1. Introduction
Diatoms are among the most common types of phytoplankton and one of the most successful clades of eukaryotic, single-celled photosynthetic organisms in the modern ocean [
1]. They are widespread in the plankton and benthos of freshwater, coastal and oceanic habitats and even in temporarily wet terrestrial environments. The ability of diatoms to thrive under many different conditions in the natural environment could depend on:
(1) their phenotypic plasticity, reflected in their physiological diversity that allows them to acclimate and face short-term environmental heterogeneity [
2,
3],
(2) intraspecific genetic variations leading to distinct populations, each with a rather narrow physiological tolerance and response (i.e., adapted populations); numerous cases of cryptic or pseudo-cryptic diatom species have recently been uncovered [
4,
5,
6], and even populations within species that show particular distributions as a result of adaptation to specific ecological niches [
7], (3) both of the above, as plasticity can be adaptive with respect to a function and may be altered by natural selection and ultimately become or facilitate adaptation.
In addition to natural environmental variability, diatoms used in experimental research are also exposed to another source of variability; they are removed from their natural environment, isolated and transferred to laboratory conditions in which evolutionary processes do not cease to occur [
8]. Many cases of novel genetic variation have been shown after years of maintenance in culture collection [
9,
10,
11]. Considering the high adaptability of diatoms, it could be hard to interpret the intra-specific algal diversity observed in related studies as the result of (a) adaptation to the natural environment from which strains were isolated or (b) evolutionary changes while being in culture.
Temperature is a very significant environmental variable that affects the physiological response of diatoms and ultimately determines their phenological patterns and geographic ranges [
12,
13,
14], as well as one of the parameters that is usually kept constant in culture conditions. An increase in temperature leads to an increase in enzyme activity in metabolic processes, including photosynthesis and respiration, so the cells are expected to grow faster. Even algae obtained from cold habitats have shown at least short-term temperature optima for photosynthesis several degrees higher than the temperature at which they were growing. Low temperatures lead to the exact opposite direction so that, in order to survive, cells respond with an increase in enzyme synthesis [
15,
16]. A recent study on the molecular mechanisms of temperature acclimation and adaptation in warm and cold adapted strains of the marine diatom
Chaetoceros concluded that evolutionary change in baseline gene expression is a key mechanism used by diatoms to adapt to different growth temperatures [
17]. However, there are temperature limits, which differ for each species and even for strains of the same species isolated from warm or cold waters [
8,
13,
14]. Temperatures close to their environmental extremes might activate stress-related pathways engaged in restoring cellular homeostasis. Temperatures that are too high may lead to denaturation and degradation of certain proteins [
18], reduced functionality of the photosynthetic machinery [
19], decreased RUBISCO activity, stimulated respiration [
20], disturbed functions of cell membranes [
21], and, thus, to reduction of cell growth. On the other hand, cold responses have been less explored and have mainly focused on the reactions and adaptations of high latitude and polar diatoms [
22,
23,
24,
25].
The cell stress response, including heat stress, is characterized initially by an increase in stress-inducible proteins [
26], followed by the activation of enhanced tolerance mechanisms involving extensive reprogramming of gene expression and further modifications of metabolism. In diatoms, specific proteins related to cold acclimation or heat stress have been identified, such as antifreeze proteins (AFPs) in polar diatoms of the genus
Fragilariopsis [
27], the heat stress-responsive protein HI-5a in the marine species
Chaetoceros compressus [
28], and the heat shock protein 20 [
29] and 70/90 [
30] in the marine species
Ditylum brightwellii. In addition, diatoms seem to have a high number of heat shock transcription factors, albeit of unknown function [
31].
Yet another possible mechanism to adapt quickly to extreme environments consists of genome re-arrangements through the action of transposable elements (TEs) [
32], which are DNA sequences with diverse structures able to move within the genome. Genes encoding reverse transcriptase, which copies RNA into cDNA codified by class 1 TEs or retrotransposons, have been found to be highly abundant and active in marine plankton assemblages [
33]. In diatoms the long terminal repeat (LTR) retrotransposons superfamily makes up 90% of TEs in the genome of
Phaeodactylum tricornutum and 58% in
Thalassiosira pseudonana, and diatom-specific TEs belong to the so called CoDi I, CoDi II and GyDi lineages [
32]. Deletions, mutations and epigenetic ‘defense’ mechanisms of the cell inactivate or silence most of the TEs, which are otherwise highly mutagenic when targeting protein coding genes [
34,
35]. However, as the “epi-transposon” hypothesis suggests [
36], changing environments, including temperature changes [
37,
38], can lead to stress-induced breakdown of the epigenetic suppression of TEs. Once reactivated, TEs can alter gene expression patterns or even restructure the genome by inserting into promoters and enhancers, causing exon shuffling, sequence expansion, gene duplication, novel gene formation and expansion [
39,
40,
41,
42,
43,
44,
45]. Such TE-induced mutations increase genetically and epigenetically-based phenotypic variation and have been associated with adaptation to the environment in numerous studies on different organisms [
46,
47], while two active, diatom-specific retrotransposons, namely Blackbeard and Surcouf, have been proposed to act as environmental sensors in the response to stress in the diatom
Phaeodactylum tricornutum [
32,
47]. Thus, TEs could substantially increase genomic diversity and be a crucial element in the acclimation and even adaptation of diatoms to the ever-changing aquatic environment but when in a constant environment such as the lab, their suppression would have no effects and could hence become permanent through genetic changes, leading to the loss of the diatom adaptability.
Leptocylindrus aporus belongs to an ancient centric diatom lineage, Leptocylindraceae, with a broad distribution, common from polar to sub-tropical coastal regions, and is often a conspicuous component of diatom blooms [
48,
49]. It grows equally efficiently at medium and high temperature but less at low temperature, where it might experience physiological stress [
50]. In the Gulf of Naples,
L. aporus has been found to bloom in summer but the species is present during other seasons as well [
6,
51]. Indications of a cold adapted population resulted from a worldwide high throughput sequencing (HTS) metabarcoding analysis on Leptocylindraceae, offering a possible explanation of the species occurrence through the year [
51]. In the current study, (i) we used RNA sequencing of multiple strains of
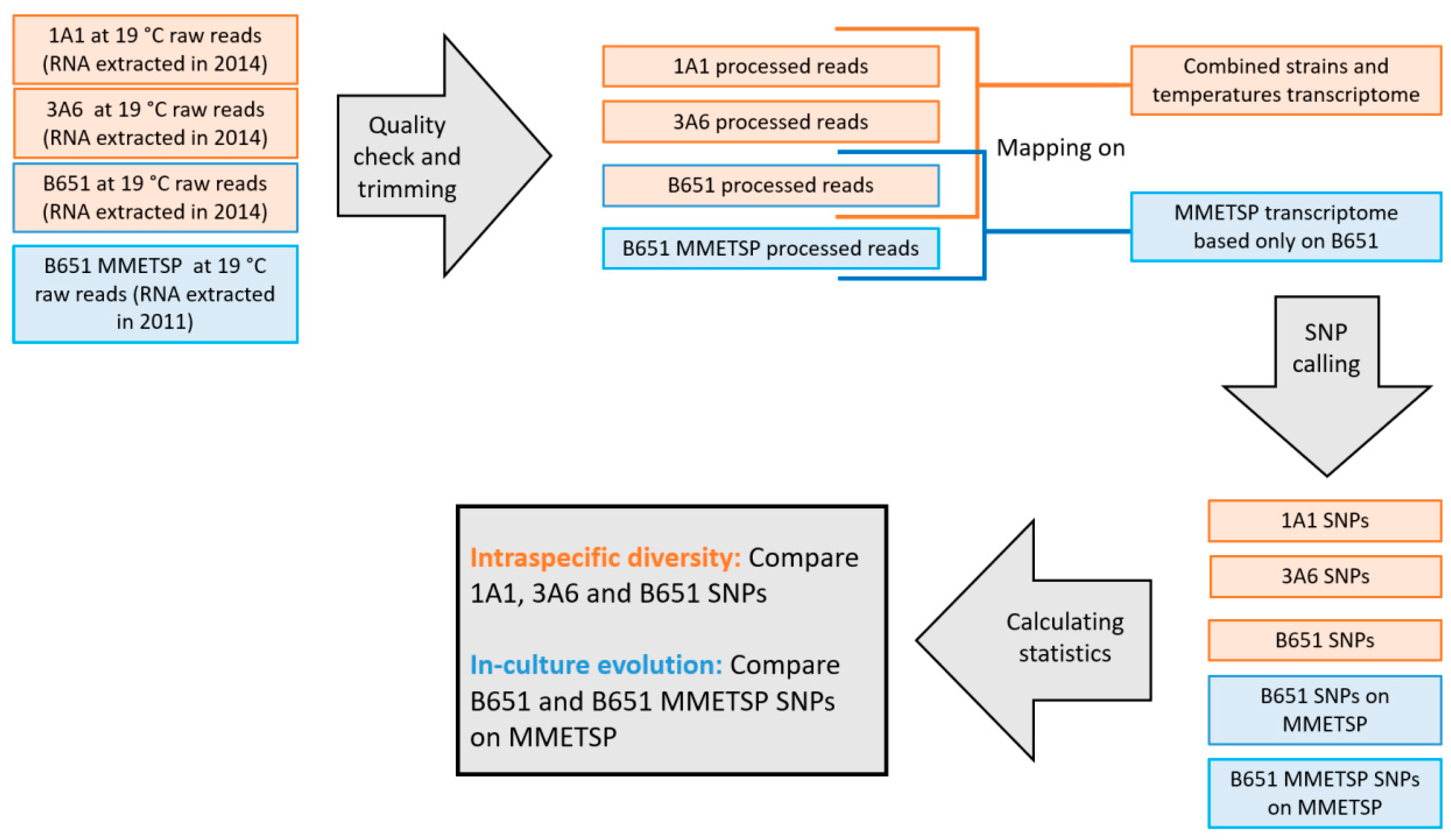
L. aporus to explore expression differences in response to temperature changes, focusing on TEs. (ii) Intraspecific genetic diversity was assessed with a single-nucleotide polymorphism (SNP) analysis. In addition, in order to detect possible genetic changes in laboratory conditions for the oldest strain, an additional SNP analysis was conducted comparing present data to the already available transcriptome in the Marine Microbial Eukaryote Transcriptome Sequencing Project (MMETSP) [
52]. (iii) The effects of experimental conditions were explored with shorter acclimation times as well as with recently isolated
L. aporus strains using qPCR.
Our overall goal was to better understand how diatoms acclimate and possibly adapt to different environmental conditions, which is a requisite to interpret their changes in distribution and phenological patterns.
3. Results
3.1. Leptocylindrus aporus Transcriptome
Preliminary growth experiments were performed on several
L. aporus strains in order to identify the best candidates for investigating the species acclimation and adaptation using RNA-Seq (
Supplementary File 1). Based on the V9 region included in the beginning of ITS marker, none of the strains belonged to the possible cold adapted population found in the worldwide HTS analysis [
51]. Ultimately, one
L. aporus strain isolated in summer 2010 (B651) and two isolated in winter 2013–2014 (1A1 and 3A6) were chosen, due to their diversity in terms of the year of isolation, seasonality and physiological characteristics. The final transcriptome of
L. aporus (ArrayExpress ID: E-MTAB-8596), obtained using all results from the three strains grown at the three different temperatures, consisted of 19,963 transcripts. Further statistics are presented in
Table S1.
The transcriptome obtained in the current study was compared to the previously available MMETSP transcriptome [
52], obtained from the oldest strain B651, in order to test whether the set of 215 protist lineage-specific single-copy genes were present in the assemblies (BUSCO analysis). This analysis gave an indication on the completeness of the assemblies as well as their redundancy. While the two assemblies contained a similar number of complete genes from the dataset (100 for the current and 103 for the older assembly), the older assembly had a higher proportion of potentially erroneous assembled transcripts and therefore were classified as duplicated by BUSCO (7 for the current and 34 for the old) (
Table S2).
3.2. Differential Expression: Heat and Cold Response
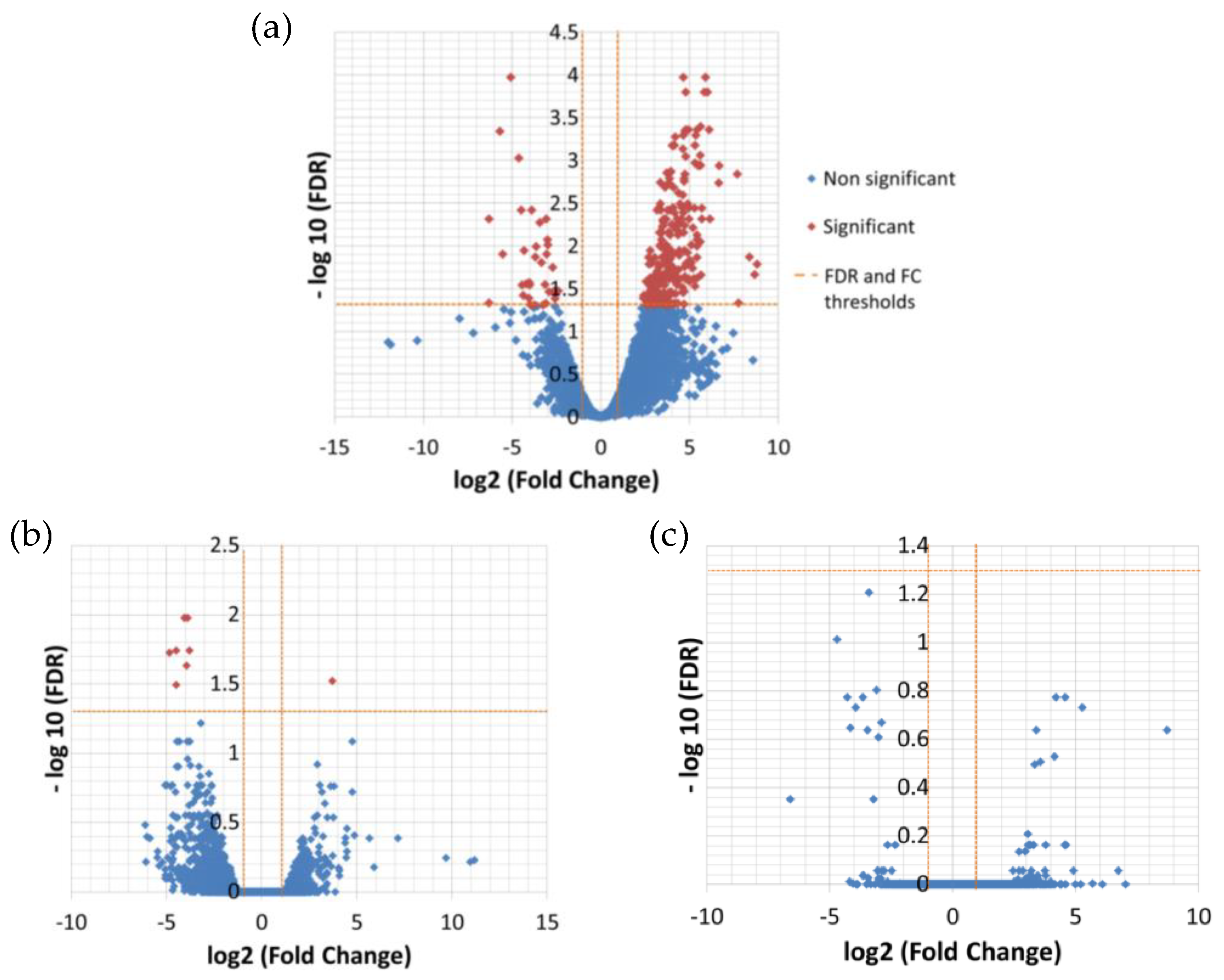
The differential expression analysis between the temperatures resulted in 276 significantly DE transcripts between low (13 °C) and high (26 °C) temperature and only nine between low and medium (19 °C) temperature (
Table 5,
Figure 2), while there was no significant difference in expression between medium and high temperature (
Supplementary File 2, 3 and 7).
All nine transcripts that were found to be significantly differentially expressed between low and medium temperature were also contained within the group of significantly differentially expressed genes in the low and high temperature contrast and followed the same direction of expression change as in the less extreme contrast.
A total of 49 significant DE transcripts, representing 17.7% of all DE transcripts, received an annotation related to temperature, referring to oxidative or any other kind of stress, heat/cold response and DNA integration, overall indicating a stress response of
L. aporus at low temperature (
Table 6).
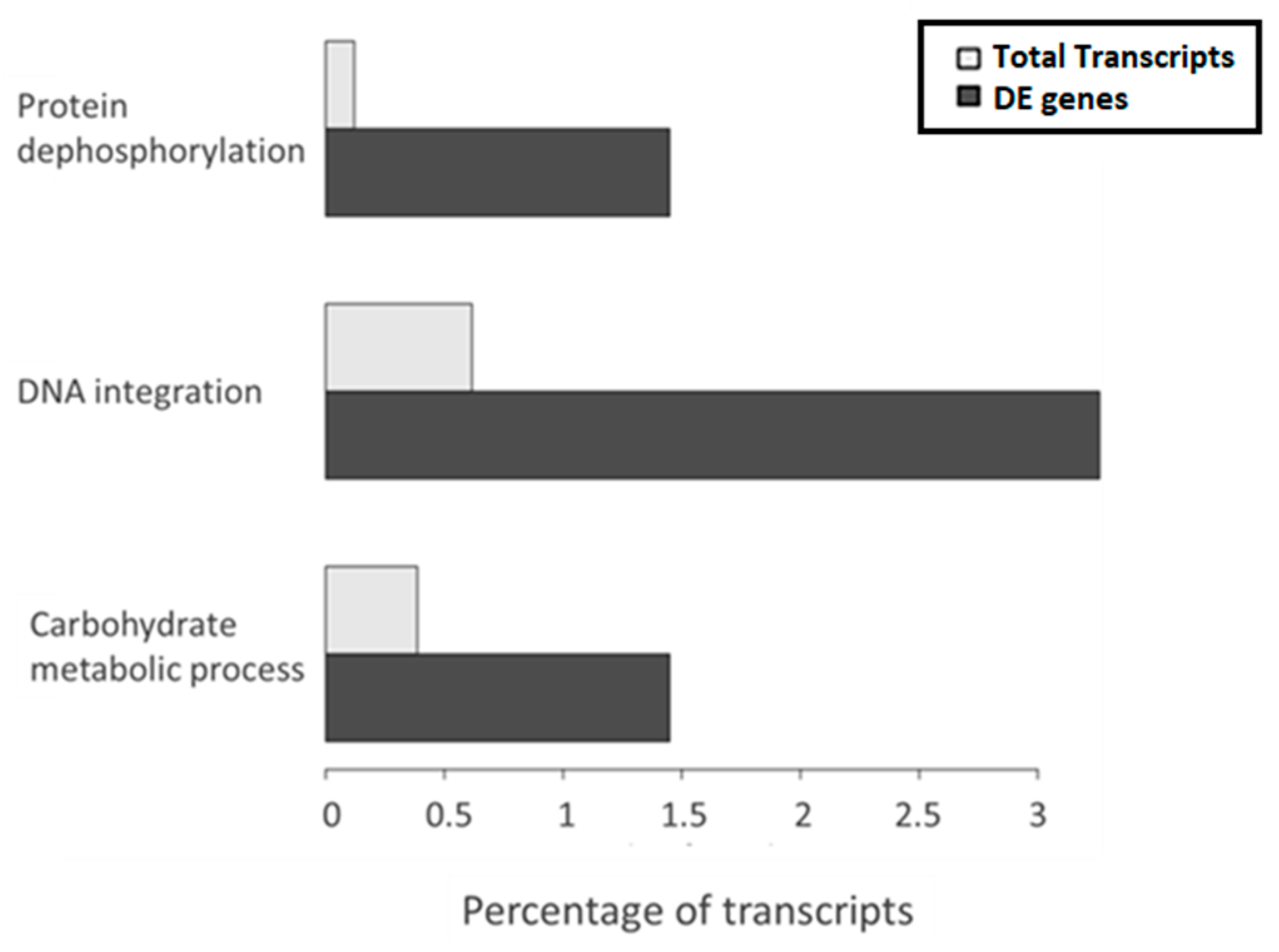
The gene ontology (GO) enrichment analysis showed the GO terms enriched with respect to the overall transcriptome. The adjusted p-value cutoff to consider a class as significant was 0.1, which means that 10% of significant classes might be false positives. The analysis indicated that three classes belonging to the biological process GO aspect were significantly enriched among the DE transcripts between high and low temperature, protein dephosphorylation, DNA integration and carbohydrate metabolic process. Interestingly, among them, the most represented was DNA integration (
Figure 3), which refers to the incorporation of a segment of DNA, including a transposon, into another, usually larger, DNA molecule such as a chromosome. Manual inspection of annotations revealed 16 DE transcripts related to DNA integration, four of which had no further annotation and 12 had specific protein annotation by at least one of the databases used in Annocript (
Table 6). These were annotated as (1) reverse transcriptase (RNA-dependent DNA polymerase), which is usually indicative of a mobile element such as a retrotransposon or retrovirus, (2) ribonuclease of_Ty1/Copia or Ty3/Gypsy family, an endonuclease that cleaves the RNA strand of an RNA/DNA hybrid, which has been observed as adjunct domains to the reverse transcriptase gene in retroviruses, long-term repeat (LTR)-bearing and non-LTR retrotransposons, (3) integrase, which mediates integration of a DNA copy into the host chromosome, or (4) a gag-pol polyprotein, encoding structural proteins and several enzymatic functions in LTR retrotransposons.
All the significantly DE TE-related transcripts except one were highly expressed at low temperature in the recently isolated strains 1A1 and 3A6, while they were very low at all temperatures for the old B651 strain. This expression pattern was also the one mostly seen in several of the other stress-related transcripts (HSFs, SYM1, AOX4, ALDH, RaiA), and in three transcripts, manually annotated by blasting in NCBI and CDD databases as likely homologs of the bacterial MAI genes related to biomineralization, which are known to undergo frequent transposition events [
81]. The observation of this expression pattern pointed towards a strain specific stress response. Indeed, in an NMDS analysis using the global gene expression matrix, the oldest strain B651 clustered apart from the other two strains, for which samples from the medium and high temperature were interspersed (
Figure S4). All the above led to further investigation of the differences among strains.
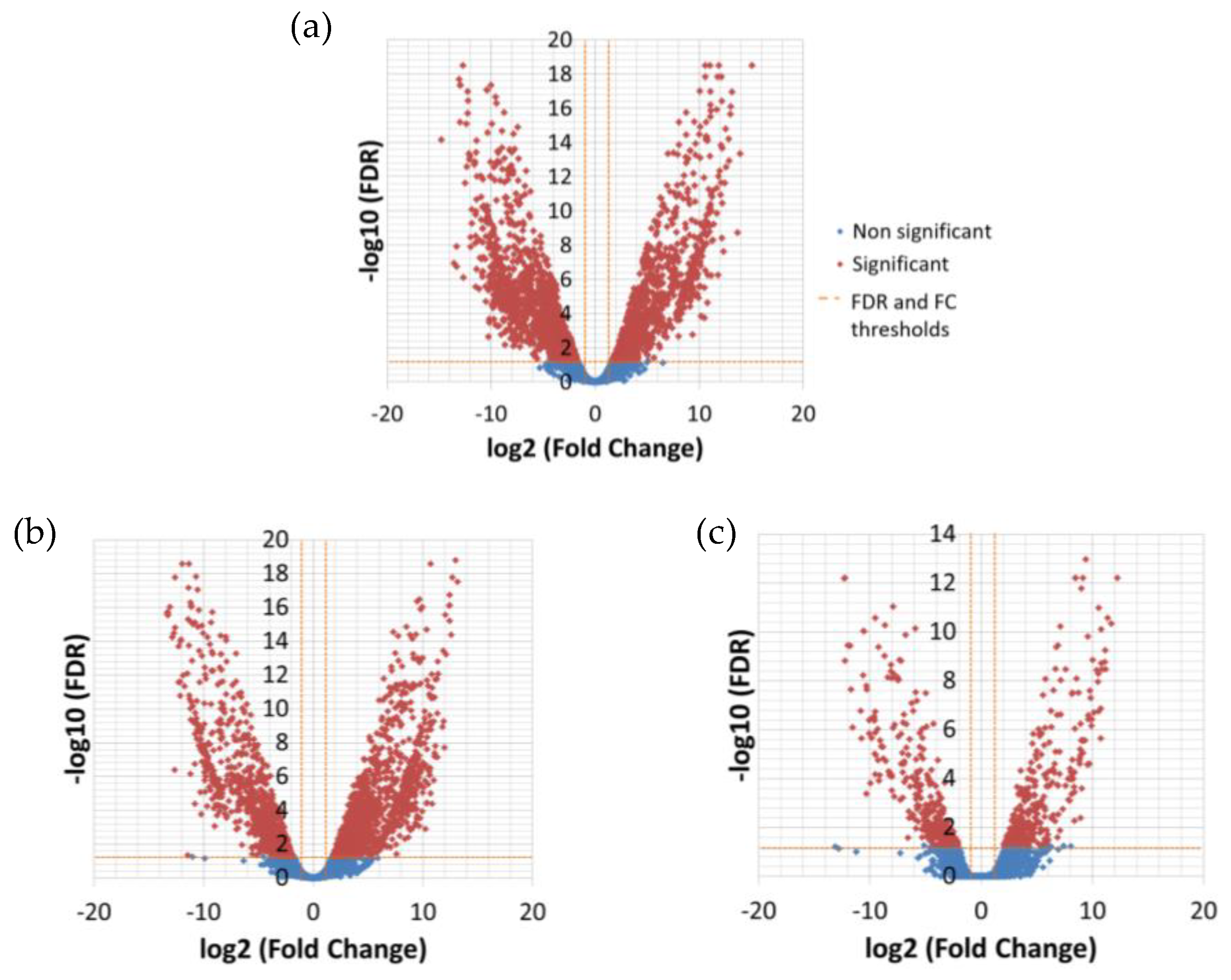
Considering couples of strains, significantly DE transcripts between 1A1 and 3A6 were 622; much less compared to the 3,015 between 1A1 and B651 and 2,418 between B651 and 3A6 (
Figure 4 and
Figure 5,
Supplementary Files 4–7).
DNA integration held a central role in this analysis as well, as it was the biological process that was significantly enriched in all three pairs of strains. The same TE-related transcripts detected in the temperature differential expression analysis were among the significantly DE ones from the strain analysis. Most of the DNA-integration transcripts were expressed in a similar manner, described above, i.e., higher at low temperature and lower in B651; there were even some transcripts completely absent from all B651 samples. Several key genes and pathways were included in the KEGG annotation of the significant DE transcripts among strains, with B651 clustering separately in the expression heatmap (
Figure S5). Overall, the differential expression analysis among strains confirmed the B651-biased, TE-related expression pattern observed in the differential expression analysis among temperatures.
3.3. Clustering of DE Transcripts
Based on genes clustered by their expression patterns, the strongest cell response was to the cold conditions (
Figure 6) except for three clusters (clusters 4, 12 and 14) showing an opposite pattern. At high temperature, cluster-4 genes were highly expressed only in the newer strains 3A6 and 1A1, whereas cluster-12 genes showed a higher level of expression in the old strain B651. Two transcripts related to TEs (transposase, ribonuclease) and a heat stress transcription factor (HSFC-1b) were present in cluster 4 and heat shock factor protein 1 in cluster 12.
Four out of the five TE-related genes of interest selected for qPCR (see below) were present in cluster 3, which, along with cluster 9, showed an increased expression at lower temperatures, but only for strains 3A6 and 1A1. At least seven more transcripts related to TEs or transposition (reverse transcriptase, ribonuclease, MAIs, gag-pol protein, integrase core domain)—eleven in total—and two heat stress-related transcripts (HSFA-1a, SYM1) were also present in these clusters. The GO terms that were significantly enriched in cluster 3 and 9 were DNA integration and antioxidant activity.
Cluster 2 included genes that showed an increase in expression while the temperature decreased but only in strain B651. The GO enrichment analysis in this cluster showed that oxidation–reduction process and DNA integration were significantly enriched terms. This cluster also included the remaining gene of interest that was investigated (TR6586) and two more TE-related transcripts.
Cluster 5 showed transcripts that were expressed more at cold temperature. This behavior was similar in all strains, but more pronounced in strain 1A1. DNA recombination and transport were the GO terms significantly enriched in this cluster.
Cluster 6 and 13 included transcripts highly expressed at low temperature for all strains. Heat stress transcription factor B-2a (HSFB-2a) was present in this cluster. The GO terms that were significantly enriched were metabolic process and catalytic activity.
Cluster 10 showed higher gene expression at low temperature for two strains B651 and 1A1, while in strain 3A6 the gene expression was lower at the extremes and higher at medium temperature. AOX was present in this cluster. The most significantly enriched GO term was oxidation–reduction process.
3.4. SNP Analysis and Genetic Diversity
The number of SNPs identified by FreeBayes was similar in 1A1 and 3A6, while it was almost double in the older strain B651 (
Table 7). The heterozygous to homozygous ratio, for both total and SNP specific, was also quite different for B651 compared to 1A1 and 3A6.
The intra-strain SNP analysis of B651 showed that there were slightly fewer SNPs in the new transcriptome compared to the original MMETSP transcriptome. Furthermore, the total and SNP specific heterozygous to homozygous ratio has been reduced to half during the three years of culturing (
Table 8).
3.5. Expression of Genes of Interest: Temperature and Strain Effects
Five of the eight target genes used in qPCR were related to TEs (
Table 9), as they were selected based on the most significantly enriched biological process GO term ‘DNA integration’.
The three remaining target genes were genes related to stress, as L. aporus seemed to go under stress at the low temperature.
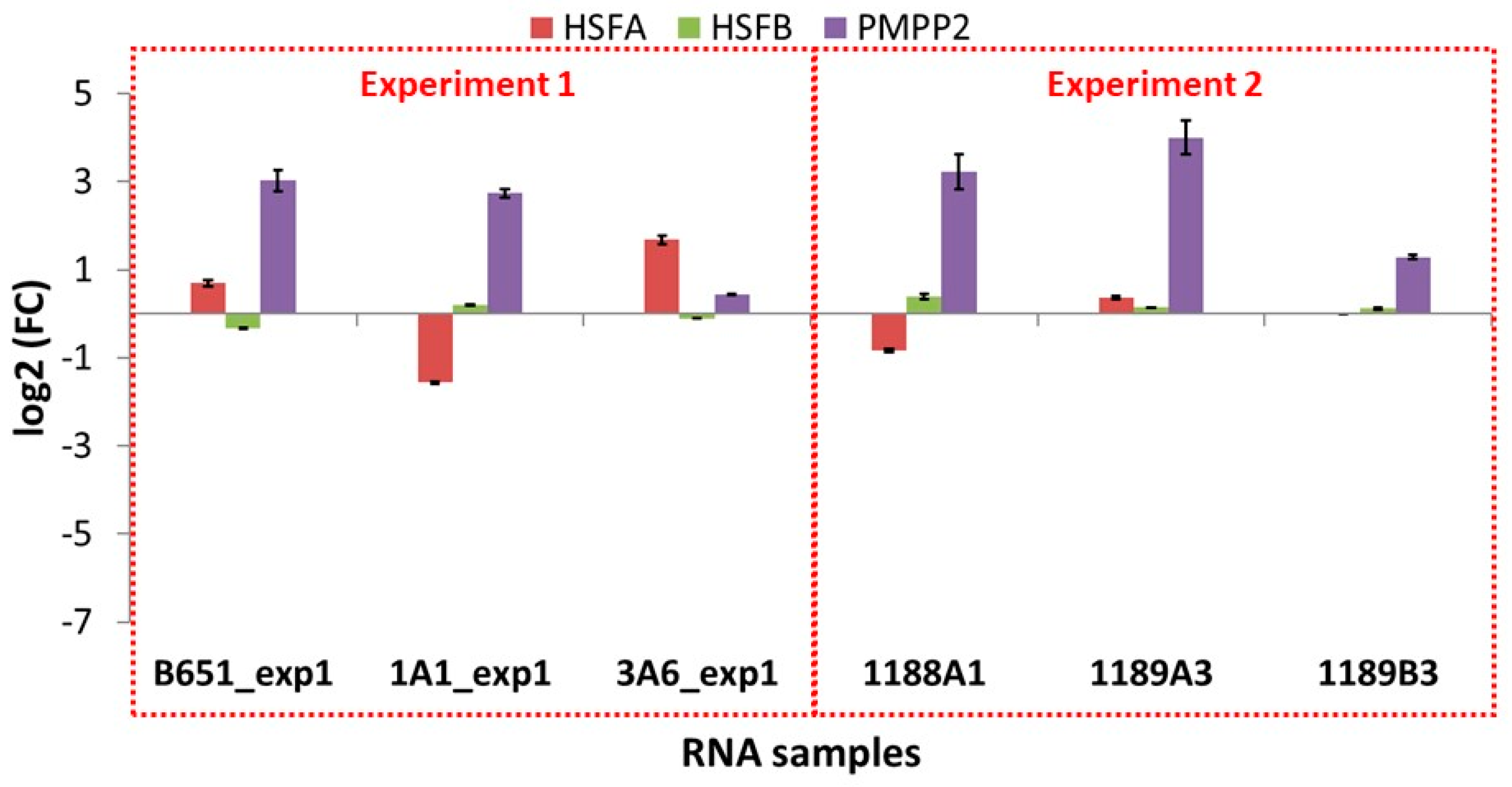
3.6. qPCR Validation Results
The eight selected transcripts were cross-validated in the same strains, grown under the same conditions (validation set), and all of the results were found to be consistent with the results from the RNA-Seq samples with the single exception of HSFB in strain 1A1, which showed almost no change in expression (
Figures S6 and S7).
As in RNA-Seq, B651 stood out based on its different regulation compared to 1A1 and 3A6. This was especially true in the case of HSFA, TR6506_i3 and TR6586: the former two were highly induced in 1A1 and 3A6 at low temperature but down or not regulated in B651, while the latter followed the opposite trend.
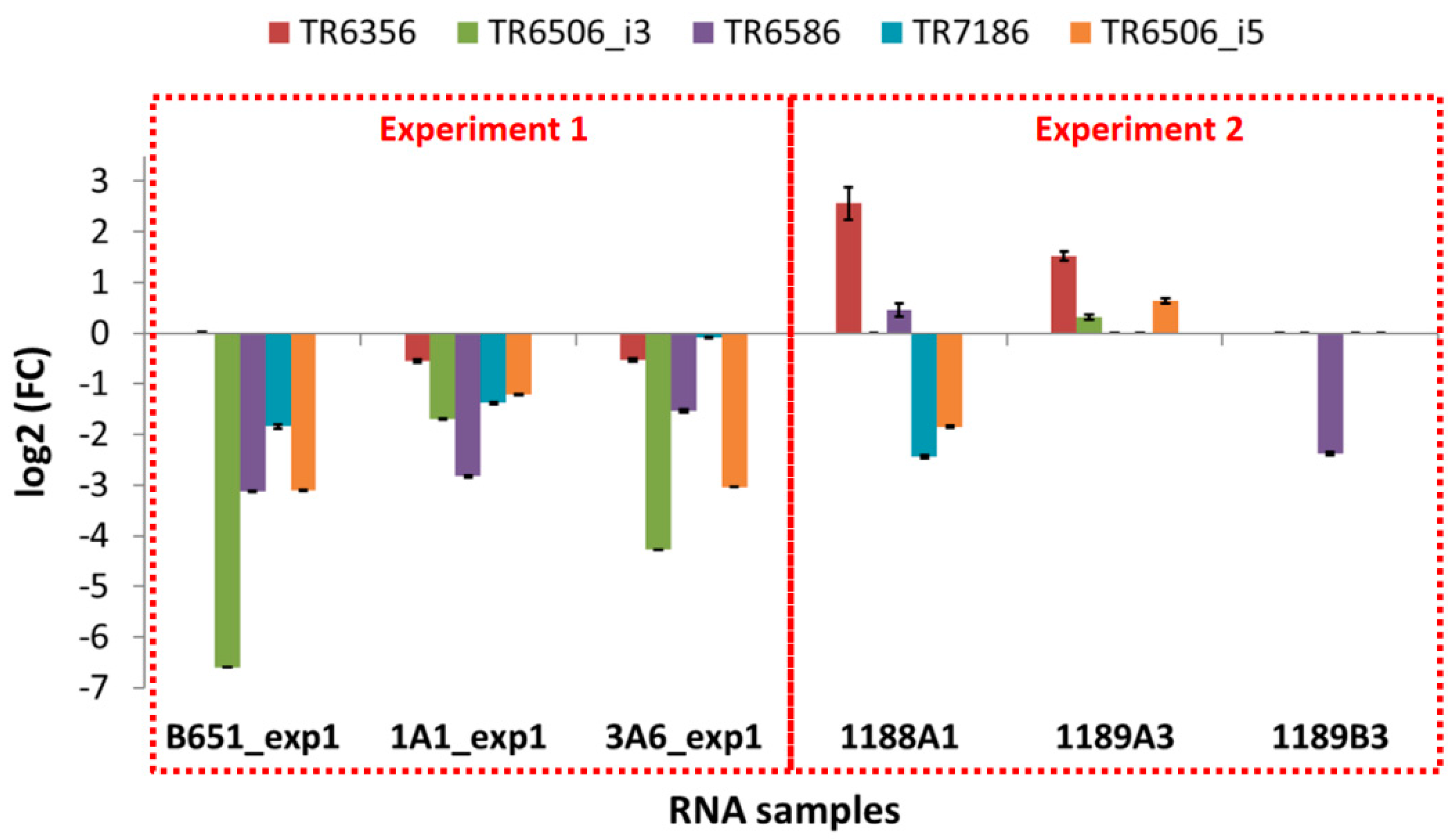
3.7. qPCR Results on Acclimation and Cultivation Time
The expression of the selected transcripts was examined in independent experiments in which the exposure to high and low temperature was applied for a shorter time (approx. 41 vs. 155 days as an average), using the same strains as well as newly isolated ones. The results of these qPCR experiments (
Figure 7 and
Figure 8 and
Table 10) were different from those of the RNA-Seq (
Figure S2).
In B651, 1A1 and 3A6, the selected transcripts were downregulated at low temperature, while in the freshly isolated strain 1189B3—acclimated for the shortest time—all genes were either expressed at low levels or not detected. The only transcript that seemed to be unaffected from the acclimation duration and the time since isolation was SYM1, which was found to always be upregulated at low temperature.
On the other hand, looking at the freshly isolated strains 1188A1 and 1189A3 that were acclimated slightly more than 1189B3, most genes were again low or not expressed (TR6506_i3, TR6586, TR7186, HSFA, HSFB) but there were also upregulated TEs (TR6356 in both 1188A1 and 1189A3, TR6586 in 1188A1, TR6506_i3 and TR6506_i5 in 1189A3).
Amplification of the gene fragments related to TEs from the genomic DNA of the strains used in these experiments revealed that some of the elements were absent from the genome of strain 1189B3 (
Figure S8), indicating that the absence of expression in this strain was due to the absence of the specific transposon in the genome. Similarly, in strain 1189A3, TR7186 appeared to be absent. In all other cases, amplification was successful.
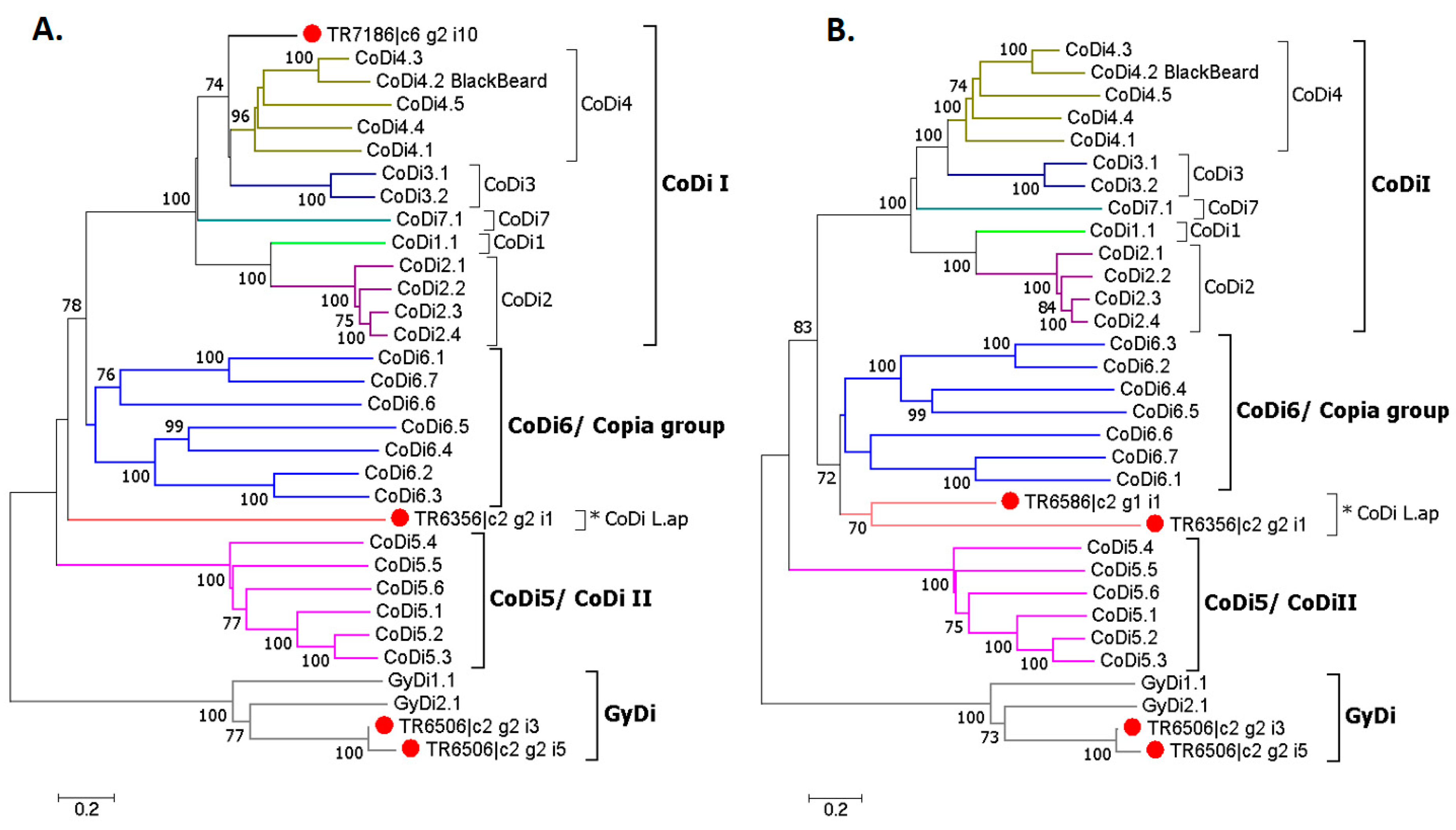
3.8. Phylogenetic Analysis
RepeatMasker analysis on the five selected TE related transcripts (
Table 10) revealed that two of them matched annotated repeats that were similar to CoDi/GyDi sequences. TR6506_i3 and GyDi2.1 both matched Gypsy3-I_TP, though at different regions each. TR7186 and the diatom-specific retrotransposon Blackbeard matched at the same region, Copia8-I_TP, but also at different ones (
Table S3).
The phylogenetic tree gave a clearer picture about the relationship between the selected TEs and CoDis/GyDis. However, it was not possible to estimate the pairwise distance between TR7186 and TR6586 due to the much shorter protein sequence of the first, so two separate NJ trees were built (
Figure 9). TR6506_i3 and TR6506_i5 clustered with the GyDi group, and TR7186 with the CoDi I group, while TR6586 and TR6356, named CoDi L.ap, were closely related to the Copia group.
4. Discussion
The analysis of L. aporus expression profiles at different temperatures based on several strains provided new information on the response of this species to cold stress, but also on the related intraspecific variability and its possible causes. In particular, we identified (i) specific genes, with a prominent role of TEs among them, that react to temperature changes and might be involved in acclimation and/or adaptation to environmental conditions, possibly underlying distinct phenological patterns, and (ii) high intraspecific variations in the species heat/cold response. The high numbers of DE genes only between the minimum and maximum temperature tested indicates that L. aporus mainly reacted to the low temperature, while it retained the same functional state at 26 °C and 19 °C. However, considering that one of the strains (B651) showed a very different reaction, this result could also be influenced by this high variance. A higher number of samples and replicates would be needed in order to completely clarify this point. In any case, the variability was much higher among strains, with 3622 DE genes, than among different experimental conditions, where only 276 DE genes were found, which highlights considerable intraspecific functional differences.
The functions related to the significant DE genes in response to temperature confirmed the stress reaction of L. aporus at low temperature. Oxidative, cold/heat, environmental or physiological stress responses, which seem to be coordinated by specific factors such as stress-activated kinases, AOX4, ALDHs, HSPs and HSFs, SYM1, MAI, FDH and transposable elements (TEs), were notable components of the species functional profile at 13 °C.
TEs proved to be the most significantly enriched units of the stress response, providing the first evidence for the possible role of TEs in the temperature stress response in
L. aporus, as a few others have done recently in diatoms [
47,
82]. All TEs were of the LTR retrotransposon superfamily, confirming the tendency of relatively high abundance of LTR retrotransposons in diatom genomes [
32]. Furthermore, the phylogenetic analysis showed that two of them belong to the GyDi group and one to the CoDi I group, which are both diatom-specific groups, while the CoDi L.ap group is related to the Copia group, which is also found in animals, plants and yeast.
When genes were grouped according to their expression profile in the different conditions, four out of the five TE-related transcripts that were selected for further analysis clustered with genes related to antioxidant activity. Antioxidant responses in plants are initiated after the accumulation of reactive oxygen species (ROS), such as hydrogen peroxide (H
2O
2) and hydroxyl free radical (·OH) [
83,
84]. The generation of numerous ROS can be linked to low temperature, when photosynthetic enzymes may be degraded and photo-damage may occur, leading to reduced photosynthetic activity and hence, the accumulation of excess energy [
85]. Similarly, the overexpression of MAI at low temperature might mean that the mobilization of this gene in
L. aporus contributes to genetic plasticity and finally adaptation to physiological stress in the same way that TEs do. Indeed, in bacteria, MAI undergoes frequent rearrangements, or else transposition events, under physiological stress conditions including prolonged storage at 4 °C or exposure to oxidative stress [
81].
From the between-strains transcriptomic analysis, TEs again held a central role, with their expression showing strain-specificity as shown for fungi and plants [
86,
87,
88]. The absence of specific TEs from the genomes of a couple of the strains (1189B3 and 1189A3), as well as the markedly different response to low, and for some genes, high, temperature of one of the three strains, B651, point to a high intraspecific variability. TE copy number variations (CNV) could also contribute to differences in expression patterns among strains, an issue that would be worth further investigation as CNV, if present, could be related to the strain response to cold. The clusters representation, among other things, allows one to appreciate the noise caused by B651. It is tempting to consider these differences as a result of adaptation: having been isolated from a summer population, strain B651 could be expected to perform worse at low temperature, with a lower expression level of genes involved in cold/heat response than the other two strains 1A1 and 3A6, belonging to winter populations. The clustering of B651 separately from 1A1 and 3A6 based on pathways including amino acid metabolism, glycolysis and gluconeogenesis, energy metabolism, cell growth and death and protein processing in endoplasmic reticulum could imply temperature adaptation changes in the baseline expression of key genes and pathways to maintain metabolic homeostasis, as highlighted in another centric diatom,
Chaetoceros spp. [
17]. The strains 1A1 and 3A6 could hence belong to a cold adapted population, though different from the one detected in the worldwide HTS study of the species [
51].
Yet another hypothesis is that strain B651 was cultivated for four years longer than the other two strains, during which constant temperature conditions could have caused a shift of baseline gene expression and the downregulation or the loss of specific cold/heat response proteins. Indeed, the SNP analysis pointed at another peculiarity of the old strain, with an important loss of heterozygosity compared to the more recent ones, but also to the same strain back in 2011. This is not the first time that loss of heterozygosity is observed in diatom strains kept in prolonged cultivation [
89]. The relevance of cultivation time would be supported by results of growth rate experiments performed on the other two strains isolated in the same year as B651 [
51], which also showed worse performance under extreme temperatures. The effect of in-culture evolution could have been a more likely reason for their distinct behavior than the season of their occurrence, because these two strains were isolated in October and November and not in summer like B651, and were kept in the same conditions as B651 for long time. Stable temperature, light conditions and culture medium, along with the absence of any kind of competition or threat, for four years (more than 1000 generations) could have led to the divergence of B651 from its original ‘natural’ state and affected its ability to respond to temperature changes and stress. The loss of certain stress response mechanisms resulting in the very low expression of TEs and stress-related genes could be one of those effects. A strain of a pennate diatom of the genus
Pseudo-nitzschia also displayed contrasting gene expression patterns under different nutrient conditions compared to earlier experiments [
90], leading to the idea of in-culture progressive physiological modifications targeting functions that are energetically costly and confer no advantage in culture. In the
L. aporus case, the SNP analysis points to a genetic modification following the physiological one, but we prefer to remain cautious about this assumption since genotypes defined only on RNA data can be a result of several confounding pre- and post-transcriptional alternative roads (monoallelic expression, allelic imbalance, DNA editing, RNA editing, etc.). Finally, in support of the in-culture evolution hypothesis, it should also be noted that after two years from isolation, the two strains 1A1 and 3A6 did not show any upregulation, whereas the freshly isolated strains 1188A1 and 1189A3 did show upregulation in a couple of their TEs, even after a short acclimation at low temperature. This small yet existent difference indicates that the absence of stress response is not strain-related to B651 alone, but, when compared with more freshly isolated strains, the same observation can be made for all strains maintained in culture.
In addition to intraspecific diversity and possible in-culture evolution, the response to a stressful environment can also vary in relation with the duration of the exposure to stressful conditions. The contrasting expression patterns noticed in the most recently cultured strains that underwent a shorter period of acclimation indicate a possible role of the exposure time to the stressing factor in
L. aporus: transcripts related to cold stress were not at all expressed (with 22 days acclimation), or, with a few exceptions, expressed at a very low level (with 26–43 days acclimation). Similar changes were seen in the cold water coralline algae
Lithothamnion glaciale, where two phases were identified in a long term experiment under the stress of elevated CO
2 treatment: the “passive” phase during the first 3 months and the “active” phase by the end of 10 months when energy was allocated from cell growth to structural support, showing a clear adaptive plasticity response of the algae [
91]. It therefore seems that the “passive” phase in
L. aporus lasts about 10–20 days, after which these TEs are expressed. Nevertheless, the response to a short and long-term changing environment may be not only species- but also strain-specific [
92], and the relative importance of strain-specific reaction versus time of exposure to stress is hard to disentangle in this study as the strains isolated more recently were also the ones that underwent the shorter acclimation.

 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}