effects—Panicle Specific Differences between DTY-IL and Swarna
4.3. qDTY1.1-Specific Contributions to Drought Tolerance
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Borah, P.; Sharma, E.; Kaur, A.; Chandel, G.; Mohapatra, T.; Kapoor, S.; Khurana, J.P. Analysis of drought-responsive signalling network in two contrasting rice cultivars using transcriptome-based approach. Sci. Rep. 2017, 7, 42131. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Cassman, K.G.; Virmani, S.S.; Sheehy, J.; Khush, G.S. Yield Potential Trends of Tropical Rice since the Release of IR8 and the Challenge of Increasing Rice Yield Potential. Crop Sci. 1999, 39, 1552–1559. [Google Scholar] [CrossRef]
- Todaka, D.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Recent advances in the dissection of drought-stress regulatory networks and strategies for development of drought-tolerant transgenic rice plants. Front. Plant Sci. 2015, 6, 84. [Google Scholar] [CrossRef] [PubMed]
- Lafitte, H.R.; Price, A.H.; Courtois, B. Yield response to water deficit in an upland rice mapping population: Associations among traits and genetic markers. Theor. Appl. Genet. 2004, 109, 1237–1246. [Google Scholar] [CrossRef]
- Venuprasad, R.; Lafitte, H.R.; Atlin, G.N. Response to Direct Selection for Grain Yield under Drought Stress in Rice. Crop Sci. 2007, 47, 285–293. [Google Scholar] [CrossRef]
- Liu, J.; Bennett, J. Reversible and Irreversible Drought-Induced Changes in the Anther Proteome of Rice (Oryza sativa L.) Genotypes IR64 and Moroberekan. Mol. Plant 2011, 4, 59–69. [Google Scholar] [CrossRef]
- Vikram, P.; Swamy, B.P.; Dixit, S.; Ahmed, H.; Cruz, M.T.; Singh, A.; Kumar, A. QDTY1.1, a major QTL for rice grain yield under reproductive-stage drought stress with a consistent effect in multiple elite genetic backgrounds. BMC Genet. 2011, 12, 89. [Google Scholar] [CrossRef]
- Jin, Y.; Yang, H.; Wei, Z.; Ma, H.; Ge, X. Rice Male Development under Drought Stress: Phenotypic Changes and Stage-Dependent Transcriptomic Reprogramming. Mol. Plant 2013, 6, 1630–1645. [Google Scholar] [CrossRef]
- Guo, C.; Ge, X.; Ma, H. The rice OsDIL gene plays a role in drought tolerance at vegetative and reproductive stages. Plant Mol. Biol. 2013, 82, 239–253. [Google Scholar] [CrossRef]
- Shankar, R.; Bhattacharjee, A.; Jain, M. Transcriptome analysis in different rice cultivars provides novel insights into desiccation and salinity stress responses. Sci. Rep. 2016, 6, 23719. [Google Scholar] [CrossRef]
- Pandey, S. Economic costs of drought and rice farmers’ coping mechanisms. Int. Rice Res. Notes 2009, 32, 1. [Google Scholar] [CrossRef]
- Kumar, A.; Dixit, S.; Ram, T.; Yadaw, R.B.; Mishra, K.K.; Mandal, N.P. Breeding high-yielding drought-tolerant rice: Genetic variations and conventional and molecular approaches. J. Exp. Bot. 2014, 65, 6265–6278. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Sandhu, N.; Dixit, S.; Yadaw, S.; Swamy, M.P.; Shamsudin, N.A. Marker-assisted selection strategy to pyramid two or more QTLs for quantitative trait-grain yield under drought. Rice 2018, 11, 35. [Google Scholar] [CrossRef] [PubMed]
- Bernier, J.; Kumar, A.; Ramaiah, V.; Spaner, D.; Atlin, G.A. Large-Effect QTL for Grain Yield under Reproductive-Stage Drought Stress in Upland Rice. Crop Sci. 2007, 47, 507. [Google Scholar] [CrossRef]
- Venuprasad, R.; Bool, M.E.; Quiatchon, L.; Atlin, G.N. A QTL for rice grain yield in aerobic environments with large effects in three genetic backgrounds. Theor. Appl. Genet. 2011, 124, 323–332. [Google Scholar] [CrossRef]
- Ghimire, K.H.; Quiatchon, L.A.; Vikram, P.; Swamy, B.M.; Dixit, S.; Ahmed, H.; Hernandez, J.; Borromeo, T.; Kumar, A. Identification and mapping of a QTL (qDTY1.1) with a consistent effect on grain yield under drought. Field Crop. Res. 2012, 131, 88–96. [Google Scholar] [CrossRef]
- Yadaw, R.B.; Dixit, S.; Raman, A.; Mishra, K.K.; Vikram, P.; Swamy, B.M.; Sta Cruz, M.T.; Maturan, P.; Pandey, M.; Kumar, A. A QTL for high grain yield under lowland drought in the background of popular rice variety Sabitri from Nepal. Field Crop. Res. 2013, 144, 281–287. [Google Scholar] [CrossRef]
- Dixit, S.; Biswal, A.K.; Min, A.; Henry, A.; Oane, R.H.; Raorane, M.L.; Longkumer, T.; Pabuayon, I.M.; Mutte, S.K.; Vardarajan, A.R.; et al. Action of multiple intra-QTL genes concerted around a co-localized transcription factor underpins a large effect QTL. Sci. Rep. 2015, 5, 15183. [Google Scholar] [CrossRef]
- Raorane, M.L.; Pabuayon, I.M.; Varadarajan, A.R.; Mutte, S.K.; Kumar, A.; Treumann, A.; Kohli, A. Proteomic insights into the role of the large-effect QTL qDTY 12.1 for rice yield under drought. Mol. Breed. 2015, 35, 6. [Google Scholar] [CrossRef]
- Degenkolbe, T.; Do, P.T.; Zuther, E.; Repsilber, D.; Walther, D.; Hincha, D.K.; Köhl, K.I. Expression profiling of rice cultivars differing in their tolerance to long-term drought stress. Plant Mol. Biol. 2009, 69, 133–153. [Google Scholar] [CrossRef]
- Lenka, S.K.; Katiyar, A.; Chinnusamy, V.; Bansal, K.C. Comparative analysis of drought-responsive transcriptome in Indica rice genotypes with contrasting drought tolerance. Plant Biotechnol. J. 2011, 9, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Dansana, P.K.; Giri, J.; Deveshwar, P.; Arora, R.; Agarwal, P.; Khurana, J.P.; Kapoor, S.; Tyagi, A.K. Modulation of transcription factor and metabolic pathway genes in response to water-deficit stress in rice. Funct. Integr. Genom. 2011, 11, 157–178. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Pan, Y.; Zhao, X.; Zhu, L.; Fu, B.; Li, Z. Genome-wide temporal-spatial gene expression profiling of drought responsiveness in rice. BMC Genom. 2011, 12, 149. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Zhang, F.; Zhang, F.; Wang, W.; Zhou, Y.; Fu, B.; Li, Z. Comparative transcriptome sequencing of tolerant rice introgression line and its parents in response to drought stress. BMC Genom. 2014, 15, 1–26. [Google Scholar] [CrossRef]
- Hu, H.; Xiong, L. Genetic Engineering and Breeding of Drought-Resistant Crops. Annu. Rev. Plant Biol. 2014, 65, 715–741. [Google Scholar] [CrossRef]
- Baldoni, E.; Bagnaresi, P.; Locatelli, F.; Mattana, M.; Genga, A. Comparative Leaf and Root Transcriptomic Analysis of two Rice Japonica Cultivars Reveals Major Differences in the Root Early Response to Osmotic Stress. Rice 2016, 9, 25. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, Y.; Xiao, B. Comparative transcriptome analysis highlights the crucial roles of photosynthetic system in drought stress adaptation in upland rice. Sci. Rep. 2008, 6, 19349. [Google Scholar] [CrossRef]
- Arvidsson, S.; Kwasniewski, M.; Riano-Pachon, D.M.; Mueller-Roeber, B. QuantPrime-a flexible tool for reliable high-throughput primer design for quantitative PCR. BMC Bioinform. 2016, 9, 465. [Google Scholar] [CrossRef]
- Umezawa, T.; Fujita, M.; Fujita, Y.; Yamaguchi-Shinozaki, K.; Shinozaki, K. Engineering drought tolerance in plants: Discovering and tailoring genes to unlock the future. Curr. Opin. Biotechnol. 2006, 17, 113–122. [Google Scholar] [CrossRef]
- Moumeni, A.; Satoh, K.; Kondoh, H.; Asano, T.; Hosaka, A.; Venuprasad, R.; Serraj, R.; Kumar, A.; Leung, H.; Kikuchi, S. Comparative analysis of root transcriptome profiles of two pairs of drought-tolerant and susceptible rice near-isogenic lines under different drought stress. BMC Plant Biol. 2011, 11, 174. [Google Scholar] [CrossRef]
- Ding, X.; Li, X.; Xiong, L. Insight into Differential Responses of Upland and Paddy Rice to Drought Stress by Comparative Expression Profiling Analysis. Int. J. Mol. Sci. 2013, 14, 5214–5238. [Google Scholar] [CrossRef]
- Weng, X.; Wang, L.; Wang, J.; Hu, Y.; Du, H.; Xu, C.; Xing, Y.; Li, X.; Xiao, J.; Zhang, Q. Grain Number, Plant Height, and Heading Date7 Is a Central Regulator of Growth, Development, and Stress Response. Plant Physiol. 2014, 164, 735–747. [Google Scholar] [CrossRef]
- Moumeni, A.; Satoh, K.; Venuprasad, R.; Serraj, R.; Kumar, A.; Leung, H.; Kikuchi, S. Transcriptional profiling of the leaves of near-isogenic rice lines with contrasting drought tolerance at the reproductive stage in response to water deficit. BMC Genom. 2015, 16, 1110. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Chen, C.; Ma, X.; Zhang, Y.; Han, J.; Mei, H.; Yu, S. Comparative Analysis of Expression Profiles of Panicle Development among Tolerant and Sensitive Rice in Response to Drought Stress. Front. Plant Sci. 2017, 8, 437. [Google Scholar] [CrossRef] [PubMed]
- Hadiarto, T.; Tran, L.P. Progress studies of drought-responsive genes in rice. Plant Cell Rep. 2010, 30, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Shojaie, A.; Michailidis, G. Analysis of Gene Sets Based on the Underlying Regulatory Network. J. Comput. Biol. 2009, 16, 407–426. [Google Scholar] [CrossRef] [PubMed]
- Dela Fuente, A. From ‘differential expression’ to ‘differential networking’–identification of dysfunctional regulatory networks in diseases. Trends Genet. 2010, 26, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Gitter, A.; Carmi, M.; Barkai, N.; Bar-Joseph, Z. Linking the signaling cascades and dynamic regulatory networks controlling stress responses. Genome Res. 2013, 23, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Xin, M.; Feldmann, K.A.; Wang, X. Machine Learning-Based Differential Network Analysis: A Study of Stress-Responsive Transcriptomes in Arabidopsis. Plant Cell 2014, 26, 520–537. [Google Scholar] [CrossRef]
- Zhang, B.; Horvath, S. A General Framework for Weighted Gene Co-Expression Network Analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 17. [Google Scholar]
- Gehan, M.A.; Greenham, K.; Mockler, T.C.; Mcclung, C.R. Transcriptional networks—crops, clocks, and abiotic stress. Curr. Opin. Plant Biol. 2015, 24, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Liseron-Monfils, C.; Ware, D. Revealing gene regulation and associations through biological networks. Curr. Plant Biol. 2015, 3–4, 30–39. [Google Scholar] [CrossRef]
- Segal, E.; Shapira, M.; Regev, A.; Peer, D.; Botstein, D.; Koller, D.; Friedman, N. Module networks: Identifying regulatory modules and their condition-specific regulators from gene expression data. Nat. Genet. 2003, 34, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Sircar, S.; Parekh, N. Functional characterization of drought-responsive modules and genes in Oryza sativa: A network-based approach. Front. Genet. 2015, 6, 256. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Dong, J. Geometric Interpretation of Gene Coexpression Network Analysis. PLoS Comput. Biol. 2008, 4, 1–27. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Shaik, R.; Ramakrishna, W. Genes and Co-Expression Modules Common to Drought and Bacterial Stress Responses in Arabidopsis and Rice. PLoS ONE 2013, 8, 10. [Google Scholar] [CrossRef]
- Zhang, L.; Yu, S.; Zuo, K.; Luo, L.; Tang, K. Identification of Gene Modules Associated with Drought Response in Rice by Network-Based Analysis. PLoS ONE 2012, 7, 5. [Google Scholar] [CrossRef]
- Counce, P.A.; Keisling, T.C.; Mitchell, A. A Uniform, Objective, and Adaptive System for Expressing Rice Development. Crop Sci. 2000, 40, 436. [Google Scholar] [CrossRef]
- Krishnan, A.; Gupta, C.; Ambavaram, M.M.R.; Pereira, A. RECoN: Rice Environment Coexpression Network for Systems Level Analysis of Abiotic-Stress Response. Front. Plant Sci. 2017, 8, 1–15. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 23 September 2020).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.; Pertea, G. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000 Res. 2016, 4, 1521. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Morales, K.Y.; Singh, N.; Perez, F.A.; Ignacio, J.C.; Thapa, R.; Arbelaez, J.D.; Tabien, R.E.; Famoso, A.; Wang, D.R.; Septiningsih, E.M.; et al. An improved 7K SNP array, the C7AIR, provides a wealth of validated SNP markers for rice breeding and genetics studies. PLoS ONE 2020, 15, e0232479. [Google Scholar] [CrossRef]
- Vikram, P.; Swamy, B.P.; Dixit, S.; Singh, R.; Singh, B.P.; Miro, B.; Kohli, A.; Henry, A.; Singh, N.K.; Kumar, A. Drought susceptibility of modern rice varieties: An effect of linkage of drought tolerance with undesirable traits. Sci. Rep. 2015, 5, 14799. [Google Scholar] [CrossRef]
- Hirota, O.; Oka, M.; Takeda, T. Sink Activity Estimation by Sink Size and Dry Matter Increase During the Ripening Stage of Barley (Hordeum vulgare) and Rice (Oryza sativa). Ann. Bot. 1990, 65, 349–353. [Google Scholar] [CrossRef]
- Biswal, A.K.; Kohli, A. Cereal flag leaf adaptations for grain yield under drought: Knowledge status and gaps. Mol. Breed. 2013, 31, 749–766. [Google Scholar] [CrossRef]
- Lawlor, D.W.; Paul, M.J. Source/sink interactions underpin crop yield: The case for trehalose 6-phosphate/SnRK1 in improvement of wheat. Front. Plant Sci. 2014, 5, 418. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Ramegowda, V.; Kumar, A.; Pereira, A. Plant adaptation to drought stress. F1000 Res. 2016, 5, 1554. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Yao, L.; You, C.; Wang, S.; Cui, J.; Ge, X.; Ma, H. MID1 plays an important role in response to drought stress during reproductive development. Plant J. 2016, 88, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Farooq, M.; Wahid, A.; Kobayashi, N.; Fujita, D.; Basra, S.M. Plant Drought Stress: Effects, Mechanisms and Management. Sustain. Agric. 2009, 29, 153–188. [Google Scholar]
- Moore, J.P.; Vicré-Gibouin, M.; Farrant, J.M.; Driouich, A. Adaptations of higher plant cell walls to water loss: Drought vs. desiccation. Physiol. Plant. 2008, 134, 237–245. [Google Scholar] [CrossRef]
- Tenhaken, R. Cell wall remodeling under abiotic stress. Front. Plant Sci. 2015, 5, 771. [Google Scholar] [CrossRef]
- Cal, A.; Sanciangco, M.; Rebolledo, M.B.; Luquet, D.; Torres, R.; McnNaly, K.; Henry, A. Leaf morphology, rather than plant water status, underlies, genetic variation of rice leaf rolling under drought. Plant Cell Environ. 2019, 42, 1532–1544. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef]
- Cosgrove, D.J. Growth of the plant cell wall. Nat. Rev. Mol. Cell Biol. 2005, 6, 850–861. [Google Scholar] [CrossRef]
- Wu, Y.; Sharp, R.E.; Durachko, D.M.; Cosgrove, D.J. Growth Maintenance of the Maize Primary Root at Low Water Potentials Involves Increases in Cell-Wall Extension Properties, Expansin Activity, and Wall Susceptibility to Expansins. Plant Physiol. 1996, 111, 765–772. [Google Scholar] [CrossRef]
- Jones, L.; Mcqueen-Mason, S. A role for expansins in dehydration and rehydration of the resurrection plantCraterostigma plantagineum. FEBS Lett. 2004, 559, 61–65. [Google Scholar] [CrossRef]
- Harb, A.; Krishnan, A.; Ambavaram, M.M.; Pereira, A. Molecular and Physiological Analysis of Drought Stress in Arabidopsis Reveals Early Responses Leading to Acclimation in Plant Growth. Plant Physiol. 2010, 154, 1254–1271. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Zhao, J.; Li, X.; Qin, L.; Yan, X.; Liao, H. A soybean β-expansin gene GmEXPB2 intrinsically involved in root system architecture responses to abiotic stresses. Plant J. 2011, 66, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Han, Y.; Feng, Y.; Xing, S.; Zhao, M.; Chen, Y.; Wang, W. Expression of wheat expansin driven by the RD29 promoter in tobacco confers water-stress tolerance without impacting growth and development. J. Biotechnol. 2013, 163, 281–291. [Google Scholar] [CrossRef]
- Dai, F.; Zhang, C.; Jiang, X.; Kang, M.; Yin, X.; Lu, P.; Zhang, X.; Zheng, Y.; Gao, J. RhNAC2 and RhEXPA4 Are Involved in the Regulation of Dehydration Tolerance during the Expansion of Rose Petals. Plant Physiol. 2012, 160, 2064–2082. [Google Scholar] [CrossRef]
- Torres, M.A.; Dangl, J.L. Functions of the respiratory burst oxidase in biotic interactions, abiotic stress and development. Curr. Opin. Plant Biol. 2005, 8, 397–403. [Google Scholar] [CrossRef]
- Shigeto, J.; Tsutsumi, Y. Diverse functions and reactions of class III peroxidases. New Phytol. 2016, 209, 1395–1402. [Google Scholar] [CrossRef]
- Raggi, S.; Ferrarini, A.; Delledonne, M.; Dunand, C.; Ranocha, P.; Lorenzo, G.D.; Cervone, F.; Ferrari, S. The Arabidopsis thaliana Class III Peroxidase AtPRX71 Negatively Regulates Growth under Physiological Conditions and in Response to Cell Wall Damage. Plant Physiol. 2015, 169, 2513–2525. [Google Scholar] [CrossRef]
- Passardi, F.; Penel, C.; Dunand, C. Performing the paradoxical: How plant peroxidases modify the cell wall. Trends Plant Sci. 2004, 9, 534–540. [Google Scholar] [CrossRef]
- Kunieda, T.; Shimada, T.; Kondo, M.; Nishimura, M.; Nishitani, K.; Hara-Nishimura, I. Spatiotemporal Secretion of PEROXIDASE36 Is Required for Seed Coat Mucilage Extrusion in Arabidopsis. Plant Cell 2013, 25, 1355–1367. [Google Scholar] [CrossRef]
- Miller, G.; Suzuki, N.; Ciftci-Yilmaz, S.; Mittler, R. Reactive oxygen species homeostasis and signalling during drought and salinity stresses. Plant Cell Environ. 2010, 33, 453–467. [Google Scholar] [CrossRef]
- You, J.; Chan, Z. ROS Regulation During Abiotic Stress Responses in Crop Plants. Front. Plant Sci. 2015, 6, 1092. [Google Scholar] [CrossRef] [PubMed]
- Patro, L.; Mohapatra, P.K.; Biswal, U.C.; Biswal, B. Dehydration induced loss of photosynthesis in Arabidopsis leaves during senescence is accompanied by the reversible enhancement in the activity of cell wall β-glucosidase. J. Photochem. Photobiol. B Biol. 2014, 137, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Cai, X.; Xu, C.; Wang, Q.; Dai, S. Drought-Responsive Mechanisms in Plant Leaves Revealed by Proteomics. Int. J. Mol. Sci. 2016, 17, 1706. [Google Scholar] [CrossRef] [PubMed]
- Voss, I.; Sunil, B.; Scheibe, R.; Raghavendra, A.S. Emerging concept for the role of photorespiration as an important part of abiotic stress response. Plant Biol. 2013, 15, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Noctor, G.; Mhamdi, A.; Foyer, C.H. The Roles of Reactive Oxygen Metabolism in Drought: Not So Cut and Dried. Plant Physiol. 2014, 164, 1636–1648. [Google Scholar] [CrossRef]
- Nakabayashi, R.; Yonekura-Sakakibara, K.; Urano, K.; Suzuki, M.; Yamada, Y.; Nishizawa, T.; Matsuda, F.; Kojima, F.; Sakakibara, H.; Shinozaki, K.; et al. Enhancement of oxidative and drought tolerance in Arabidopsis by overaccumulation of antioxidant flavonoids. Plant J. 2014, 77, 367–379. [Google Scholar] [CrossRef]
- Wang, J.; Lan, P.; Gao, H.; Zheng, L.; Li, W.; Schmidt, W. Expression changes of ribosomal proteins in phosphate-and iron-deficient Arabidopsis roots predict stress-specific alterations in ribosome composition. BMC Genom. 2013, 14, 783. [Google Scholar] [CrossRef]
- Miazek, A.; Zagdańska, B. Involvement of exopeptidases in dehydration tolerance of spring wheat seedlings. Biologia Plant. 2008, 52, 687–694. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Q.; Wu, J.; Zheng, X.; Zheng, S.; Sun, X.; Qiu, Q.; Lu, T. Gene Knockout Study Reveals That Cytosolic Ascorbate Peroxidase 2(OsAPX2) Plays a Critical Role in Growth and Reproduction in Rice under Drought, Salt and Cold Stresses. PLoS ONE 2013, 8, 2. [Google Scholar] [CrossRef]
- Cramer, G.R.; Van Sluyter, S.C.; Hopper, D.W.; Pascovici, D.; Keighley, T.; Haynes, P.A. Proteomic analysis indicates massive changes in metabolism prior to the inhibition of growth and photosynthesis of grapevine (Vitis vinifera L.) in response to water deficit. BMC Plant Biol. 2013, 13, 49. [Google Scholar] [CrossRef]
- Xu, J.; Xing, X.; Tian, Y.; Peng, R.; Xue, Y.; Zhao, W.; Yao, Q. Transgenic Arabidopsis Plants Expressing Tomato Glutathione S-Transferase Showed Enhanced Resistance to Salt and Drought Stress. PLoS ONE 2015, 10, 9. [Google Scholar] [CrossRef] [PubMed]
- Islam, T.; Manna, M.; Reddy, M.K. Glutathione Peroxidase of Pennisetum glaucum (PgGPx) Is a Functional Cd2 Dependent Peroxiredoxin that Enhances Tolerance against Salinity and Drought Stress. PLoS ONE 2015, 10, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Ma, X.; Guo, H.; Sukiran, N.L.; Guo, B.; Assmann, S.M.; Ma, H. Flower Development under Drought Stress: Morphological and Transcriptomic Analyses Reveal Acute Responses and Long-Term Acclimation in Arabidopsis. Plant Cell 2013, 25, 3785–3807. [Google Scholar] [CrossRef] [PubMed]
- Bhaskara, G.B.; Nguyen, T.T.; Verslues, P.E. Unique Drought Resistance Functions of the Highly ABA-Induced Clade A Protein Phosphatase 2Cs. Plant Physiol. 2012, 160, 379–395. [Google Scholar] [CrossRef] [PubMed]
- Gosti, F. ABI1 Protein Phosphatase 2C Is a Negative Regulator of Abscisic Acid Signaling. Plant Cell Online 1999, 11, 1897–1910. [Google Scholar] [CrossRef] [PubMed]
- Mandava, N.B. Plant Growth-Promoting Brassinosteroids. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1988, 39, 23–52. [Google Scholar] [CrossRef]
- Neff, M.M.; Nguyen, S.M.; Malancharuvil, E.J.; Fujioka, S.; Noguchi, T.; Seto, H.; Tsukubi, M.; Takatsuto, S.; Yoshida, S.; Chory, J. BAS1: A gene regulating brassinosteroid levels and light responsiveness in Arabidopsis. Proc. Natl. Acad. Sci. USA 1999, 96, 15316–15323. [Google Scholar] [CrossRef] [PubMed]
- Belkhadir, Y.; Chory, J. Brassinosteroid Signaling: A Paradigm for Steroid Hormone Signaling from the Cell Surface. Science 2006, 314, 1410–1411. [Google Scholar] [CrossRef] [PubMed]
- Clouse, S.D. Brassinosteroid Signal Transduction: From Receptor Kinase Activation to Transcriptional Networks Regulating Plant Development. Plant Cell 2011, 23, 1219–1230. [Google Scholar] [CrossRef] [PubMed]
- Uga, Y.; Sugimoto, K.; Ogawa, S.; Rane, J.; Ishitani, M.; Hara, N.; Kitomi, Y.; Inukai, Y.; Ono, K.; Kanno, N.; et al. Control of root system architecture by DEEPER ROOTING 1 increases rice yield under drought conditions. Nat. Genet. 2013, 45, 1097–1102. [Google Scholar] [CrossRef]
- Verelst, W.; Asard, H. A phylogenetic study of cytochrome b561 proteins. Genome Biol. 2003, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Asard, H.; Barbaro, R.; Trost, P.; Bérczi, A. Cytochromes b561: Ascorbate-mediated trans-membrane electron transport. Antioxid. Redox Signal. 2013, 19, 1026–1035. [Google Scholar] [CrossRef] [PubMed]
- Nanasato, Y.; Akashi, K.; Yokota, A. Co-expression of Cytochrome b561 and Ascorbate Oxidase in Leaves of Wild Watermelon under Drought and High Light Conditions. Plant Cell Physiol. 2005, 46, 1515–1524. [Google Scholar] [CrossRef]
- Lyer, L.M.; Anantharaman, V.; Aravind, L. The DOMON domains are involved in heme and sugar recognition. Bioinformatics 2007, 23, 2660–2664. [Google Scholar]
- Choudhury, F.K.; Rivero, R.M.; Blumwald, E.; Mittler, R. Reactive oxygen species, abiotic stress and stress combination. Plant J. 2016, 90, 856–867. [Google Scholar] [CrossRef] [PubMed]
- Mittler, R. ROS Are Good. Trends Plant. Sci. 2017, 22, 11–19. [Google Scholar] [CrossRef]
- Oszvald, M.; Primavesi, L.F.; Griffiths, C.A.; Cohn, J.; Basu, S.S.; Nuccio, M.L.; Paul, M.J. Trehalose 6-Phosphate Regulates Photosynthesis and Assimilate Partitioning in Reproductive Tissue. Plant Physiol. 2018, 176, 2623–2638. [Google Scholar] [CrossRef]
- Sánchez-Barrena, M.J.; Fujii, H.; Angulo, I.; Martínez-Ripoll, M.; Zhu, J.; Albert, A. The Structure of the C-terminal Domain of the Protein Kinase AtSOS2 Bound to the Calcium Sensor AtSOS3. Mol. Cell 2007, 26, 427–435. [Google Scholar] [CrossRef]
- Hirabak, E.M.; Chan, C.W.; Gribskov, M.; Harper, J.F.; Choi, J.H.; Halford, N.; Kudla, J.; Luan, S.; Nimmo, H.G.; Sussman, M.R.; et al. The Arabidopsis CDPK-SnRK Superfamily of Protein Kinases. Plant Physiol. 2003, 132, 666–680. [Google Scholar] [CrossRef]
- Wang, Y.; Yan, H.; Qiu, Z.; Hu, B.; Zeng, B.; Zhong, C.; Fan, C. Comprehensive Analysis of SnRK Gene Family and their Responses to Salt Stress in Eucalyptus grandis. Int. J. Mol. Sci. 2019, 20, 2786. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DEGs of Interest | Expression | Genes | GO Terms | Significant GO Terms | |||
|---|---|---|---|---|---|---|---|
| Flag-Leaf | Panicle | Flag-Leaf | Panicle | Flag-Leaf | Panicle | ||
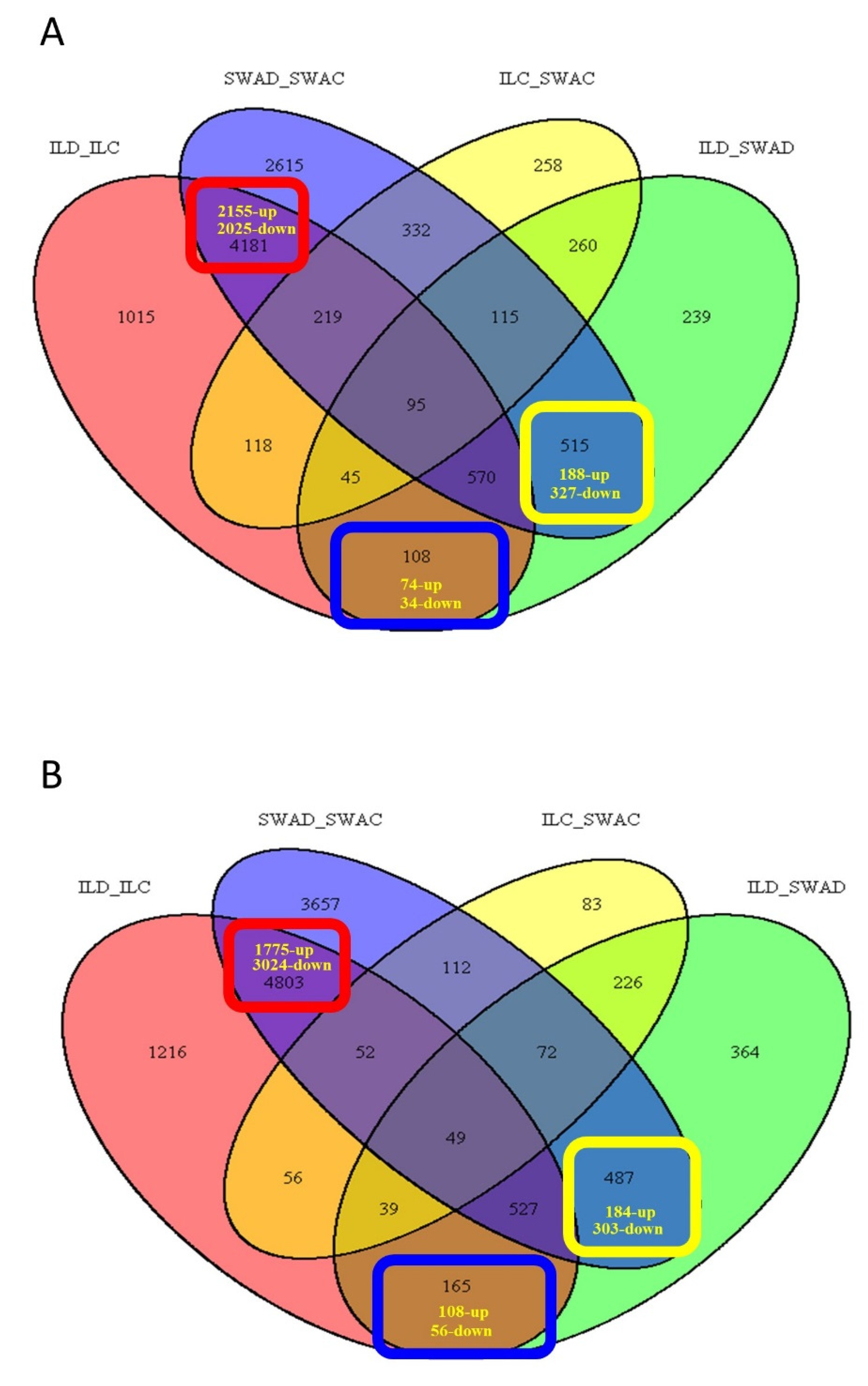
| Common responses of DTY-IL and Swarna DEGs to drought | Upregulated | 2155 | 1775 | 409 | 324 | 88 | 110 |
| Downregulated | 2025 | 3024 | 482 | 525 | 171 | 135 | |
| Sub-total | 4180 | 4799 | 891 | 849 | 259 | 245 | |
| Unique responses of Swarna DEGs to drought | Upregulated | 188 | 184 | 92 | 89 | 0 | 0 |
| Downregulated | 327 | 303 | 134 | 141 | 35 | 13 | |
| Sub-total | 515 | 487 | 226 | 230 | 35 | 13 | |
| Unique responses of DTY-IL DEGs to drought | Upregulated | 74 | 108 | 31 | 58 | 4 | 36 |
| Downregulated | 34 | 56 | 19 | 22 | 0 | 0 | |
| Sub-total | 108 | 164 | 50 | 80 | 4 | 36 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tarun, J.A.; Mauleon, R.; Arbelaez, J.D.; Catausan, S.; Dixit, S.; Kumar, A.; Brown, P.; Kohli, A.; Kretzschmar, T. Comparative Transcriptomics and Co-Expression Networks Reveal Tissue- and Genotype-Specific Responses of qDTYs to Reproductive-Stage Drought Stress in Rice (Oryza sativa L.). Genes 2020, 11, 1124. https://doi.org/10.3390/genes11101124
Tarun JA, Mauleon R, Arbelaez JD, Catausan S, Dixit S, Kumar A, Brown P, Kohli A, Kretzschmar T. Comparative Transcriptomics and Co-Expression Networks Reveal Tissue- and Genotype-Specific Responses of qDTYs to Reproductive-Stage Drought Stress in Rice (Oryza sativa L.). Genes. 2020; 11(10):1124. https://doi.org/10.3390/genes11101124
Chicago/Turabian StyleTarun, Jeshurun Asher, Ramil Mauleon, Juan David Arbelaez, Sheryl Catausan, Shalabh Dixit, Arvind Kumar, Patrick Brown, Ajay Kohli, and Tobias Kretzschmar. 2020. "Comparative Transcriptomics and Co-Expression Networks Reveal Tissue- and Genotype-Specific Responses of qDTYs to Reproductive-Stage Drought Stress in Rice (Oryza sativa L.)" Genes 11, no. 10: 1124. https://doi.org/10.3390/genes11101124
APA StyleTarun, J. A., Mauleon, R., Arbelaez, J. D., Catausan, S., Dixit, S., Kumar, A., Brown, P., Kohli, A., & Kretzschmar, T. (2020). Comparative Transcriptomics and Co-Expression Networks Reveal Tissue- and Genotype-Specific Responses of qDTYs to Reproductive-Stage Drought Stress in Rice (Oryza sativa L.). Genes, 11(10), 1124. https://doi.org/10.3390/genes11101124