Mechanisms of Photoreceptor Death in Retinitis Pigmentosa


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
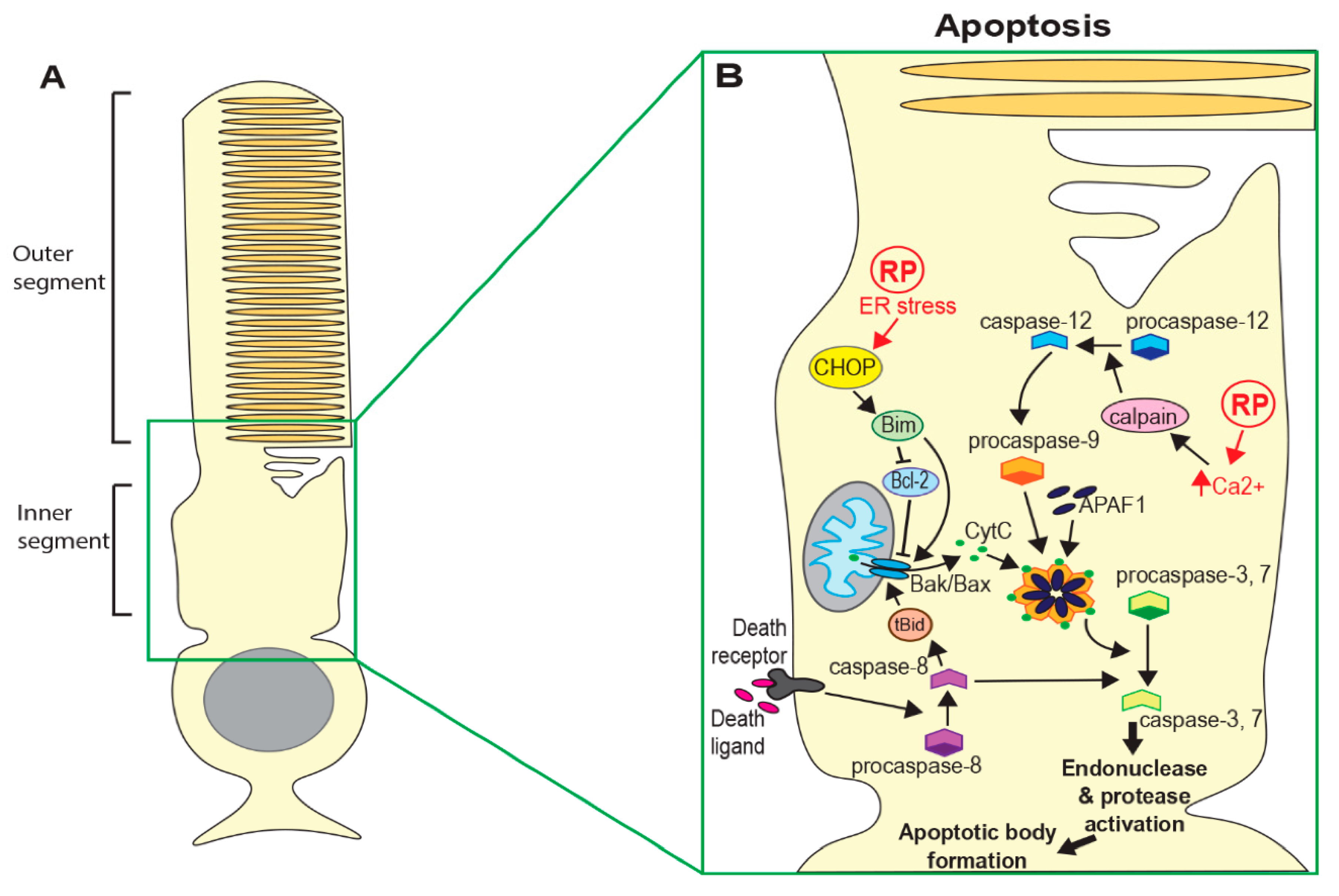
2. Cell Death Mechanisms
2.1. Apoptosis
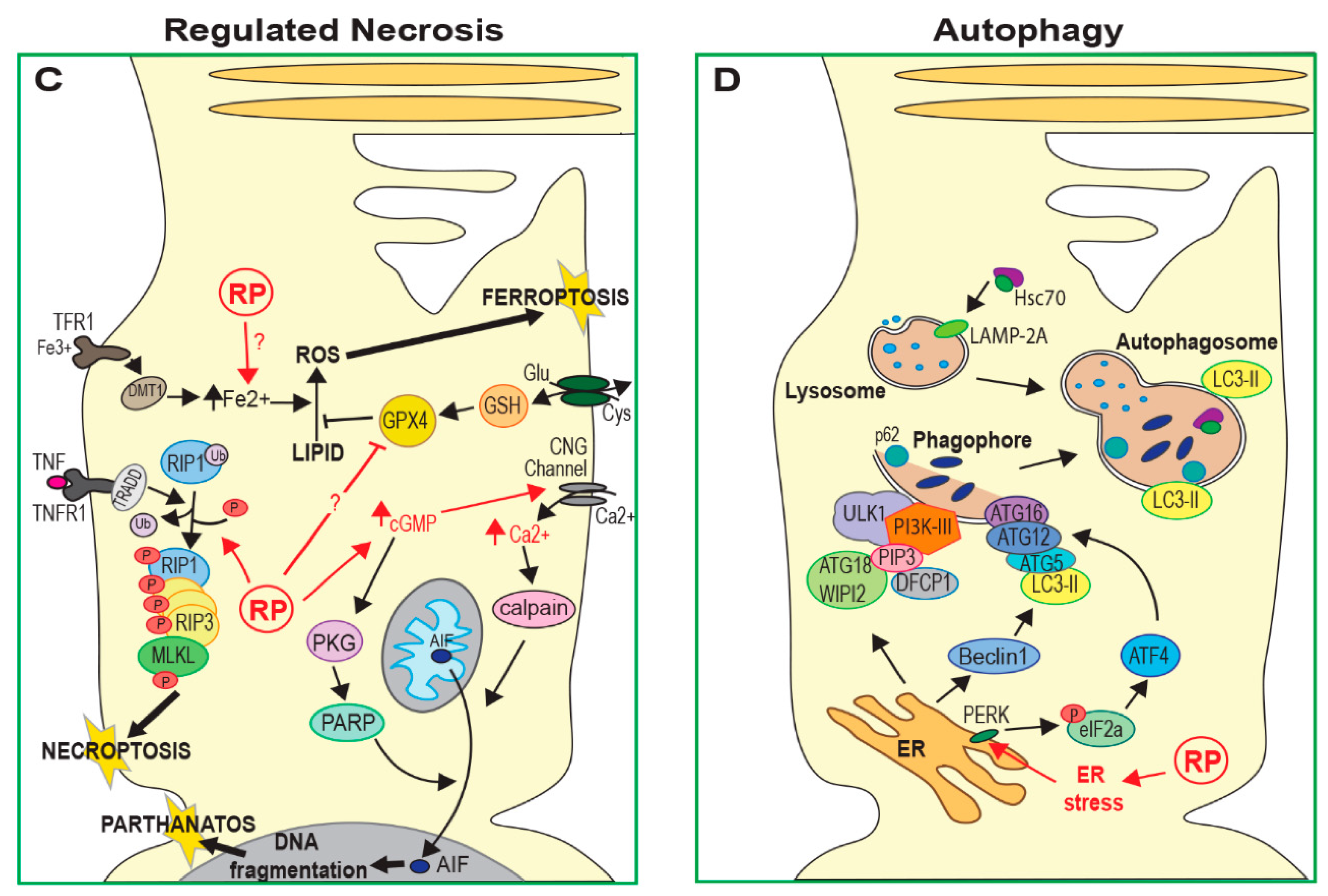
2.2. Regulated Necrosis
2.3. Autophagy
3. Pathways Leading to the Death of Rod Photoreceptors in RP
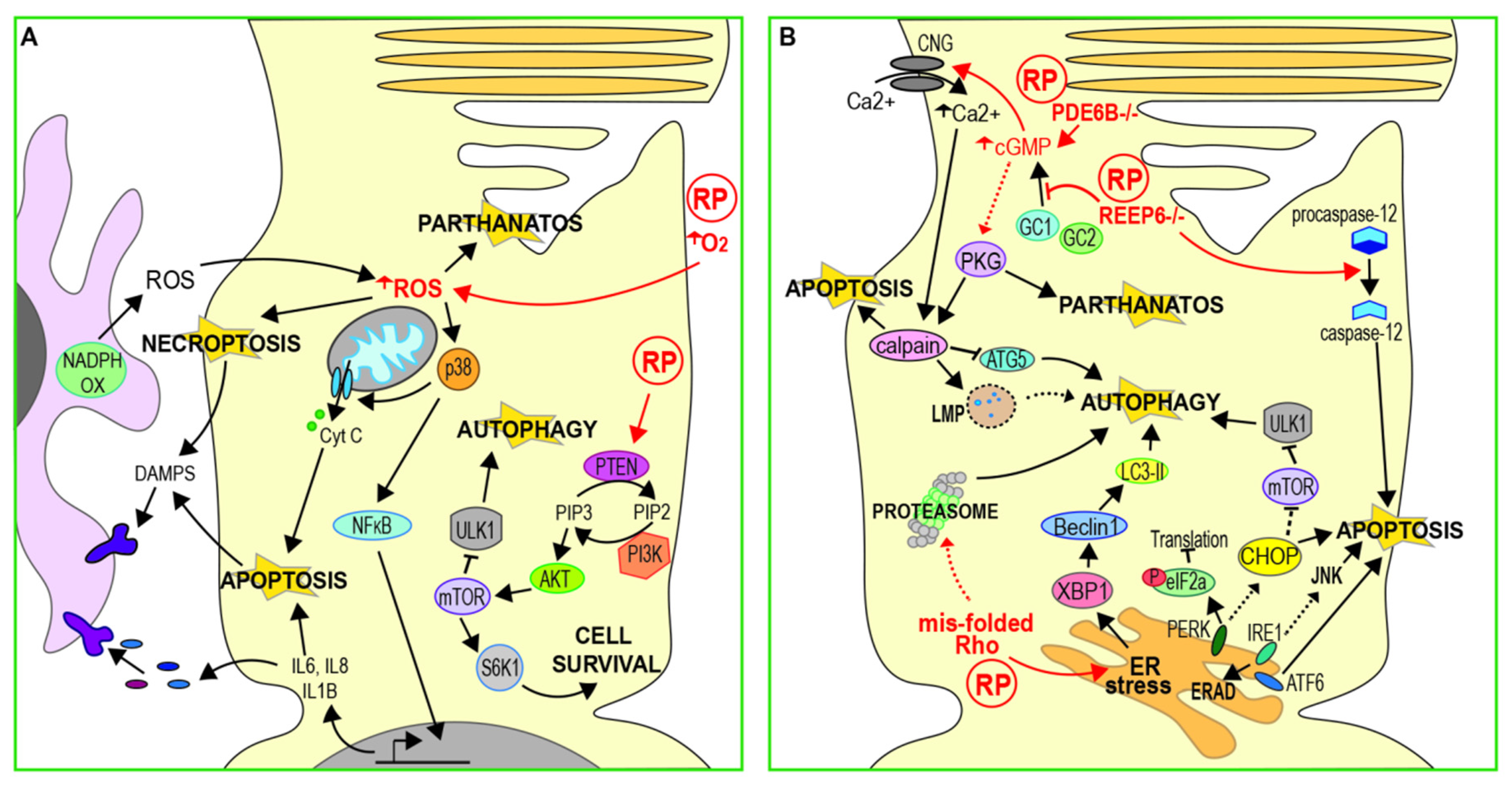
3.1. Oxidative Stress
3.2. Metabolic Stress
3.3. ER Stress and Calcium Regulation
3.3.1. cGMP Regulation of Calcium
3.3.2. Calcium Regulates Apoptosis Independent of ER Stress
3.3.3. Calcium and Non-Apoptotic Cell Death
3.4. Autophagy and Proteasome Regulation
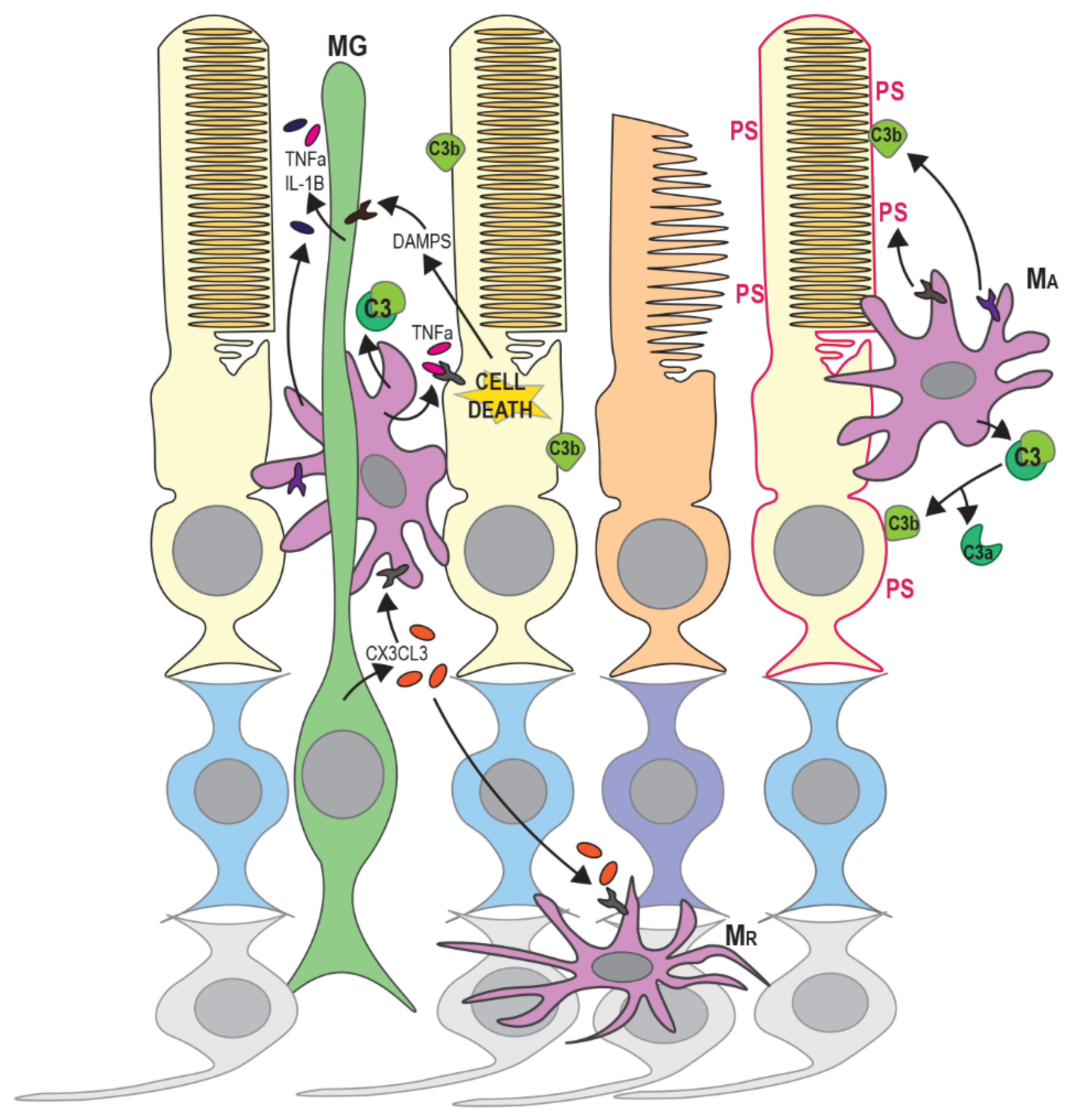
4. Recruitment and Activation of Microglia and Monocytes
5. Mechanisms of Secondary Cone Death in RP
6. Clinical Trials
6.1. Neuroprotective Agents
6.2. Anti-Inflammatory Agents
6.3. Targeting ER Stress and Protein Homeostasis
6.4. Novel Therapeutics for RP
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wright, A.F.; Chakarova, C.F.; El-Aziz, M.M.A.; Bhattacharya, S.S. Photoreceptor degeneration: Genetic and mechanistic dissection of a complex trait. Nat. Rev. Genet. 2010, 11, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Sahel, J.-A.; Marazova, K.; Audo, I. Clinical Characteristics and Current Therapies for Inherited Retinal Degenerations. Cold Spring Harb. Perspect. Med. 2014, 5, a017111. [Google Scholar] [CrossRef] [PubMed]
- Megaw, R.; Soares, D.C.; Wright, A.F. RPGR: Its role in photoreceptor physiology, human disease, and future therapies. Exp. Eye Res. 2015, 138, 32–41. [Google Scholar] [CrossRef] [PubMed]
- E McLaughlin, M.; Sandberg, M.A.; Berson, E.L.; Dryja, T.P. Recessive mutations in the gene encoding the β–subunit of rod phosphodiesterase in patients with retinitis pigmentosa. Nat. Genet. 1993, 4, 130–134. [Google Scholar] [CrossRef]
- Hollingsworth, T.; Gross, A.K. Defective Trafficking of Rhodopsin and Its Role in Retinal Degenerations. Int. Rev. Cell Mol. Biol. 2012, 293, 1–44. [Google Scholar] [CrossRef] [PubMed]
- Radi, E.; Formichi, P.; Battisti, C.; Federico, A. Apoptosis and Oxidative Stress in Neurodegenerative Diseases. J. Alzheimer’s Dis. 2014, 42, S125–S152. [Google Scholar] [CrossRef]
- Choudhury, S.; Bhootada, Y.; Gorbatyuk, O.; Gorbatyuk, M. Caspase-7 ablation modulates UPR, reprograms TRAF2-JNK apoptosis and protects T17M rhodopsin mice from severe retinal degeneration. Cell Death Dis. 2013, 4, e528. [Google Scholar] [CrossRef]
- Comitato, A.; Sanges, D.; Rossi, A.; Humphries, M.M.; Marigo, V. Activation of Bax in Three Models of Retinitis Pigmentosa. Investig. Opthalmol. Vis. Sci. 2014, 55, 3555–3561. [Google Scholar] [CrossRef]
- Elmore, S.A. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Igney, F.H.; Krammer, P.H. Death and anti-death: Tumour resistance to apoptosis. Nat. Rev. Cancer 2002, 2, 277–288. [Google Scholar] [CrossRef]
- Kunte, M.M.; Choudhury, S.; Manheim, J.F.; Shinde, V.M.; Miura, M.; Chiodo, V.A.; Hauswirth, W.W.; Gorbatyuk, O.S.; Gorbatyuk, M. ER Stress Is Involved in T17M Rhodopsin-Induced Retinal Degeneration. Investig. Opthalmol. Vis. Sci. 2012, 53, 3792–3800. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yang, M.; Wang, Z.-J. (Z)-7,4′-Dimethoxy-6-hydroxy-aurone-4-O-β-glucopyranoside mitigates retinal degeneration in Rd10 mouse model through inhibiting oxidative stress and inflammatory responses. Cutan. Ocul. Toxicol. 2019, 39, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Arango-Gonzalez, B.; Trifunović, D.; Sahaboglu, A.; Kranz, K.; Michalakis, S.; Farinelli, P.; Koch, S.; Koch, F.; Cottet, S.; Janssen-Bienhold, U.; et al. Identification of a Common Non-Apoptotic Cell Death Mechanism in Hereditary Retinal Degeneration. PLoS ONE 2014, 9, e112142. [Google Scholar] [CrossRef]
- Viringipurampeer, I.A.; Gregory-Evans, C.Y.; Metcalfe, A.L.; Bashar, E.; Moritz, O.L.; Gregory-Evans, K. Cell Death Pathways in Mutant Rhodopsin Rat Models Identifies Genotype-Specific Targets Controlling Retinal Degeneration. Mol. Neurobiol. 2018, 56, 1637–1652. [Google Scholar] [CrossRef]
- Vandenberghe, T.; Linkermann, A.; Jouan, E.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef]
- Ying, Y.; Padanilam, B.J. Regulation of necrotic cell death: p53, PARP1 and cyclophilin D-overlapping pathways of regulated necrosis? Cell. Mol. Life Sci. 2016, 73, 2309–2324. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Galluzzi, L.; Vandenberghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; A Moskowitz, M.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Methods 2005, 1, 112–119. [Google Scholar] [CrossRef]
- Dondelinger, Y.; Declercq, W.; Montessuit, S.; Roelandt, R.; Gonçalves, A.; Bruggeman, I.; Hulpiau, P.; Weber, K.; Sehon, C.A.; Marquis, R.W.; et al. MLKL Compromises Plasma Membrane Integrity by Binding to Phosphatidylinositol Phosphates. Cell Rep. 2014, 7, 971–981. [Google Scholar] [CrossRef]
- Murakami, Y.; Matsumoto, H.; Roh, M.; Suzuki, J.; Hisatomi, T.; Ikeda, Y.; Miller, J.W.; Vavvas, D.G. Receptor interacting protein kinase mediates necrotic cone but not rod cell death in a mouse model of inherited degeneration. Proc. Natl. Acad. Sci. USA 2012, 109, 14598–14603. [Google Scholar] [CrossRef]
- Sato, K.; Li, S.; Gordon, W.C.; He, J.; Liou, G.I.; Hill, J.M.; Travis, G.H.; Bazan, N.G.; Jin, M. Receptor interacting protein kinase-mediated necrosis contributes to cone and rod photoreceptor degeneration in the retina lacking interphotoreceptor retinoid-binding protein. J. Neurosci. 2013, 33, 17458–17468. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Fu, Y.; Liu, X.; Shahi, P.K.; Mavlyutov, T.A.; Li, J.; Yao, A.; Guo, S.Z.-W.; Pattnaik, B.R.; Guo, L.-W. Role of the sigma-1 receptor chaperone in rod and cone photoreceptor degenerations in a mouse model of retinitis pigmentosa. Mol. Neurodegener. 2017, 12, 68. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Obolensky, A.; Berenshtein, E.; Lederman, M.; Bulvik, B.; Alper-Pinus, R.; Yaul, R.; DeLeon, E.; Chowers, I.; Chevion, M.; Banin, E. Zinc–desferrioxamine attenuates retinal degeneration in the rd10 mouse model of retinitis pigmentosa. Free. Radic. Biol. Med. 2011, 51, 1482–1491. [Google Scholar] [CrossRef]
- Lu, L.; Oveson, B.C.; Jo, Y.; Lauer, T.W.; Usui, S.; Komeima, K.; Xie, B.; Campochiaro, P.A. Increased Expression of Glutathione Peroxidase 4 Strongly Protects Retina from Oxidative Damage. Antioxid. Redox Signal. 2009, 11, 715–724. [Google Scholar] [CrossRef]
- Usui, S.; Oveson, B.C.; Iwase, T.; Lu, L.; Lee, S.Y.; Jo, Y.-J.; Wu, Z.; Choi, E.-Y.; Samulski, R.J.; Campochiaro, P.A. Overexpression of SOD in retina: Need for increase in H2O2-detoxifying enzyme in same cellular compartment. Free. Radic. Biol. Med. 2011, 51, 1347–1354. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Yumnamcha, T.; Devi, T.S.; Singh, L.P. Auranofin Mediates Mitochondrial Dysregulation and Inflammatory Cell Death in Human Retinal Pigment Epithelial Cells: Implications of Retinal Neurodegenerative Diseases. Front. Mol. Neurosci. 2019, 13. [Google Scholar] [CrossRef]
- Fatokun, A.A.; Dawson, V.L.; Dawson, T.M. Parthanatos: Mitochondrial-linked mechanisms and therapeutic opportunities. Br. J. Pharmacol. 2014, 171, 2000–2016. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef]
- Boya, P.; Esteban, L.; Serrano-Puebla, A.; Gómez-Sintes, R.; Villarejo-Zori, B. Autophagy in the eye: Development, degeneration, and aging. Prog. Retin. Eye Res. 2016, 55, 206–245. [Google Scholar] [CrossRef] [PubMed]
- Dice, J.F.; Terlecky, S.R.; Chiang, H.L.; Olson, T.S.; Isenman, L.D.; Short-Russell, S.R.; Freundlieb, S.; Terlecky, L.J. A selective pathway for degradation of cytosolic proteins by lysosomes. Semin. Cell Biol. 1990, 1, 449–455. [Google Scholar]
- Sahu, R.; Kaushik, S.; Clement, C.C.; Cannizzo, E.S.; Scharf, B.; Follenzi, A.; Potolicchio, I.; Nieves, E.; Cuervo, A.M.; Santambrogio, L. Microautophagy of Cytosolic Proteins by Late Endosomes. Dev. Cell 2011, 20, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Panda, P.K.; Sinha, N.; Das, D.N.; Bhutia, S.K. Autophagy and apoptosis: Where do they meet? Apoptosis 2014, 19, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Muela, N.; Hernández-Pinto, A.M.; Serrano-Puebla, A.; García-Ledo, L.; Latorre, S.H.; De La Rosa, E.J.; Boya, P. Lysosomal membrane permeabilization and autophagy blockade contribute to photoreceptor cell death in a mouse model of retinitis pigmentosa. Cell Death Differ. 2014, 22, 476–487. [Google Scholar] [CrossRef]
- Yao, J.; Qiu, Y.; Frontera, E.; Jia, L.; Khan, N.W.; Klionsky, D.J.; Ferguson, T.A.; Thompson, D.A.; Zacks, D.N. Inhibiting autophagy reduces retinal degeneration caused by protein misfolding. Autophagy 2018, 14, 1226–1238. [Google Scholar] [CrossRef]
- Qiu, Y.; Yao, J.; Jia, L.; Thompson, D.A.; Zacks, D.N. Shifting the balance of autophagy and proteasome activation reduces proteotoxic cell death: A novel therapeutic approach for restoring photoreceptor homeostasis. Cell Death Dis. 2019, 10, 547. [Google Scholar] [CrossRef]
- Li, Y.; Wang, C.; Liu, Y.; You, J.; Su, G.-F. Autophagy, lysosome dysfunction and mTOR inhibition in MNU-induced photoreceptor cell damage. Tissue Cell 2019, 61, 98–108. [Google Scholar] [CrossRef]
- Wellard, J.W.; Lee, D.; Valter, K.; Stone, J. Photoreceptors in the rat retina are specifically vulnerable to both hypoxia and hyperoxia. Vis. Neurosci. 2005, 22, 501–507. [Google Scholar] [CrossRef]
- Lange, C.A.; Bainbridge, J.W. Oxygen Sensing in Retinal Health and Disease. Ophthalmologica 2012, 227, 115–131. [Google Scholar] [CrossRef]
- Natoli, R.; Valter, K.; Chrysostomou, V.; Stone, J.; Provis, J. Morphological, functional and gene expression analysis of the hyperoxic mouse retina. Exp. Eye Res. 2011, 92, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.-Y.; Cringle, S.J.; Su, E.N.; Yu, P.K. Intraretinal oxygen levels before and after photoreceptor loss in the RCS rat. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3999–4006. [Google Scholar]
- Yu, D.-Y.; Cringle, S.; Valter, K.; Walsh, N.; Lee, D.; Stone, J. Photoreceptor Death, Trophic Factor Expression, Retinal Oxygen Status, and Photoreceptor Function in the P23H Rat. Investig. Opthalmol. Vis. Sci. 2004, 45, 2013–2019. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Ding, M.; Chen, X.-X.; Lu, Q. Microglial NADPH oxidase activation mediates rod cell death in the retinal degeneration in rd mice. Neuroscience 2014, 275, 54–61. [Google Scholar] [CrossRef]
- Tao, Y.; Cai, L.; Zhou, D.; Wang, C.; Ma, Z.; Dong, X.; Peng, G. CoPP-Induced-Induced HO-1 Overexpression Alleviates Photoreceptor Degeneration With Rapid Dynamics: A Therapeutic Molecular Against Retinopathy. Investig. Opthalmol. Vis. Sci. 2019, 60, 5080–5094. [Google Scholar] [CrossRef]
- Noguchi, T.; Takeda, K.; Matsuzawa, A.; Saegusa, K.; Nakano, H.; Gohda, J.; Inoue, J.-I.; Ichijo, H. Recruitment of Tumor Necrosis Factor Receptor-associated Factor Family Proteins to Apoptosis Signal-regulating Kinase 1 Signalosome Is Essential for Oxidative Stress-induced Cell Death. J. Biol. Chem. 2005, 280, 37033–37040. [Google Scholar] [CrossRef]
- Hanuš, J.; Zhang, H.; Wang, Z.; Liu, Q.; Zhou, Q.; Wang, S. Induction of necrotic cell death by oxidative stress in retinal pigment epithelial cells. Cell Death Dis. 2013, 4, e965. [Google Scholar] [CrossRef]
- Noguchi, T.; Suzuki, M.; Mutoh, N.; Hirata, Y.; Tsuchida, M.; Miyagawa, S.; Hwang, G.-W.; Aoki, J.; Matsuzawa, A. Nuclear-accumulated SQSTM1/p62-based ALIS act as microdomains sensing cellular stresses and triggering oxidative stress-induced parthanatos. Cell Death Dis. 2018, 9, 1193. [Google Scholar] [CrossRef]
- Zhang, L.; Justus, S.; Xu, Y.; Pluchenik, T.; Hsu, C.-W.; Yang, J.; Duong, J.K.; Lin, C.-S.; Jia, Y.; Bassuk, A.G.; et al. Reprogramming towards anabolism impedes degeneration in a preclinical model of retinitis pigmentosa. Hum. Mol. Genet. 2016, 25, 4244–4255. [Google Scholar] [CrossRef]
- Lin, B.; Xiong, G.; Yang, W. Ribosomal protein S6 kinase 1 promotes the survival of photoreceptors in retinitis pigmentosa. Cell Death Dis. 2018, 9, 1141. [Google Scholar] [CrossRef]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef]
- Shore, G.C.; Papa, F.R.; Oakes, S.A. Signaling cell death from the endoplasmic reticulum stress response. Curr. Opin. Cell Biol. 2011, 23, 143–149. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of Stress in the ER to Activation of JNK Protein Kinases by Transmembrane Protein Kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef]
- Lerner, A.G.; Upton, J.-P.; Praveen, P.; Ghosh, R.; Nakagawa, Y.; Igbaria, A.; Shen, S.; Nguyen, V.; Backes, B.J.; Heiman, M.; et al. IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 2012, 16, 250–264. [Google Scholar] [CrossRef] [PubMed]
- McCullough, K.D.; Martindale, J.L.; Klotz, L.-O.; Aw, T.-Y.; Holbrook, N.J. Gadd153 Sensitizes Cells to Endoplasmic Reticulum Stress by Down-Regulating Bcl2 and Perturbing the Cellular Redox State. Mol. Cell. Biol. 2001, 21, 1249–1259. [Google Scholar] [CrossRef]
- Lin, J.H.; Li, H.; Yasumura, U.; Cohen, H.R.; Zhang, C.; Panning, B.; Shokat, K.M.; Lavail, M.M.; Walter, P. IRE1 Signaling Affects Cell Fate During the Unfolded Protein Response. Science 2007, 318, 944–949. [Google Scholar] [CrossRef]
- Comitato, A.; Schiroli, D.; La Marca, C.; Marigo, V. Differential Contribution of Calcium-Activated Proteases and ER-Stress in Three Mouse Models of Retinitis Pigmentosa Expressing P23H Mutant RHO. Adv. Exp. Med. Biol. 2019, 1185, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Chiang, W.-C.; Kroeger, H.; Sakami, S.; Messah, C.; Yasumura, U.; Matthes, M.T.; Coppinger, J.A.; Palczewski, K.; Lavail, M.M.; Lin, J.H. Robust Endoplasmic Reticulum-Associated Degradation of Rhodopsin Precedes Retinal Degeneration. Mol. Neurobiol. 2014, 52, 679–695. [Google Scholar] [CrossRef]
- Athanasiou, D.; Aguila, M.; Bellingham, J.; Kanuga, N.; Adamson, P.; Cheetham, M.E. The role of the ER stress-response protein PERK in rhodopsin retinitis pigmentosa. Hum. Mol. Genet. 2017, 26, 4896–4905. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Butler, M.R.; Thapa, A.; Belcher, J.; Yang, F.; Baehr, W.; Biel, M.; Michalakis, S.; Ding, X.-Q. cGMP/Protein Kinase G Signaling Suppresses Inositol 1,4,5-Trisphosphate Receptor Phosphorylation and Promotes Endoplasmic Reticulum Stress in Photoreceptors of Cyclic Nucleotide-gated Channel-deficient Mice. J. Biol. Chem. 2015, 290, 20880–20892. [Google Scholar] [CrossRef]
- Paquet-Durand, F.; Beck, S.; Michalakis, S.; Goldmann, T.; Huber, G.; Mühlfriedel, R.; Trifunović, D.; Fischer, M.D.; Fahl, E.; Duetsch, G.; et al. A key role for cyclic nucleotide gated (CNG) channels in cGMP-related retinitis pigmentosa. Hum. Mol. Genet. 2010, 20, 941–947. [Google Scholar] [CrossRef]
- Fox, D.A.; Poblenz, A.T.; He, L. Calcium overload triggers rod photoreceptor apoptotic cell death in chemical-induced and inherited retinal degenerations. Ann. N. Y. Acad. Sci. 1999, 893, 282–285. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, A.R.; Giordano, F.; Gerlo, S.; Segura, I.; Van Eygen, S.; Molenberghs, G.; Rocha, S.; Houcine, A.; Derua, R.; Verfaillie, T.; et al. The ER Stress Sensor PERK Coordinates ER-Plasma Membrane Contact Site Formation through Interaction with Filamin-A and F-Actin Remodeling. Mol. Cell 2017, 65, 885–899.e6. [Google Scholar] [CrossRef] [PubMed]
- Xiong, K.; Cheng, S.-Y.; Wang, S.-C.; Lei, M.; Wang, Z. Regulatory role of calpain in neuronal death. Neural Regen. Res. 2018, 13, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Farber, D.B.; Lolley, R.N. Cyclic Guanosine Monophosphate: Elevation in Degenerating Photoreceptor Cells of the C3H Mouse Retina. Science 1974, 186, 449–451. [Google Scholar] [CrossRef] [PubMed]
- Kjellström, U.; Veiga-Crespo, P.; Andréasson, S.; Ekström, P. Increased Plasma cGMP in a Family with Autosomal Recessive Retinitis Pigmentosa Due to Homozygous Mutations in the PDE6A Gene. Investig. Opthalmol. Vis. Sci. 2016, 57, 6048–6057. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.-L.; Lei, X.-L.; Han, F.; He, K.-W.; Jin, S.-Q.; Zhang, Y.-Y.; Jin, Z.-B. Patient-Specific Retinal Organoids Recapitulate Disease Features of Late-Onset Retinitis Pigmentosa. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef]
- Wang, T.; Tsang, S.H.; Chen, J. Two pathways of rod photoreceptor cell death induced by elevated cGMP. Hum. Mol. Genet. 2017, 26, 2299–2306. [Google Scholar] [CrossRef]
- Paquet-Durand, F.; Hauck, S.M.; Van Veen, T.; Ueffing, M.; Ekström, P. PKG activity causes photoreceptor cell death in two retinitis pigmentosa models. J. Neurochem. 2009, 108, 796–810. [Google Scholar] [CrossRef]
- Arno, G.; Agrawal, S.A.; Eblimit, A.; Bellingham, J.; Xu, M.; Wang, F.; Chakarova, C.; Parfitt, D.A.; Lane, A.; Burgoyne, T.; et al. Mutations in REEP6 Cause Autosomal-Recessive Retinitis Pigmentosa. Am. J. Hum. Genet. 2016, 99, 1305–1315. [Google Scholar] [CrossRef]
- Agrawal, S.A.; Burgoyne, T.; Eblimit, A.; Bellingham, J.; Parfitt, D.A.; Lane, A.; Nichols, R.; Asomugha, C.; Hayes, M.J.; Munro, P.M.; et al. REEP6 deficiency leads to retinal degeneration through disruption of ER homeostasis and protein trafficking. Hum. Mol. Genet. 2017, 26, 2667–2677. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Zhu, H.; Morishima, N.; Li, E.; Xu, J.; Yankner, B.A.; Yuan, J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature 2000, 403, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.R.; Ma, H.; Yang, F.; Belcher, J.; Le, Y.-Z.; Mikoshiba, K.; Biel, M.; Michalakis, S.; Iuso, A.; Krizaj, D.; et al. Endoplasmic reticulum (ER) Ca2+—Channel activity contributes to ER stress and cone death in cyclic nucleotide-gated channel deficiency. J. Biol. Chem. 2017, 292, 11189–11205. [Google Scholar] [CrossRef] [PubMed]
- Sanges, D.; Comitato, A.; Tammaro, R.; Marigo, V. Apoptosis in retinal degeneration involves cross-talk between apoptosis-inducing factor (AIF) and caspase-12 and is blocked by calpain inhibitors. Proc. Natl. Acad. Sci. USA 2006, 103, 17366–17371. [Google Scholar] [CrossRef]
- Comitato, A.; Schiroli, D.; Montanari, M.; Marigo, V. Calpain Activation Is the Major Cause of Cell Death in Photoreceptors Expressing a Rhodopsin Misfolding Mutation. Mol. Neurobiol. 2019, 57, 589–599. [Google Scholar] [CrossRef]
- Venkatesh, A.; Cheng, S.-Y.; Punzo, C. Loss of the cone-enriched caspase-7 does not affect secondary cone death in retinitis pigmentosa. Mol. Vis. 2017, 23, 944–951. [Google Scholar] [PubMed]
- Kocaturk, N.M.; Gozuacik, D. Crosstalk Between Mammalian Autophagy and the Ubiquitin-Proteasome System. Front. Cell Dev. Biol. 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-J.; Chen, S.; Huang, K.-X.; Le, W. Why should autophagic flux be assessed? Acta Pharmacol. Sin. 2013, 34, 595–599. [Google Scholar] [CrossRef]
- Wang, J.; Saul, A.; Cui, X.; Roon, P.; Smith, S.B. Absence of Sigma 1 Receptor Accelerates Photoreceptor Cell Death in a Murine Model of Retinitis Pigmentosa. Investig. Opthalmol. Vis. Sci. 2017, 58, 4545–4558. [Google Scholar] [CrossRef]
- Aits, S.; Jäättelä, M. Lysosomal cell death at a glance. J. Cell Sci. 2013, 126, 1905–1912. [Google Scholar] [CrossRef]
- Sahara, S.; Yamashima, T. Calpain-mediated Hsp70.1 cleavage in hippocampal CA1 neuronal death. Biochem. Biophys. Res. Commun. 2010, 393, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Norman, J.M.; Cohen, G.M.; Bampton, E.T. The in vitro cleavage of the hAtg proteins by cell death proteases. Autophagy 2010, 6, 1042–1056. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Ishikawa, K.; Nakao, S.; Sonoda, K.-H. Innate immune response in retinal homeostasis and inflammatory disorders. Prog. Retin. Eye Res. 2020, 74, 100778. [Google Scholar] [CrossRef]
- Gupta, N.; E Brown, K.; Milam, A.H. Activated microglia in human retinitis pigmentosa, late-onset retinal degeneration, and age-related macular degeneration. Exp. Eye Res. 2003, 76, 463–471. [Google Scholar] [CrossRef]
- Zeiss, C.J.; Johnson, E.A. Proliferation of microglia, but not photoreceptors, in the outer nuclear layer of the rd-1 mouse. Investig. Opthalmol. Vis. Sci. 2004, 45, 971–976. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zabel, M.K.; Wang, X.; Ma, W.; Shah, P.; Fariss, R.; Qian, H.; Parkhurst, C.N.; Gan, W.-B.; Wong, W.T. Microglial phagocytosis of living photoreceptors contributes to inherited retinal degeneration. EMBO Mol. Med. 2015, 7, 1179–1197. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B. Innate immunity: An overview. Mol. Immunol. 2004, 40, 845–859. [Google Scholar] [CrossRef] [PubMed]
- Ajami, B.; Bennett, J.L.; Krieger, C.; Tetzlaff, W.; Rossi, F.M. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat. Neurosci. 2007, 10, 1538–1543. [Google Scholar] [CrossRef]
- Rashid, K.; Akhtar-Schaefer, I.; Langmann, T. Microglia in Retinal Degeneration. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, S.; Gong, W.; Zhu, G.; Wang, S.; Wang, Y.; Halim, M.; Wang, K.; Zhou, G.; Liu, Q. Müller Cell Regulated Microglial Activation and Migration in Rats with N-Methyl-N-Nitrosourea-Induced Retinal Degeneration. Front. Mol. Neurosci. 2018, 12. [Google Scholar] [CrossRef]
- Rutar, M.; Natoli, R.; Valter, K.; Provis, J. Early Focal Expression of the Chemokine Ccl2 by Müller Cells during Exposure to Damage-Inducing Bright Continuous Light. Investig. Opthalmol. Vis. Sci. 2011, 52, 2379–2388. [Google Scholar] [CrossRef] [PubMed]
- Vecino, E.; Rodriguez, F.; Ruzafa, N.; Pereiro, X.; Sharma, S.C. Glia–neuron interactions in the mammalian retina. Prog. Retin. Eye Res. 2016, 51, 1–40. [Google Scholar] [CrossRef]
- Reichenbach, A.; Bringmann, A. New functions of Müller cells. Glia 2013, 61, 651–678. [Google Scholar] [CrossRef] [PubMed]
- Feldman, N.; Rotter-Maskowitz, A.; Okun, E. DAMPs as mediators of sterile inflammation in aging-related pathologies. Ageing Res. Rev. 2015, 24, 29–39. [Google Scholar] [CrossRef]
- Jiao, H.; Natoli, R.; Valter, K.; Provis, J.; Rutar, M. Spatiotemporal Cadence of Macrophage Polarisation in a Model of Light-Induced Retinal Degeneration. PLoS ONE 2015, 10, e0143952. [Google Scholar] [CrossRef]
- Natoli, R.; Fernando, N.; Madigan, M.C.; Chu-Tan, J.A.; Valter, K.; Provis, J.; Rutar, M. Microglia-derived IL-1β promotes chemokine expression by Müller cells and RPE in focal retinal degeneration. Mol. Neurodegener. 2017, 12, 31. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Wang, X.; Liu, T.; Zhang, M.; Xu, G.; Ni, Y. Expression of CCL2 and its receptor in activation and migration of microglia and monocytes induced by photoreceptor apoptosis. Mol. Vis. 2017, 23, 765–777. [Google Scholar] [PubMed]
- Platón-Corchado, M.; Barcelona, P.F.; Jmaeff, S.; Marchena, M.; Hernández-Pinto, A.M.; Hernandez-Sanchez, C.; Saragovi, H.U.; De La Rosa, E.J. p75NTR antagonists attenuate photoreceptor cell loss in murine models of retinitis pigmentosa. Cell Death Dis. 2017, 8, e2922. [Google Scholar] [CrossRef]
- Hughes, E.H.; Schlichtenbrede, F.C.; Murphy, C.C.; Sarra, G.-M.; Luthert, P.J.; Ali, R.R.; Dick, A.D. Generation of activated sialoadhesin-positive microglia during retinal degeneration. Investig. Opthalmology Vis. Sci. 2003, 44, 2229–2234. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.-Y.; Zhu, X.-A.; Zhang, C.; Yang, L.-P.; Wu, L.-M.; Tso, M.O.M. Identification of Sequential Events and Factors Associated with Microglial Activation, Migration, and Cytotoxicity in Retinal Degeneration in rd Mice. Investig. Opthalmol. Vis. Sci. 2005, 46, 2992–2999. [Google Scholar] [CrossRef] [PubMed]
- Blank, T.; Goldmann, T.; Koch, M.; Amann, L.; Schön, C.; Bonin, M.; Pang, S.; Prinz, M.; Burnet, M.; Wagner, J.E.; et al. Early Microglia Activation Precedes Photoreceptor Degeneration in a Mouse Model of CNGB1-Linked Retinitis Pigmentosa. Front. Immunol. 2018, 8. [Google Scholar] [CrossRef]
- Megaw, R.; Abu-Arafeh, I.; Jungnickel, M.; Mellough, C.B.; Gurniak, C.; Witke, W.; Zhang, W.; Khanna, H.; Mill, P.; Dhillon, B.; et al. Gelsolin dysfunction causes photoreceptor loss in induced pluripotent cell and animal retinitis pigmentosa models. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef]
- Guo, C.; Otani, A.; Oishi, A.; Kojima, H.; Makiyama, Y.; Nakagawa, S.; Yoshimura, N. Knockout of ccr2 alleviates photoreceptor cell death in a model of retinitis pigmentosa. Exp. Eye Res. 2012, 104, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, M.; Pino, P.A.; Saederup, N.; Charo, I.F.; Ransohoff, R.M.; Cardona, A.E. The fractalkine receptor but not CCR2 is present on microglia from embryonic development throughout adulthood. J. Immunol. 2011, 188, 29–36. [Google Scholar] [CrossRef]
- O’Koren, E.G.; Yu, C.; Klingeborn, M.; Wong, A.Y.; Prigge, C.L.; Mathew, R.; Kalnitsky, J.; Msallam, R.A.; Silvin, A.; Kay, J.N.; et al. Microglial Function Is Distinct in Different Anatomical Locations during Retinal Homeostasis and Degeneration. Immunity 2019, 50, 723–737.e7. [Google Scholar] [CrossRef] [PubMed]
- Ronning, K.E.; Karlen, S.J.; Miller, E.B.; Burns, M.E. Molecular profiling of resident and infiltrating mononuclear phagocytes during rapid adult retinal degeneration using single-cell RNA sequencing. Sci. Rep. 2019, 9, 4858. [Google Scholar] [CrossRef]
- Wang, Y.; Yin, Z.; Gao, L.; Sun, D.; Hu, X.; Xue, L.; Dai, J.; Zeng, Y.; Chen, S.; Pan, B.; et al. Curcumin Delays Retinal Degeneration by Regulating Microglia Activation in the Retina of rd1 Mice. Cell. Physiol. Biochem. 2017, 44, 479–493. [Google Scholar] [CrossRef]
- Xu, X.-J.; Wang, S.-M.; Jin, Y.; Hu, Y.-T.; Feng, K.; Ma, Z.-Z. Melatonin delays photoreceptor degeneration in a mouse model of autosomal recessive retinitis pigmentosa. J. Pineal Res. 2017, 63, e12428. [Google Scholar] [CrossRef]
- Karali, M.; Guadagnino, I.; Marrocco, E.; De Cegli, R.; Carissimo, A.; Pizzo, M.; Casarosa, S.; Conte, I.; Surace, E.M.; Banfi, S. AAV-miR-204 Protects from Retinal Degeneration by Attenuation of Microglia Activation and Photoreceptor Cell Death. Mol. Ther. Nucleic Acids 2020, 19, 144–156. [Google Scholar] [CrossRef]
- Yoshida, N.; Ikeda, Y.; Notomi, S.; Ishikawa, K.; Murakami, Y.; Hisatomi, T.; Enaida, H.; Ishibashi, T. Laboratory Evidence of Sustained Chronic Inflammatory Reaction in Retinitis Pigmentosa. Ophthalmol. 2013, 120, e5–e12. [Google Scholar] [CrossRef]
- Brown, G.C.; Neher, J.J. Microglial phagocytosis of live neurons. Nat. Rev. Neurosci. 2014, 15, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, T.; Hisatomi, T.; Nakazawa, C.; Noda, K.; Maruyama, K.; She, H.; Matsubara, A.; Miyahara, S.; Nakao, S.; Yin, Y.; et al. Monocyte chemoattractant protein 1 mediates retinal detachment-induced photoreceptor apoptosis. Proc. Natl. Acad. Sci. USA 2007, 104, 2425–2430. [Google Scholar] [CrossRef]
- Eastlake, K.; Banerjee, P.J.; Angbohang, A.; Charteris, D.G.; Khaw, P.T.; Limb, G.A. Müller glia as an important source of cytokines and inflammatory factors present in the gliotic retina during proliferative vitreoretinopathy. Glia 2015, 64, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Zabel, M.K.; Zhao, L.; Zhang, Y.; Gonzalez, S.R.; Ma, W.; Wang, X.; Fariss, R.; Wong, W.T. Microglial phagocytosis and activation underlying photoreceptor degeneration is regulated by CX3CL1-CX3CR1 signaling in a mouse model of retinitis pigmentosa. Glia 2016, 64, 1479–1491. [Google Scholar] [CrossRef]
- Okunuki, Y.; Mukai, R.; Pearsall, E.A.; Klokman, G.; Husain, D.; Park, D.-H.; Korobkina, E.; Weiner, H.L.; Butovsky, O.; Ksander, B.R.; et al. Microglia inhibit photoreceptor cell death and regulate immune cell infiltration in response to retinal detachment. Proc. Natl. Acad. Sci. USA 2018, 115, E6264–E6273. [Google Scholar] [CrossRef]
- Ma, W.; Silverman, S.M.; Zhao, L.; Villasmil, R.; Campos, M.M.; Amaral, J.; Wong, W.T. Absence of TGFβ signaling in retinal microglia induces retinal degeneration and exacerbates choroidal neovascularization. eLife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Paschalis, E.I.; Lei, F.; Zhou, C.; Chen, X.; Kapoulea, V.; Hui, P.-C.; Dana, R.; Chodosh, J.; Vavvas, D.G.; Dohlman, C.H.; et al. Microglia Regulate Neuroglia Remodeling in Various Ocular and Retinal Injuries. J. Immunol. 2018, 202, 539–549. [Google Scholar] [CrossRef]
- Sakami, S.; Imanishi, Y.; Palczewski, K. Müller glia phagocytose dead photoreceptor cells in a mouse model of retinal degenerative disease. FASEB J. 2018, 33, 3680–3692. [Google Scholar] [CrossRef]
- Kassa, E.; Ciulla, T.A.; Hussain, R.M.; Dugel, P.U. Complement inhibition as a therapeutic strategy in retinal disorders. Expert Opin. Biol. Ther. 2019, 19, 335–342. [Google Scholar] [CrossRef]
- Turner, M. The lectin pathway of complement activation. Res. Immunol. 1996, 147, 110–115. [Google Scholar] [CrossRef]
- Taylor, P.R.; Carugati, A.; Fadok, V.A.; Cook, H.T.; Andrews, M.; Carroll, M.C.; Savill, J.S.; Henson, P.M.; Botto, M.; Walport, M.J. A Hierarchical Role for Classical Pathway Complement Proteins in the Clearance of Apoptotic Cells In Vivo. J. Exp. Med. 2000, 192, 359–366. [Google Scholar] [CrossRef]
- Luo, C.; Chen, M.; Xu, H. Complement gene expression and regulation in mouse retina and retinal pigment epithelium/choroid. Mol. Vis. 2011, 17, 1588–1597. [Google Scholar] [PubMed]
- Rutar, M.; Natoli, R.; Kozulin, P.; Valter, K.; Gatenby, P.; Provis, J. Analysis of Complement Expression in Light-Induced Retinal Degeneration: Synthesis and Deposition of C3 by Microglia/Macrophages Is Associated with Focal Photoreceptor Degeneration. Investig. Opthalmol. Vis. Sci. 2011, 52, 5347–5358. [Google Scholar] [CrossRef]
- Silverman, S.M.; Ma, W.; Wang, X.; Zhao, L.; Wong, W.T. C3- and CR3-dependent microglial clearance protects photoreceptors in retinitis pigmentosa. J. Exp. Med. 2019, 216, 1925–1943. [Google Scholar] [CrossRef]
- Garijo, A.P.; Fuchs, Y.; Steller, H. Apoptotic cells can induce non-autonomous apoptosis through the TNF pathway. eLife 2013, 2. [Google Scholar] [CrossRef]
- Ripps, H. Cell Death in Retinitis Pigmentosa: Gap Junctions and the ‘Bystander’ Effect. Exp. Eye Res. 2002, 74, 327–336. [Google Scholar] [CrossRef]
- Peng, B.; Xiao, J.; Wang, K.; So, K.-F.; Tipoe, G.L.; Lin, B. Suppression of Microglial Activation Is Neuroprotective in a Mouse Model of Human Retinitis Pigmentosa. J. Neurosci. 2014, 34, 8139–8150. [Google Scholar] [CrossRef]
- Léveillard, T.; Mohand-Saïd, S.; Lorentz, O.; Hicks, D.; Fintz, A.-C.; Clérin, E.; Simonutti, M.; Forster, V.; Cavusoglu, N.; Chalmel, F.; et al. Identification and characterization of rod-derived cone viability factor. Nat. Genet. 2004, 36, 755–759. [Google Scholar] [CrossRef]
- Byrne, L.C.; Dalkara, D.; Luna, G.; Fisher, S.K.; Clérin, E.; Sahel, J.-A.; Léveillard, T.; Flannery, J.G. Viral-mediated RdCVF and RdCVFL expression protects cone and rod photoreceptors in retinal degeneration. J. Clin. Investig. 2014, 125, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Cronin, T.; Raffelsberger, W.; Lee-Rivera, I.; Jaillard, C.; Niepon, M.-L.; Kinzel, B.; Clérin, E.; Petrosian, A.; Picaud, S.; Poch, O.; et al. The disruption of the rod-derived cone viability gene leads to photoreceptor dysfunction and susceptibility to oxidative stress. Cell Death Differ. 2010, 17, 1199–1210. [Google Scholar] [CrossRef] [PubMed]
- ElAchouri, G.; Lee-Rivera, I.; Clerin, E.; Argentini, M.; Fridlich, R.; Blond, F.; Ferracane, V.; Yang, Y.; Raffelsberger, W.; Wan, J.; et al. Thioredoxin rod-derived cone viability factor protects against photooxidative retinal damage. Free. Radic. Biol. Med. 2015, 81, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Aït-Ali, N.; Fridlich, R.; Millet-Puel, G.; Clérin, E.; Delalande, F.; Jaillard, C.; Blond, F.; Perrocheau, L.; Reichman, S.; Byrne, L.C.; et al. Rod-Derived Cone Viability Factor Promotes Cone Survival by Stimulating Aerobic Glycolysis. Cell 2015, 161, 817–832. [Google Scholar] [CrossRef] [PubMed]
- Komeima, K.; Rogers, B.S.; Campochiaro, P.A. Antioxidants slow photoreceptor cell death in mouse models of retinitis pigmentosa. J. Cell. Physiol. 2007, 213, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Sánchez, L.; Lax, P.; Esquiva, G.; Martín-Nieto, J.; Pinilla, I.; Cuenca, N. Safranal, a Saffron Constituent, Attenuates Retinal Degeneration in P23H Rats. PLoS ONE 2012, 7, e43074. [Google Scholar] [CrossRef]
- Wang, J.; Saul, A.; Roon, P.; Smith, S.B. Activation of the molecular chaperone, sigma 1 receptor, preserves cone function in a murine model of inherited retinal degeneration. Proc. Natl. Acad. Sci. USA 2016, 113, E3764–E3772. [Google Scholar] [CrossRef]
- Venkatesh, A.; Ma, S.; Le, Y.Z.; Hall, M.N.; Rüegg, M.A.; Punzo, C. Activated mTORC1 promotes long-term cone survival in retinitis pigmentosa mice. J. Clin. Investig. 2015, 125, 1446–1458. [Google Scholar] [CrossRef]
- Punzo, C.; Kornacker, K.; Cepko, C.L. Stimulation of the insulin/mTOR pathway delays cone death in a mouse model of retinitis pigmentosa. Nat. Neurosci. 2008, 12, 44–52. [Google Scholar] [CrossRef]
- Dias, M.F.; Joo, K.; Kemp, J.A.; Fialho, S.L.; Cunha-Jr, A.D.S.; Woo, S.J.; Kwon, Y.J. Molecular genetics and emerging therapies for retinitis pigmentosa: Basic research and clinical perspectives. Prog. Retin. Eye Res. 2018, 63, 107–131. [Google Scholar] [CrossRef]
- Garafalo, A.V.; Cideciyan, A.V.; Heon, E.; Sheplock, R.; Pearson, A.; Yu, C.W.; Sumaroka, A.; Aguirre, G.D.; Jacobson, S.G. Progress in treating inherited retinal diseases: Early subretinal gene therapy clinical trials and candidates for future initiatives. Prog. Retin. Eye Res. 2019, 100827, 100827. [Google Scholar] [CrossRef]
- Yanik, M.; Müller, B.; Song, F.; Gall, J.; Wagner, F.; Wende, W.; Lorenz, B.; Stieger, K. In vivo genome editing as a potential treatment strategy for inherited retinal dystrophies. Prog. Retin. Eye Res. 2017, 56, 1–18. [Google Scholar] [CrossRef]
- Terrell, D.; Comander, J. Current Stem-Cell Approaches for the Treatment of Inherited Retinal Degenerations. Semin. Ophthalmol. 2019, 34, 287–292. [Google Scholar] [CrossRef]
- Collins, M.K.L.; Perkins, G.R.; Rodriguez-Tarduchy, G.; Nieto, M.A.; López-Rivas, A. Growth factors as survival factors: Regulation of apoptosis. BioEssays 1994, 16, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Mnich, K.; A Carleton, L.; Kavanagh, E.T.; Doyle, K.M.; Samali, A.; Gorman, A.M. Nerve growth factor-mediated inhibition of apoptosis post-caspase activation is due to removal of active caspase-3 in a lysosome-dependent manner. Cell Death Dis. 2014, 5, e1202. [Google Scholar] [CrossRef] [PubMed]
- Tsybko, A.S.; Il’Chibaeva, T.V.; Naumenko, V.S. The effects of the glial cell line-derived neurotrophic factor (GDNF) on the levels of mRNA of apoptotic genes Bax and Bcl-xl in the brain of mice genetically predisposed to pathological behavior. Russ. J. Genet. Appl. Res. 2015, 5, 407–412. [Google Scholar] [CrossRef]
- Chen, S.-D.; Wu, C.-L.; Hwang, W.-C.; Yang, D.-I. More Insight into BDNF against Neurodegeneration: Anti-Apoptosis, Anti-Oxidation, and Suppression of Autophagy. Int. J. Mol. Sci. 2017, 18, 545. [Google Scholar] [CrossRef]
- Machida, S.; Tanaka, M.; Ishii, T.; Ohtaka, K.; Takahashi, T.; Tazawa, Y. Neuroprotective Effect of Hepatocyte Growth Factor against Photoreceptor Degeneration in Rats. Investig. Opthalmol. Vis. Sci. 2004, 45, 4174–4182. [Google Scholar] [CrossRef]
- Di Pierdomenico, J.; Scholz, R.; Valiente-Soriano, F.J.; Sánchez-Migallón, M.C.; Salinas-Navarro, M.; Langmann, T.; Agudo-Barriuso, M.; García-Ayuso, D.; Villegas-Pérez, M.P. Neuroprotective Effects of FGF2 and Minocycline in Two Animal Models of Inherited Retinal Degeneration. Investig. Opthalmol. Vis. Sci. 2018, 59, 4392–4403. [Google Scholar] [CrossRef]
- García-Caballero, C.; Lieppman, B.; Arranz-Romera, A.; Molina-Martínez, I.T.; Bravo-Osuna, I.; Young, M.; Baranov, P.; Herrero-Vanrell, R. Photoreceptor preservation induced by intravitreal controlled delivery of GDNF and GDNF/melatonin in rhodopsin knockout mice. Mol. Vis. 2018, 24, 733–745. [Google Scholar]
- Amable, P.R.; Carias, R.B.V.; Teixeira, M.V.T.; da Cruz Pacheco, Í.; Amaral, R.J.F.C.D.; Granjeiro, J.M.; Borojevic, R. Platelet-rich plasma preparation for regenerative medicine: Optimization and quantification of cytokines and growth factors. Stem Cell Res. Ther. 2013, 4, 67. [Google Scholar] [CrossRef]
- Kieb, M.; Sander, F.; Prinz, C.; Adam, S.; Mau-Möller, A.; Bader, R.; Peters, K.; Tischer, T. Platelet-Rich Plasma Powder: A New Preparation Method for the Standardization of Growth Factor Concentrations. Am. J. Sports Med. 2016, 45, 954–960. [Google Scholar] [CrossRef]
- Arslan, U.; Özmert, E.; Demirel, S.; Örnek, F.; Şermet, F. Effects of subtenon-injected autologous platelet-rich plasma on visual functions in eyes with retinitis pigmentosa: Preliminary clinical results. Graefe’s Arch. Clin. Exp. Ophthalmol. 2018, 256, 893–908. [Google Scholar] [CrossRef] [PubMed]
- Falsini, B.; Iarossi, G.; Chiaretti, A.; Ruggiero, A.; Manni, L.; Manni, L.; Galli-Resta, L.; Corbo, G.; Abed, E. NGF eye-drops topical administration in patients with retinitis pigmentosa, a pilot study. J. Transl. Med. 2016, 14, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, D.M.; Barnard, A.R.; Singh, M.S.; Martin, C.; Lee, E.J.; Davies, W.I.L.; MacLaren, R.E. CNTF Gene Therapy Confers Lifelong Neuroprotection in a Mouse Model of Human Retinitis Pigmentosa. Mol. Ther. 2015, 23, 1308–1319. [Google Scholar] [CrossRef]
- Birch, D.; Bennett, L.D.; Duncan, J.L.; Weleber, R.G.; Pennesi, M.E. Long-term Follow-up of Patients with Retinitis Pigmentosa Receiving Intraocular Ciliary Neurotrophic Factor Implants. Am. J. Ophthalmol. 2016, 170, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Berson, E.L.; Rosner, B.; Sandberg, M.A.; Hayes, K.C.; Nicholson, B.W.; Weigel-DiFranco, C.; Willett, W. A Randomized Trial of Vitamin A and Vitamin E Supplementation for Retinitis Pigmentosa. Arch. Ophthalmol. 1993, 111, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Sandberg, M.A.; Pawlyk, B.S.; Rosner, B.; Hayes, K.C.; Dryja, T.P.; Berson, E.L. Effect of vitamin A supplementation on rhodopsin mutants threonine-17 -> methionine and proline-347 -> serine in transgenic mice and in cell cultures. Proc. Natl. Acad. Sci. USA 1998, 95, 11933–11938. [Google Scholar] [CrossRef]
- Chen, Y.; Okano, K.; Maeda, T.; Chauhan, V.; Golczak, M.; Maeda, A.; Palczewski, K. Mechanism of All-trans-retinal Toxicity with Implications for Stargardt Disease and Age-related Macular Degeneration. J. Biol. Chem. 2011, 287, 5059–5069. [Google Scholar] [CrossRef]
- Yum, H.-W.; Na, H.-K.; Surh, Y.-J. Anti-inflammatory effects of docosahexaenoic acid: Implications for its cancer chemopreventive potential. Semin. Cancer Biol. 2016, 40, 141–159. [Google Scholar] [CrossRef]
- Berson, E.L.; Rosner, B.; Sandberg, M.A.; Weigel-DiFranco, C.; Moser, A.; Brockhurst, R.J.; Hayes, K.C.; Johnson, C.A.; Anderson, E.J.; Gaudio, A.R.; et al. Further Evaluation of Docosahexaenoic Acid in Patients With RetinitisPigmentosa Receiving Vitamin A Treatment. Arch. Ophthalmol. 2004, 122, 1306–1314. [Google Scholar] [CrossRef]
- Hoffman, D.R.; Hughbanks-Wheaton, D.K.; Spencer, R.; Fish, G.E.; Pearson, N.S.; Wang, Y.-Z.; Klein, M.; Takacs, A.; Locke, K.G.; Birch, D.G. Docosahexaenoic Acid Slows Visual Field Progression in X-Linked Retinitis Pigmentosa: Ancillary Outcomes of the DHAX Trial. Investig. Opthalmol. Vis. Sci. 2015, 56, 6646–6653. [Google Scholar] [CrossRef]
- Chang, R.C.-C.; So, K.-F. Use of Anti-aging Herbal Medicine, Lycium barbarum, Against Aging-associated Diseases. What Do We Know So Far? Cell. Mol. Neurobiol. 2007, 28, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Olatunji, O.J.; Chen, H.; Zhou, Y. Lycium chinensis Mill attenuates glutamate induced oxidative toxicity in PC12 cells by increasing antioxidant defense enzymes and down regulating ROS and Ca2+ generation. Neurosci. Lett. 2016, 616, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Xiao, J.; Peng, B.; Xing, F.; So, K.-F.; Tipoe, G.L.; Lin, B. Retinal structure and function preservation by polysaccharides of wolfberry in a mouse model of retinal degeneration. Sci. Rep. 2014, 4, 7601. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhao, Q.; Gao, H.; Peng, X.; Wen, Y.; Dai, G.-D. Lycium barbarum polysaccharides attenuates N-methy-N-nitrosourea-induced photoreceptor cell apoptosis in rats through regulation of poly (ADP-ribose) polymerase and caspase expression. J. Ethnopharmacol. 2016, 191, 125–134. [Google Scholar] [CrossRef]
- Chan, H.H.-L.; Lam, C.H.-I.; Choi, K.-Y.; Li, S.Z.-C.; Lakshmanan, Y.; Yu, W.-Y.; Chang, R.C.-C.; Lai, J.S.-M.; So, K.-F. Delay of cone degeneration in retinitis pigmentosa using a 12-month treatment with Lycium barbarum supplement. J. Ethnopharmacol. 2019, 236, 336–344. [Google Scholar] [CrossRef]
- Maccarone, R.; Di Marco, S.; Bisti, S. Saffron Supplement Maintains Morphology and Function after Exposure to Damaging Light in Mammalian Retina. Investig. Opthalmol. Vis. Sci. 2008, 49, 1254–1261. [Google Scholar] [CrossRef]
- Assimopoulou, A.N.; Sinakos, Z.; Papageorgiou, V.P. Radical scavenging activity ofCrocus sativus L. extract and its bioactive constituents. Phytother. Res. 2005, 19, 997–1000. [Google Scholar] [CrossRef]
- Kanakis, C.; Tarantilis, P.A.; Pappas, C.; Bariyanga, J.; Tajmir-Riahi, H.A.; Polissiou, M. An overview of structural features of DNA and RNA complexes with saffron compounds: Models and antioxidant activity. J. Photochem. Photobiol. B Biol. 2009, 95, 204–212. [Google Scholar] [CrossRef]
- Mehri, S.; Abnous, K.; Mousavi, S.H.; Shariaty, V.M.; Hosseinzadeh, H. Neuroprotective Effect of Crocin on Acrylamide-induced Cytotoxicity in PC12 cells. Cell. Mol. Neurobiol. 2011, 32, 227–235. [Google Scholar] [CrossRef]
- Sepahi, S.; Mohajeri, S.A.; Hosseini, S.M.; Khodaverdi, E.; Shoeibi, N.; Namdari, M.; Tabassi, S.A.S. Effects of Crocin on Diabetic Maculopathy: A Placebo-Controlled Randomized Clinical Trial. Am. J. Ophthalmol. 2018, 190, 89–98. [Google Scholar] [CrossRef]
- Piccardi, M.; Fadda, A.; Martelli, F.; Marangoni, D.; Magli, A.; Minnella, A.M.; Bertelli, M.; Di Marco, S.; Bisti, S.; Falsini, B. Antioxidant Saffron and Central Retinal Function in ABCA4-Related Stargardt Macular Dystrophy. Nutrients 2019, 11, 2461. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Ohguro, H.; Maeda, T.; Maruyama, I.; Takano, Y.; Metoki, T.; Nakazawa, M.; Sawada, H.; Dezawa, M. Preservation of retinal morphology and functions in royal college surgeons rat by nilvadipine, a Ca2+ antagonist. Investig. Ophthalmol. Vis. Sci. 2002, 43, 919–926. [Google Scholar]
- Takano, Y.; Ohguro, H.; Dezawa, M.; Ishikawa, H.; Yamazaki, H.; Ohguro, I.; Mamiya, K.; Metoki, T.; Ishikawa, F.; Nakazawa, M. Study of drug effects of calcium channel blockers on retinal degeneration of rd mouse. Biochem. Biophys. Res. Commun. 2004, 313, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Nakazawa, M.; Mizukoshi, S. Systemic administration of nilvadipine delays photoreceptor degeneration of heterozygous retinal degeneration slow (rds) mouse. Exp. Eye Res. 2008, 86, 60–69. [Google Scholar] [CrossRef]
- Nakazawa, M.; Suzuki, Y.; Ito, T.; Metoki, T.; Kudo, T.; Ohguro, H. Long-Term Effects of Nilvadipine against Progression of the Central Visual Field Defect in Retinitis Pigmentosa: An Extended Study. BioMed Res. Int. 2013, 2013, 1–6. [Google Scholar] [CrossRef]
- Haller, J.A.; Bandello, F.; Belfort, R.; Blumenkranz, M.S.; Gillies, M.C.; Heier, J.; Loewenstein, A.; Yoon, Y.H.; Jiao, J.; Ulinska, M.; et al. Dexamethasone Intravitreal Implant in Patients with Macular Edema Related to Branch or Central Retinal Vein Occlusion. Ophthalmology 2011, 118, 2453–2460. [Google Scholar] [CrossRef]
- Mansour, A.M.; Sheheitli, H.; Kucukerdonmez, C.; Sisk, R.A.; Moura, R.; Moschos, M.M.; Lima, L.H.; Al-Shaar, L.; Arevalo, J.F.; Maia, M.; et al. INTRAVITREAL DEXAMETHASONE IMPLANT IN RETINITIS PIGMENTOSA–RELATED CYSTOID MACULAR EDEMA. Retina 2018, 38, 416–423. [Google Scholar] [CrossRef]
- Ahn, S.J.; Kim, K.E.; Woo, S.J.; Park, K.H.; Joon, A.S.; Eun, K.K.; Hyung, P.K. The Effect of an Intravitreal Dexamethasone Implant for Cystoid Macular Edema in Retinitis Pigmentosa: A Case Report and Literature Review. Ophthalmic Surg. Lasers Imaging Retin. 2014, 45, 160–164. [Google Scholar] [CrossRef]
- Wang, J.; Xu, J.; Zhao, X.; Xie, W.; Wang, H.; Kong, H. Fasudil inhibits neutrophil-endothelial cell interactions by regulating the expressions of GRP78 and BMPR2. Exp. Cell Res. 2018, 365, 97–105. [Google Scholar] [CrossRef]
- Nourinia, R.; Nakao, S.; Zandi, S.; Safi, S.; Hafezi-Moghadam, A.; Ahmadieh, H. ROCK inhibitors for the treatment of ocular diseases. Br. J. Ophthalmol. 2017, 102, 1–5. [Google Scholar] [CrossRef]
- Zhang, T.; Wei, Y.; Jiang, X.; Li, J.; Qiu, S.; Zhang, S. Protection of photoreceptors by intravitreal injection of the Y-27632 Rho-associated protein kinase inhibitor in Royal College of Surgeons rats. Mol. Med. Rep. 2015, 12, 3655–3661. [Google Scholar] [CrossRef] [PubMed]
- Ahmadieh, H.; Nourinia, R.; Hafezi-Moghadam, A.; Sabbaghi, H.; Nakao, S.; Zandi, S.; Yaseri, M.; Tofighi, Z.; Akbarian, S. Intravitreal injection of a Rho-kinase inhibitor (fasudil) combined with bevacizumab versus bevacizumab monotherapy for diabetic macular oedema: A pilot randomised clinical trial. Br. J. Ophthalmol. 2018, 103, 922–927. [Google Scholar] [CrossRef] [PubMed]
- Gale, J.D.; Berger, B.; Gilbert, S.; Popa, S.; Sultan, M.B.; Schachar, R.A.; Girgenti, D.; Perros-Huguet, C. A CCR2/5 Inhibitor, PF-04634817, Is Inferior to Monthly Ranibizumab in the Treatment of Diabetic Macular Edema. Investig. Opthalmol. Vis. Sci. 2018, 59, 2659–2669. [Google Scholar] [CrossRef]
- Mendes, H.F.; Cheetham, M.E. Pharmacological manipulation of gain-of-function and dominant-negative mechanisms in rhodopsin retinitis pigmentosa. Hum. Mol. Genet. 2008, 17, 3043–3054. [Google Scholar] [CrossRef] [PubMed]
- Krebs, M.P.; Holden, D.C.; Joshi, P.; Clark, C.L.; Lee, A.H.; Kaushal, S.; Iii, C.L.C. Molecular Mechanisms of Rhodopsin Retinitis Pigmentosa and the Efficacy of Pharmacological Rescue. J. Mol. Biol. 2010, 395, 1063–1078. [Google Scholar] [CrossRef]
- Opefi, C.A.; South, K.; Reynolds, C.A.; Smith, S.O.; Reeves, P.J. Retinitis Pigmentosa Mutants Provide Insight into the Role of the N-terminal Cap in Rhodopsin Folding, Structure, and Function. J. Biol. Chem. 2013, 288, 33912–33926. [Google Scholar] [CrossRef]
- Athanasiou, D.; Aguila, M.; Bellingham, J.; Li, W.; McCulley, C.; Reeves, P.J.; Cheetham, M.E. The molecular and cellular basis of rhodopsin retinitis pigmentosa reveals potential strategies for therapy. Prog. Retin. Eye Res. 2018, 62, 1–23. [Google Scholar] [CrossRef]
- Athanasiou, D.; Aguila, M.; Opefi, C.A.; South, K.; Bellingham, J.; Bevilacqua, D.; Munro, P.M.; Kanuga, N.; MacKenzie, F.E.; Dubis, A.M.; et al. Rescue of mutant rhodopsin traffic by metformin-induced AMPK activation accelerates photoreceptor degeneration. Hum. Mol. Genet. 2017, 26. [Google Scholar] [CrossRef]
- Athanasiou, D.; Bevilacqua, D.; Aguila, M.; McCulley, C.; Kanuga, N.; Iwawaki, T.; Chapple, J.P.; Cheetham, M.E. The co-chaperone and reductase ERdj5 facilitates rod opsin biogenesis and quality control. Hum. Mol. Genet. 2014, 23, 6594–6606. [Google Scholar] [CrossRef]
- Kosmaoglou, M.; Kanuga, N.; Aguilà, M.; Garriga, P.; Cheetham, M.E. A dual role for EDEM1 in the processing of rod opsin. J. Cell Sci. 2009, 122, 4465–4472. [Google Scholar] [CrossRef]
- Athanasiou, D.; Kosmaoglou, M.; Kanuga, N.; Novoselov, S.S.; Paton, A.W.; Paton, J.C.; Chapple, J.P.; Cheetham, M.E. BiP prevents rod opsin aggregation. Mol. Biol. Cell 2012, 23, 3522–3531. [Google Scholar] [CrossRef] [PubMed]
- Gorbatyuk, M.; Knox, T.; Lavail, M.M.; Gorbatyuk, O.S.; Noorwez, S.M.; Hauswirth, W.W.; Lin, J.H.; Muzyczka, N.; Lewin, A.S. Restoration of visual function in P23H rhodopsin transgenic rats by gene delivery of BiP/Grp78. Proc. Natl. Acad. Sci. USA 2010, 107, 5961–5966. [Google Scholar] [CrossRef] [PubMed]
- Aguilà, M.; Bellingham, J.; Athanasiou, D.; Bevilacqua, D.; Duran, Y.; Maswood, R.; A Parfitt, D.; Iwawaki, T.; Spyrou, G.; Smith, A.J.; et al. AAV-mediated ERdj5 overexpression protects against P23H rhodopsin toxicity. Hum. Mol. Genet. 2020, 29, 1310–1318. [Google Scholar] [CrossRef]
- Mandal, N.A.; Patlolla, J.M.; Zheng, L.; Agbaga, M.-P.; Tran, J.-T.A.; Wicker, L.; Kasus-Jacobi, A.; Elliott, M.H.; Rao, C.V.; Anderson, R.E. Curcumin protects retinal cells from light-and oxidant stress-induced cell death. Free. Radic. Biol. Med. 2009, 46, 672–679. [Google Scholar] [CrossRef]
- Park, S.-I.; Lee, E.H.; Kim, S.R.; Jang, Y.P. Anti-apoptotic effects ofCurcuma longaL. extract and its curcuminoids against blue light-induced cytotoxicity in A2E-laden human retinal pigment epithelial cells. J. Pharm. Pharmacol. 2017, 69, 334–340. [Google Scholar] [CrossRef]
- Vasireddy, V.; Chavali, V.R.M.; Joseph, V.T.; Kadam, R.; Lin, J.H.; Jamison, J.A.; Kompella, U.B.; Reddy, G.B.; Ayyagari, R. Rescue of Photoreceptor Degeneration by Curcumin in Transgenic Rats with P23H Rhodopsin Mutation. PLoS ONE 2011, 6, e21193. [Google Scholar] [CrossRef] [PubMed]
- Pullakhandam, R.; Srinivas, P.; Nair, M.K.; Reddy, G.B. Binding and stabilization of transthyretin by curcumin. Arch. Biochem. Biophys. 2009, 485, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Mazzolani, F.; Togni, S.; Giacomelli, L.; Eggenhoffner, R.; Franceschi, F. Oral administration of a curcumin-phospholipid formulation (Meriva®) for treatment of chronic diabetic macular edema: A pilot study. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3617–3625. [Google Scholar] [PubMed]
- Boatright, J.H.; Nickerson, J.M.; Moring, A.G.; Pardue, M.T. Bile acids in treatment of ocular disease. J. Ocul. Biol. Dis. Inform. 2009, 2, 149–159. [Google Scholar] [CrossRef]
- Elia, A.E.; Lalli, S.; Monsurro, M.R.; Sagnelli, A.; Taiello, A.C.; Reggiori, B.; La Bella, V.; Tedeschi, G.; Albanese, A. Tauroursodeoxycholic acid in the treatment of patients with amyotrophic lateral sclerosis. Eur. J. Neurol. 2015, 23, 45–52. [Google Scholar] [CrossRef]
- Phillips, M.J.; Walker, T.A.; Choi, H.-Y.; Faulkner, A.E.; Kim, M.K.; Sidney, S.S.; Boyd, A.P.; Nickerson, J.M.; Boatright, J.H.; Pardue, M.T. Tauroursodeoxycholic acid preservation of photoreceptor structure and function in the rd10 mouse through postnatal day 30. Investig. Opthalmol. Vis. Sci. 2008, 49, 2148–2155. [Google Scholar] [CrossRef] [PubMed]
- Drack, A.V.; Dumitrescu, A.V.; Bhattarai, S.; Gratie, D.; Stone, E.M.; Mullins, R.F.; Sheffield, V.C. TUDCA slows retinal degeneration in two different mouse models of retinitis pigmentosa and prevents obesity in Bardet-Biedl syndrome type 1 mice. Investig. Opthalmol. Vis. Sci. 2012, 53, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Lawson, E.C.; Bhatia, S.K.; Han, M.K.; Aung, M.H.; Ciavatta, V.; Boatright, J.H.; Pardue, M.T. Tauroursodeoxycholic Acid Protects Retinal Function and Structure in rd1 Mice. Single Mol. Single Cell Seq. 2015, 854, 431–436. [Google Scholar] [CrossRef]
- Fernández-Sánchez, L.; Lax, P.; Pinilla, I.; Martín-Nieto, J.; Cuenca, N. Tauroursodeoxycholic Acid Prevents Retinal Degeneration in Transgenic P23H Rats. Investig. Opthalmol. Vis. Sci. 2011, 52, 4998–5008. [Google Scholar] [CrossRef]
- A Parfitt, D.; Aguila, M.; McCulley, C.H.; Bevilacqua, D.; Mendes, H.F.; Athanasiou, D.; Novoselov, S.S.; Kanuga, N.; Munro, P.M.; Coffey, P.J.; et al. The heat-shock response co-inducer arimoclomol protects against retinal degeneration in rhodopsin retinitis pigmentosa. Cell Death Dis. 2014, 5, e1236. [Google Scholar] [CrossRef]
- Fernández-Sánchez, L.; Bravo-Osuna, I.; Lax, P.; Arranz-Romera, A.; Maneu, V.; Esteban-Pérez, S.; Pinilla, I.; Puebla-González, M.D.M.; Herrero-Vanrell, R.; Cuenca, N. Controlled delivery of tauroursodeoxycholic acid from biodegradable microspheres slows retinal degeneration and vision loss in P23H rats. PLoS ONE 2017, 12, e0177998. [Google Scholar] [CrossRef]
- Bainbridge, J.W.; Mehat, M.S.; Sundaram, V.; Robbie, S.J.; Barker, S.E.; Ripamonti, C.; Georgarias, A.; Mowat, F.M.; Beattie, S.G.; Gardner, P.J.; et al. Long-Term Effect of Gene Therapy on Leber’s Congenital Amaurosis. N. Engl. J. Med. 2015, 372, 1887–1897. [Google Scholar] [CrossRef]
- Russell, S.; Bennett, J.; A Wellman, J.; Chung, D.C.; Yu, Z.-F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef]
- MacLaren, R.E.; Xue, K.; De La Camara, C.M.-F.; Nanda, A.; Davies, A.; Wood, L.J.; Salvetti, A.P.; Fischer, M.D.; Aylward, J.W.; Barnard, A.R.; et al. Initial results from a first-in-human gene therapy trial on X-linked retinitis pigmentosa caused by mutations in RPGR. Nat. Med. 2020, 26, 354–359. [Google Scholar] [CrossRef]
- Mehat, M.S.; Sundaram, V.; Ripamonti, C.; Robson, A.G.; Smith, A.J.; Borooah, S.; Robinson, M.; Rosenthal, A.N.; Innes, W.; Weleber, R.G.; et al. Transplantation of Human Embryonic Stem Cell-Derived Retinal Pigment Epithelial Cells in Macular Degeneration. Ophthalmology 2018, 125, 1765–1775. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Newton, F.; Megaw, R. Mechanisms of Photoreceptor Death in Retinitis Pigmentosa. Genes 2020, 11, 1120. https://doi.org/10.3390/genes11101120
Newton F, Megaw R. Mechanisms of Photoreceptor Death in Retinitis Pigmentosa. Genes. 2020; 11(10):1120. https://doi.org/10.3390/genes11101120
Chicago/Turabian StyleNewton, Fay, and Roly Megaw. 2020. "Mechanisms of Photoreceptor Death in Retinitis Pigmentosa" Genes 11, no. 10: 1120. https://doi.org/10.3390/genes11101120
APA StyleNewton, F., & Megaw, R. (2020). Mechanisms of Photoreceptor Death in Retinitis Pigmentosa. Genes, 11(10), 1120. https://doi.org/10.3390/genes11101120