Population Genetics of the Highly Polymorphic RPP8 Gene Family

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Genotyping
2.3. Sequencing
2.4. Data Analysis
2.5. Forward-in-Time Simulations of Polymorphism
3. Results
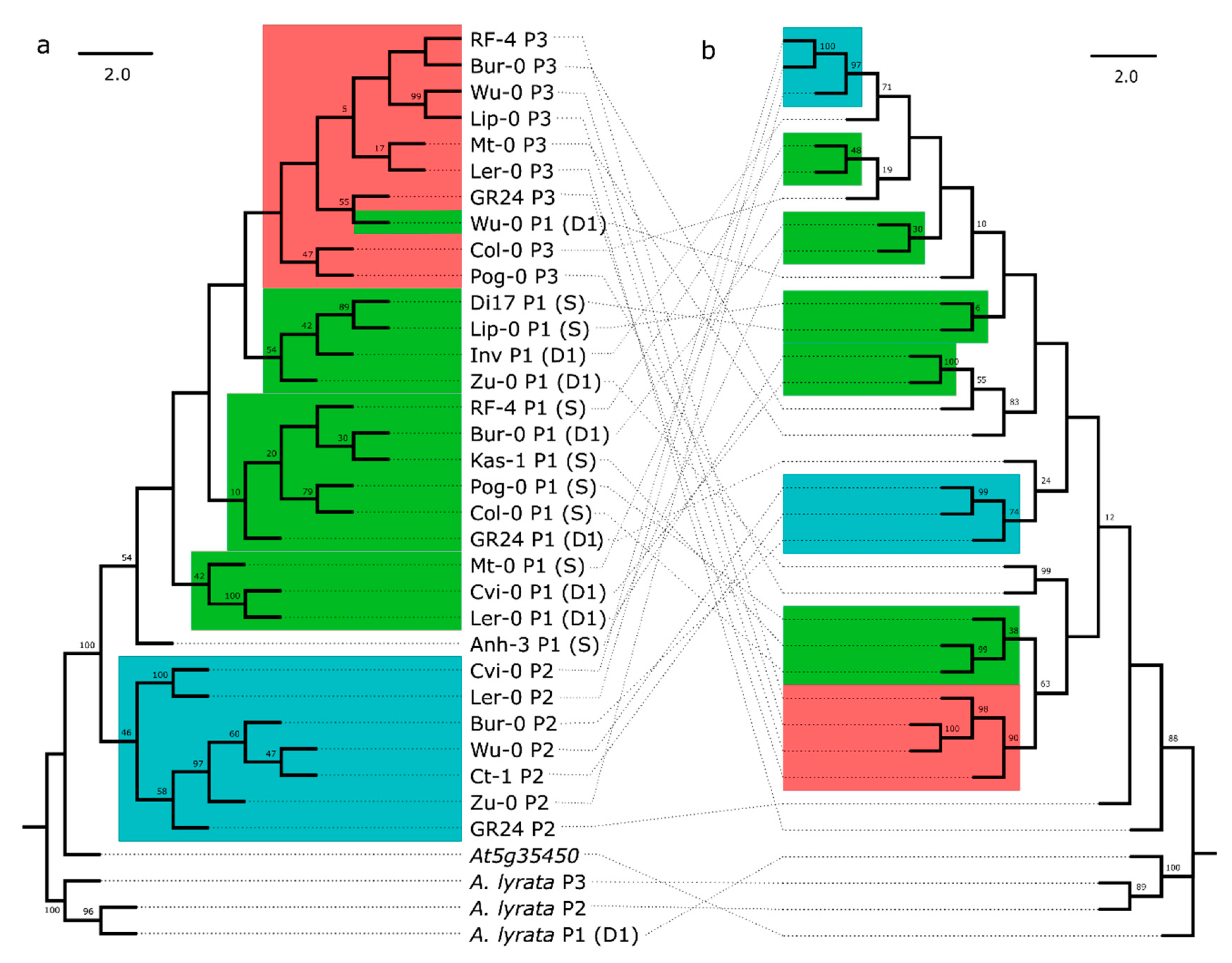
3.1. Variation in RPP8 Genomic Architecture, Phenotypes, and Sequence
3.2. Rates of IGC between RPP8 Paralogs
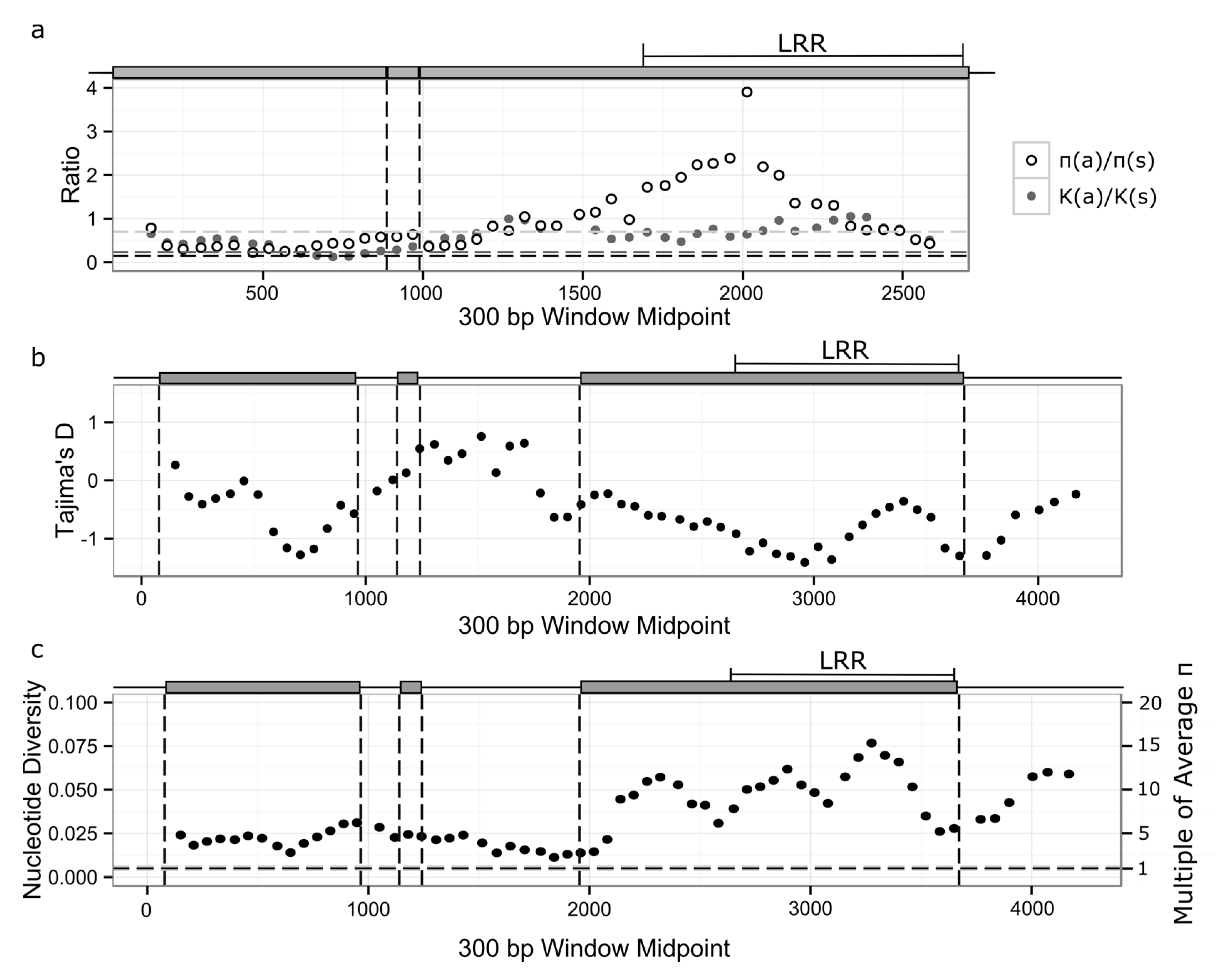
3.3. Signatures of Selection in RPP8 Paralogs
3.4. Nucleotide Diversity
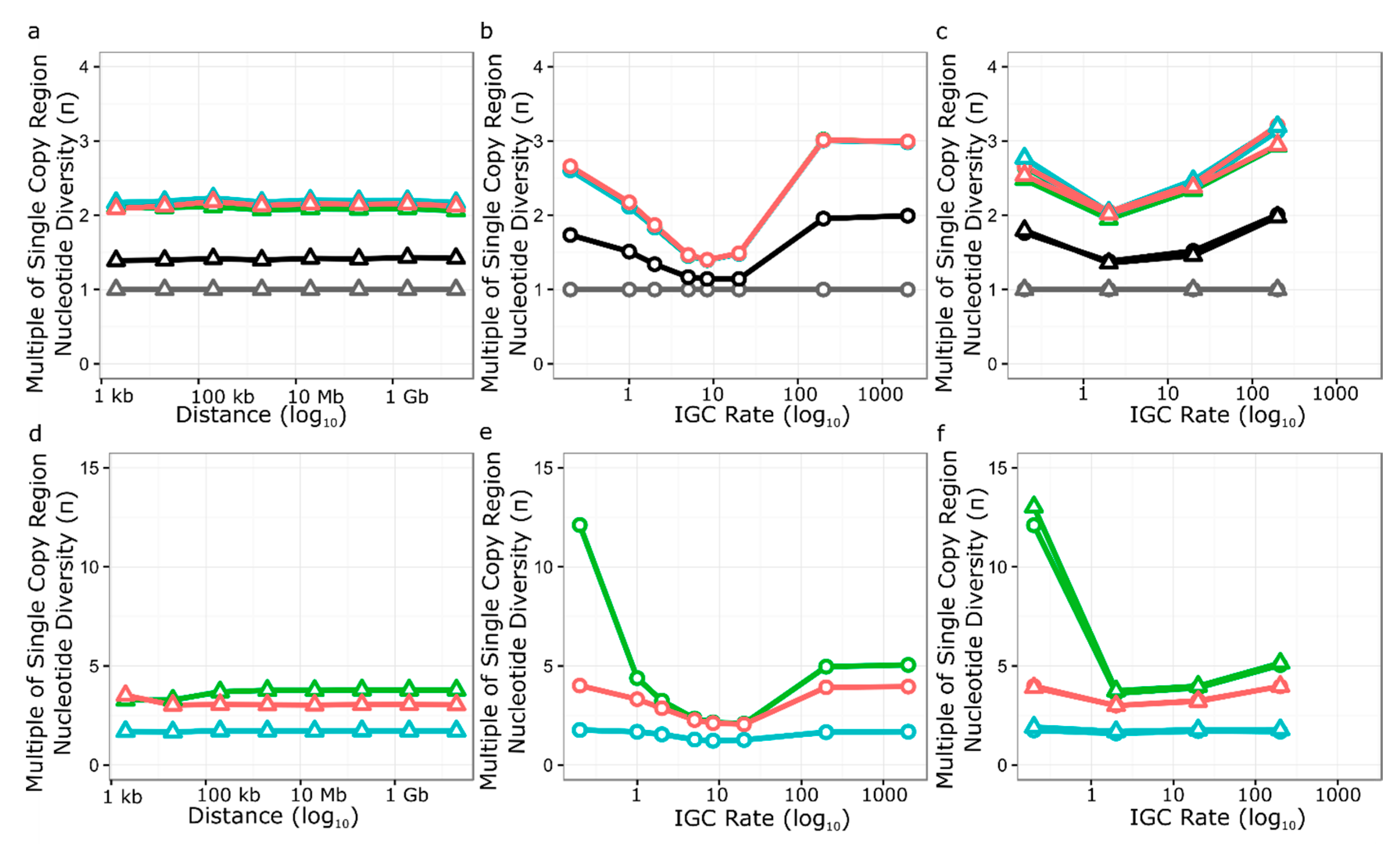
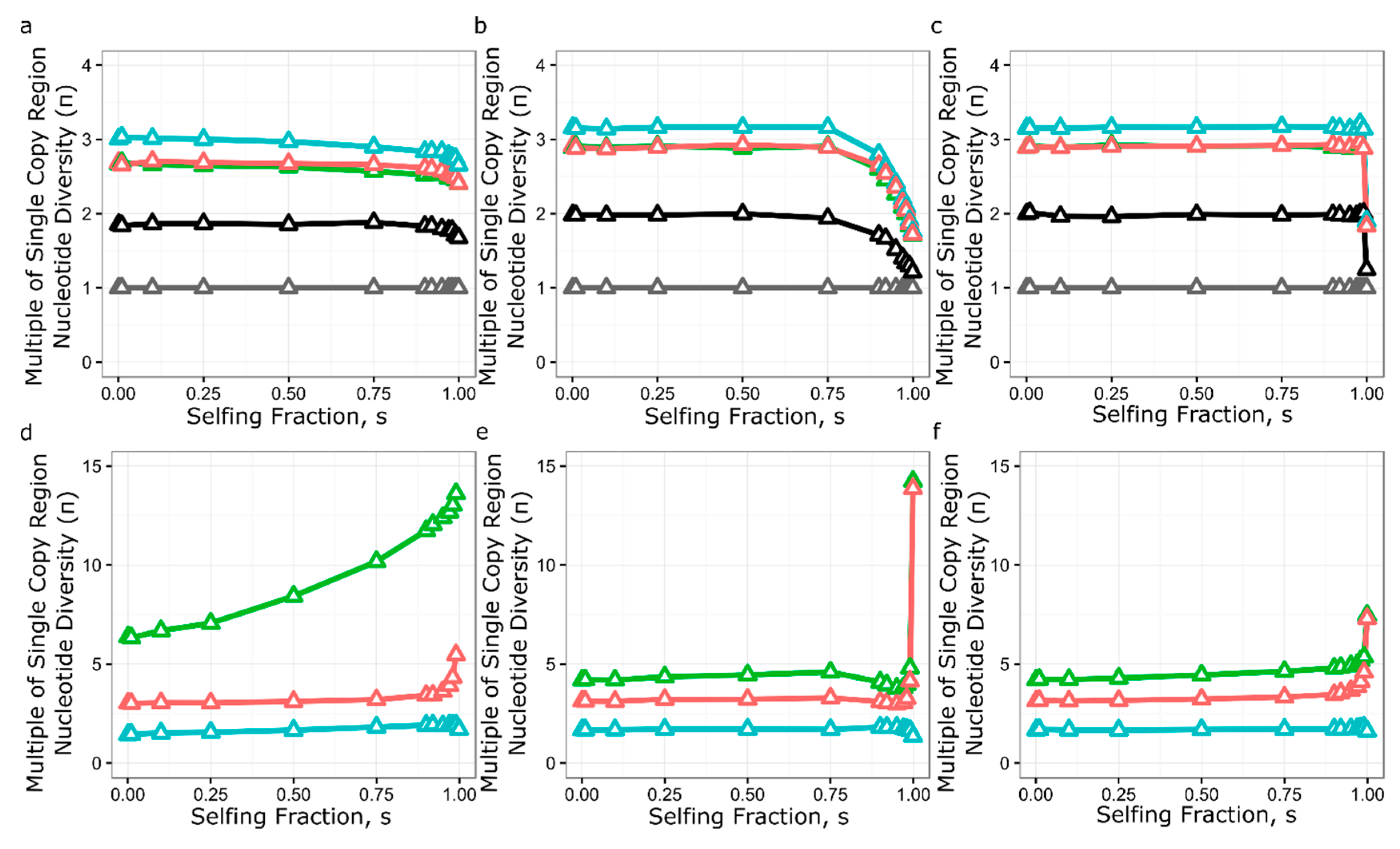
3.5. Simulation of Neutral Evolution and Balancing Selection at RPP8
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Van de Weyer, A.-L.; Monteiro, F.; Furzer, O.J.; Nishimura, M.T.; Cevik, V.; Witek, K.; Jones, J.D.G.; Dangl, J.L.; Weigel, D.; Bemm, F. The Arabidopsis thaliana pan-NLRome. bioRxiv 2019. [Google Scholar] [CrossRef]
- Michelmore, R.; Meyers, B. Clusters of Resistance Genes in Plants Evolve by Divergent Selection and a Birth-and-Death Process. Genome Res. 1998, 8, 1113–1130. [Google Scholar] [CrossRef] [PubMed]
- Gan, X.; Stegle, O.; Behr, J.; Steffen, J.G.; Drewe, P.; Hildebrand, K.L.; Lyngsoe, R.; Schultheiss, S.J.; Osborne, E.J.; Sreedharan, V.T.; et al. Multiple reference genomes and transcriptomes for Arabidopsis thaliana. Nature 2011, 477, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Catanzariti, A.-M.; Dodds, P.N.; Ve, T.; Kobe, B.; Ellis, J.G.; Staskawicz, B.J. The AvrM effector from flax rust has a structured C-terminal domain and interacts directly with the M resistance protein. Mol. Plant Microbe Interact. MPMI 2010, 23, 49–57. [Google Scholar] [CrossRef]
- Dodds, P.N.; Lawrence, G.J.; Catanzariti, A.-M.; Teh, T.; Wang, C.-I.A.; Ayliffe, M.A.; Kobe, B.; Ellis, J.G. Direct protein interaction underlies gene-for-gene specificity and coevolution of the flax resistance genes and flax rust avirulence genes. Proc. Natl. Acad. Sci. USA 2006, 103, 8888. [Google Scholar] [CrossRef]
- Krasileva, K.V.; Dahlbeck, D.; Staskawicz, B.J. Activation of an Arabidopsis Resistance Protein Is Specified by the in Planta Association of Its Leucine-Rich Repeat Domain with the Cognate Oomycete Effector. Plant Cell 2010, 22, 2444. [Google Scholar] [CrossRef]
- McDowell, J.M.; Dhandaydham, M.; Long, T.A.; Aarts, M.G.M.; Goff, S.; Holub, E.B.; Dangl, J.L. Intragenic Recombination and Diversifying Selection Contribute to the Evolution of Downy Mildew Resistance at the RPP8 Locus of Arabidopsis. Plant Cell 1998, 10, 1861–1874. [Google Scholar] [CrossRef]
- Mackey, D.; Holt, B.F., III; Wiig, A.; Dangl, J.L. RIN4 Interacts with Pseudomonas syringae Type III Effector Molecules and Is Required for RPM1-Mediated Resistance in Arabidopsis. Cell 2002, 108, 743–754. [Google Scholar] [CrossRef]
- Qi, D.; Dubiella, U.; Kim, S.H.; Sloss, D.I.; Dowen, R.H.; Dixon, J.E.; Innes, R.W. Recognition of the protein kinase AVRPPHB SUSCEPTIBLE1 by the disease resistance protein RESISTANCE TO PSEUDOMONAS SYRINGAE5 is dependent on s-acylation and an exposed loop in AVRPPHB SUSCEPTIBLE1. Plant Physiol. 2014, 164, 340–351. [Google Scholar] [CrossRef]
- Wang, G.; Roux, B.; Feng, F.; Guy, E.; Li, L.; Li, N.; Zhang, X.; Lautier, M.; Jardinaud, M.-F.; Chabannes, M.; et al. The Decoy Substrate of a Pathogen Effector and a Pseudokinase Specify Pathogen-Induced Modified-Self Recognition and Immunity in Plants. Cell Host Microbe 2015, 18, 285–295. [Google Scholar] [CrossRef]
- Cesari, S.; Bernoux, M.; Moncuquet, P.; Kroj, T.; Dodds, P.N. A novel conserved mechanism for plant NLR protein pairs: The “integrated decoy” hypothesis. Front. Plant Sci. 2014, 5, 606. [Google Scholar] [CrossRef] [PubMed]
- Cesari, S.; Thilliez, G.; Ribot, C.; Chalvon, V.; Michel, C.; Jauneau, A.; Rivas, S.; Alaux, L.; Kanzaki, H.; Okuyama, Y.; et al. The rice resistance protein pair RGA4/RGA5 recognizes the Magnaporthe oryzae effectors AVR-Pia and AVR1-CO39 by direct binding. Plant Cell 2013, 25, 1463–1481. [Google Scholar] [CrossRef] [PubMed]
- Le Roux, C.; Huet, G.; Jauneau, A.; Camborde, L.; Trémousaygue, D.; Kraut, A.; Zhou, B.; Levaillant, M.; Adachi, H.; Yoshioka, H.; et al. A Receptor Pair with an Integrated Decoy Converts Pathogen Disabling of Transcription Factors to Immunity. Cell 2015, 161, 1074–1088. [Google Scholar] [CrossRef] [PubMed]
- Bergelson, J.; Kreitman, M.; Stahl, E.A.; Tian, D. Evolutionary Dynamics of Plant R-Genes. Science 2001, 292, 2281. [Google Scholar] [CrossRef] [PubMed]
- Schmutz, J.; McClean, P.E.; Mamidi, S.; Wu, G.A.; Cannon, S.B.; Grimwood, J.; Jenkins, J.; Shu, S.; Song, Q.; Chavarro, C.; et al. A reference genome for common bean and genome-wide analysis of dual domestications. Nat. Genet. 2014, 46, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Meyers, B.C. Genome-Wide Analysis of NBS-LRR-Encoding Genes in Arabidopsis. Plant Cell Online 2003, 15, 809–834. [Google Scholar] [CrossRef]
- Guo, Y.-L.; Fitz, J.; Schneeberger, K.; Ossowski, S.; Cao, J.; Weigel, D. Genome-Wide Comparison of Nucleotide-Binding Site-Leucine-Rich Repeat-Encoding Genes in Arabidopsis. Plant Physiol. 2011, 157, 757. [Google Scholar] [CrossRef]
- Die, J.V.; Castro, P.; Millán, T.; Gil, J. Segmental and Tandem Duplications Driving the Recent NBS-LRR Gene Expansion in the Asparagus Genome. Genes 2018, 9, 568. [Google Scholar] [CrossRef]
- Zhang, Y.-M.; Shao, Z.-Q.; Wang, Q.; Hang, Y.-Y.; Xue, J.-Y.; Wang, B.; Chen, J.-Q. Uncovering the dynamic evolution of nucleotide-binding site-leucine-rich repeat (NBS-LRR) genes in Brassicaceae. J. Integr. Plant Biol. 2016, 58, 165–177. [Google Scholar] [CrossRef]
- Meyers, B.C.; Chin, D.B.; Shen, K.A.; Sivaramakrishnan, S.; Lavelle, D.O.; Zhang, Z.; Michelmore, R.W. The major resistance gene cluster in lettuce is highly duplicated and spans several megabases. Plant Cell 1998, 10, 1817–1832. [Google Scholar] [CrossRef]
- Cheng, Y.; Li, X.; Jiang, H.; Ma, W.; Miao, W.; Yamada, T.; Zhang, M. Systematic analysis and comparison of nucleotide-binding site disease resistance genes in maize. FEBS J. 2012, 279, 2431–2443. [Google Scholar] [CrossRef] [PubMed]
- Leister, D. Tandem and segmental gene duplication and recombination in the evolution of plant disease resistance genes. Trends Genet. 2004, 20, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Perazzolli, M.; Malacarne, G.; Baldo, A.; Righetti, L.; Bailey, A.; Fontana, P.; Velasco, R.; Malnoy, M. Characterization of resistance gene analogues (RGAs) in apple (Malus × domestica Borkh.) and their evolutionary history of the Rosaceae family. PLoS ONE 2014, 9, e83844. [Google Scholar] [CrossRef] [PubMed]
- Stein, J.C.; Yu, Y.; Copetti, D.; Zwickl, D.J.; Zhang, L.; Zhang, C.; Chougule, K.; Gao, D.; Iwata, A.; Goicoechea, J.L.; et al. Genomes of 13 domesticated and wild rice relatives highlight genetic conservation, turnover and innovation across the genus Oryza. Nat. Genet. 2018, 50, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Seo, E.; Kim, S.; Yeom, S.-I.; Choi, D. Genome-Wide Comparative Analyses Reveal the Dynamic Evolution of Nucleotide-Binding Leucine-Rich Repeat Gene Family among Solanaceae Plants. Front. Plant Sci. 2016, 7, 1205. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.L.; Bittner-Eddy, P.D.; Grenville-Briggs, L.J.; Meitz, J.C.; Rehmany, A.P.; Rose, L.E.; Beynon, J.L. Host-Parasite Coevolutionary Conflict Between Arabidopsis and Downy Mildew. Science 2004, 306, 1957–1960. [Google Scholar] [CrossRef] [PubMed]
- Rose, L.E.; Bittner-Eddy, P.D.; Langley, C.H.; Holub, E.B.; Michelmore, R.W.; Beynon, J.L. The maintenance of extreme amino acid diversity at the disease resistance gene, RPP13 in Arabidopsis Thaliana. Genetics 2004, 166, 1517–1527. [Google Scholar] [CrossRef]
- Hörger, A.C.; Ilyas, M.; Stephan, W.; Tellier, A.; van der Hoorn, R.A.L.; Rose, L.E. Balancing Selection at the Tomato RCR3 Guardee Gene Family Maintains Variation in Strength of Pathogen Defense. PLoS Genet. 2012, 8, e1002813. [Google Scholar] [CrossRef]
- Mauricio, R.; Stahl, E.A.; Korves, T.; Tian, D.; Kreitman, M.; Bergelson, J. Natural selection for polymorphism in the disease resistance gene Rps2 of Arabidopsis Thaliana. Genetics 2003, 163, 735–746. [Google Scholar]
- Stahl, E.A.; Dwyer, G.; Mauricio, R.; Kreitman, M.; Bergelson, J. Dynamics of disease resistance polymorphism at the Rpm1 locus of Arabidopsis. Nature 1999, 400, 667–671. [Google Scholar] [CrossRef]
- Rose, L.E.; Grzeskowiak, L.; Hörger, A.C.; Groth, M.; Stephan, W. Targets of selection in a disease resistance network in wild tomatoes. Mol. Plant Pathol. 2011, 12, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Tian, D.; Traw, M.B.; Chen, J.Q.; Kreitman, M.; Bergelson, J. Fitness costs of R-gene-mediated resistance in Arabidopsis thaliana. Nature 2003, 423, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Karasov, T.L.; Kniskern, J.M.; Gao, L.; DeYoung, B.J.; Ding, J.; Dubiella, U.; Lastra, R.O.; Nallu, S.; Roux, F.; Innes, R.W.; et al. The long-term maintenance of a resistance polymorphism through diffuse interactions. Nature 2014, 512, 436–440. [Google Scholar] [CrossRef] [PubMed]
- Stam, R.; Silva-Arias, G.A.; Tellier, A. Subsets of NLR genes show differential signatures of adaptation during colonization of new habitats. New Phytol. 2019, 224, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Bakker, E.G.; Toomajian, C.; Kreitman, M.; Bergelson, J. A genome-wide survey of R gene polymorphisms in Arabidopsis. Plant Cell 2006, 18, 1803–1818. [Google Scholar] [CrossRef] [PubMed]
- Cridland, J.M.; Thornton, K.R. Validation of rearrangement break points identified by paired-end sequencing in natural populations of Drosophila melanogaster. Genome Biol. Evol. 2010, 2, 83–101. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.; Conery, J.S. The Evolutionary Fate and Consequences of Duplicate Genes. Science 2000, 290, 1151–1155. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.A.; Gu, Z.; Clark, R.A.; Reinert, K.; Samonte, R.V.; Schwartz, S.; Adams, M.D.; Myers, E.W.; Li, P.W.; Eichler, E.E. Recent Segmental Duplications in the Human Genome. Science 2002, 297, 1003–1007. [Google Scholar] [CrossRef]
- Chen, J.-M.; Cooper, D.N.; Chuzhanova, N.; Férec, C.; Patrinos, G.P. Gene conversion: Mechanisms, evolution and human disease. Nat. Rev. Genet. 2007, 8, 762. [Google Scholar] [CrossRef]
- Marques-Bonet, T.; Kidd, J.M.; Ventura, M.; Graves, T.A.; Cheng, Z.; Hillier, L.W.; Jiang, Z.; Baker, C.; Malfavon-Borja, R.; Fulton, L.A.; et al. A burst of segmental duplications in the genome of the African great ape ancestor. Nature 2009, 457, 877. [Google Scholar] [CrossRef]
- Innan, H. A method for estimating the mutation, gene conversion and recombination parameters in small multigene families. Genetics 2002, 161, 865–872. [Google Scholar] [PubMed]
- Innan, H. The Coalescent and Infinite-Site Model of a Small Multigene Family. Genetics 2003, 163, 803. [Google Scholar] [PubMed]
- Teshima, K.M.; Innan, H. The coalescent with selection on copy number variants. Genetics 2012, 190, 1077–1086. [Google Scholar] [CrossRef] [PubMed]
- Ohta, T. On the evolution of multigene families. Theor. Popul. Biol. 1983, 23, 216–240. [Google Scholar] [CrossRef]
- Brown, J.K.M.; Tellier, A. Plant-Parasite Coevolution: Bridging the Gap between Genetics and Ecology. Annu. Rev. Phytopathol. 2011, 49, 345–367. [Google Scholar] [CrossRef] [PubMed]
- Laine, A.-L.; Tellier, A. Heterogeneous selection promotes maintenance of polymorphism in host–parasite interactions. Oikos 2008, 117, 1281–1288. [Google Scholar] [CrossRef]
- Kuang, H.; Caldwell, K.S.; Meyers, B.C.; Michelmore, R.W. Frequent sequence exchanges between homologs of RPP8 in Arabidopsis are not necessarily associated with genomic proximity. Plant J. 2008, 54, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Cooley, M.B.; Pathirana, S.; Wu, H.J.; Kachroo, P.; Klessig, D.F. Members of the Arabidopsis HRT/RPP8 family of resistance genes confer resistance to both viral and oomycete pathogens. Plant Cell 2000, 12, 663–676. [Google Scholar] [CrossRef]
- Takahashi, H.; Miller, J.; Nozaki, Y.; Sukamto; Takeda, M.; Shah, J.; Hase, S.; Ikegami, M.; Ehara, Y.; Dinesh-Kumar, S.P. RCY1, an Arabidopsis thaliana RPP8/HRT family resistance gene, conferring resistance to cucumber mosaic virus requires salicylic acid, ethylene and a novel signal transduction mechanism. Plant J. 2002, 32, 655–667. [Google Scholar] [CrossRef]
- Hartasánchez, D.A.; Brasó-Vives, M.; Fuentes-Díaz, J.; Vallès-Codina, O.; Navarro, A. SeDuS: Segmental duplication simulator. Bioinformatics 2015, 32, 148–150. [Google Scholar] [CrossRef]
- Kim, S.; Plagnol, V.; Hu, T.T.; Toomajian, C.; Clark, R.M.; Ossowski, S.; Ecker, J.R.; Weigel, D.; Nordborg, M. Recombination and linkage disequilibrium in Arabidopsis thaliana. Nat. Genet. 2007, 39, 1151. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.J. DNA Protocols for Plants; Springer: Berlin/Heidelberg, Germany, 1991. [Google Scholar]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Mansai, S.P.; Innan, H. The power of the methods for detecting interlocus gene conversion. Genetics 2010, 184, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Kolaczkowski, B.; Thornton, J.W. Performance of maximum parsimony and likelihood phylogenetics when evolution is heterogeneous. Nature 2004, 431, 980–984. [Google Scholar] [CrossRef] [PubMed]
- Hartasánchez, D.A.; Valles-Codina, O.; Braso-Vives, M.; Navarro, A. Interplay of interlocus gene conversion and crossover in segmental duplications under a neutral scenario. G3 2014, 4, 1479–1489. [Google Scholar] [CrossRef]
- Swofford, D.L. PAUP*. Phylogenetic Analysis Using Parsimony (*and other Methods) Version 4. Sinauer, Sunderland, Massachusetts, USA. Nat. Biotechnol. 2003, 18, 233–234. [Google Scholar]
- Nordborg, M. Linkage disequilibrium, gene trees and selfing: An ancestral recombination graph with partial self-fertilization. Genetics 2000, 154, 923–929. [Google Scholar]
- Platt, A.; Horton, M.; Huang, Y.S.; Li, Y.; Anastasio, A.E.; Mulyati, N.W.; Agren, J.; Bossdorf, O.; Byers, D.; Donohue, K.; et al. The scale of population structure in Arabidopsis thaliana. PLoS Genet. 2010, 6, e1000843. [Google Scholar] [CrossRef]
- Remington, D.L. Alleles versus mutations: Understanding the evolution of genetic architecture requires a molecular perspective on allelic origins. Evolution 2015, 69, 3025–3038. [Google Scholar] [CrossRef]
- Benovoy, D.; Morris, R.T.; Morin, A.; Drouin, G. Ectopic Gene Conversions Increase the G + C Content of Duplicated Yeast and Arabidopsis Genes. Mol. Biol. Evol. 2005, 22, 1865–1868. [Google Scholar] [CrossRef]
- Teshima, K.M.; Innan, H. Effect of Gene Conversion on Divergence Between Duplicated Genes. Genetics 2003, 166, 1553–1560. [Google Scholar] [CrossRef]
- Choi, K.; Henderson, I.R. Meiotic recombination hotspots—A comparative view. Plant J. 2015, 83, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Pyhäjärvi, T.; Weber, A.L.; Dawe, R.K.; Glaubitz, J.C.; González, J.d.J.S.; Ross-Ibarra, C.; Doebley, J.; Morrell, P.L.; Ross-Ibarra, J. Megabase-scale inversion polymorphism in the wild ancestor of maize. Genetics 2012, 191, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Fransz, P.; Linc, G.; Lee, C.-R.; Aflitos, S.A.; Lasky, J.R.; Toomajian, C.; Ali, H.; Peters, J.; van Dam, P.; Ji, X.; et al. Molecular, genetic and evolutionary analysis of a paracentric inversion in Arabidopsis thaliana. Plant J. 2016, 88, 159–178. [Google Scholar] [CrossRef] [PubMed]
- Serra, H.; Choi, K.; Zhao, X.; Blackwell, A.R.; Kim, J.; Henderson, I.R. Interhomolog polymorphism shapes meiotic crossover within the Arabidopsis RAC1 and RPP13 disease resistance genes. PLoS Genet. 2018, 14, e1007843. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.T.; Pattyn, P.; Bakker, E.G.; Cao, J.; Cheng, J.F.; Clark, R.M.; Fahlgren, N.; Fawcett, J.A.; Grimwood, J.; Gundlach, H.; et al. The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nat. Genet. 2011, 43, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Nordborg, M.; Hu, T.T.; Ishino, Y.; Jhaveri, J.; Toomajian, C.; Zheng, H.; Bakker, E.; Calabrese, P.; Gladstone, J.; Goyal, R.; et al. The pattern of polymorphism in Arabidopsis thaliana. PLoS Biol. 2005, 3, e196. [Google Scholar] [CrossRef]
- Coyne, J.; Orr, H. Speciation; Oxford University Press: Sunderland, UK, 2004; p. 545. [Google Scholar]
- Innan, H. A two-locus gene conversion model with selection and its application to the human RHCE and RHD genes. Proc. Natl. Acad. Sci. USA 2003, 100, 8793–8798. [Google Scholar] [CrossRef]
- Shen, P.; Huang, H.V. Homologous recombination in Escherichia coli: Dependence on substrate length and homology. Genetics 1986, 112, 441–457. [Google Scholar]
- Semple, C.H.; Wolfe, K. Gene Duplication and Gene Conversion in the Caenorhabditis elegans Genome. J. Mol. Evol. 1999, 48, 555–564. [Google Scholar] [CrossRef]
- Mondragon-Palomino, M.; Gaut, B.S. Gene Conversion and the Evolution of Three Leucine-Rich Repeat Gene Families in Arabidopsis thaliana. Mol. Biol. Evol. 2005, 22, 2444–2456. [Google Scholar] [CrossRef] [PubMed]
- Novikova, P.Y.; Hohmann, N.; Nizhynska, V.; Tsuchimatsu, T.; Ali, J.; Muir, G.; Guggisberg, A.; Paape, T.; Schmid, K.; Fedorenko, O.M.; et al. Sequencing of the genus Arabidopsis identifies a complex history of nonbifurcating speciation and abundant trans-specific polymorphism. Nat. Genet. 2016, 48, 1077. [Google Scholar] [CrossRef] [PubMed]
- Korves, T.; Bergelson, J. A Novel Cost of R-gene Resistance in the Presence of Disease. Am. Nat. 2004, 163, 489–504. [Google Scholar] [CrossRef] [PubMed]
- MacQueen, A.; Sun, X.; Bergelson, J. Genetic architecture and pleiotropy shape costs of Rps2-mediated resistance in Arabidopsis thaliana. Nat. Plants 2016, 2, 16110. [Google Scholar] [CrossRef] [PubMed]
- Croze, M.; Živković, D.; Stephan, W.; Hutter, S. Balancing selection on immunity genes: Review of the current literature and new analysis in Drosophila melanogaster. Zoology 2016, 119, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Spurgin, L.G.; Richardson, D.S. How pathogens drive genetic diversity: MHC, mechanisms and misunderstandings. Proc. Biol. Sci. 2010, 277, 979–988. [Google Scholar] [CrossRef] [PubMed]
- Lewontin, R.C.; Ginzburg, L.R.; Tuljapurkar, S.D. Heterosis as an Explanation for Large Amounts of Genic Polymorphism. Genetics 1978, 88, 149. [Google Scholar]
- Spencer, H.G.; Mitchell, C. The Selective Maintenance of Allelic Variation Under Generalized Dominance. G3 Genes Genomes Genet. 2016, 6, 3725–3732. [Google Scholar] [CrossRef]
- Fowler, K.R.; Hyppa, R.W.; Cromie, G.A.; Smith, G.R. Physical basis for long-distance communication along meiotic chromosomes. Proc. Natl. Acad. Sci. USA 2018, 115, E9333–E9342. [Google Scholar] [CrossRef]
- Parniske, M.; Jones, J.D. Recombination between diverged clusters of the tomato Cf-9 plant disease resistance gene family. Proc. Natl. Acad. Sci. USA 1999, 96, 5850–5855. [Google Scholar] [CrossRef]
- Parniske, M.; Hammond-Kosack, K.E.; Golstein, C.; Thomas, C.M.; Jones, D.A.; Harrison, K.; Wulff, B.B.H.; Jones, J.D.G. Novel Disease Resistance Specificities Result from Sequence Exchange between Tandemly Repeated Genes at the Cf-4/9 Locus of Tomato. Cell 1997, 91, 821–832. [Google Scholar] [CrossRef]
- Collins, N.; Drake, J.; Ayliffe, M.; Sun, Q.; Ellis, J.; Hulbert, S.; Pryor, T. Molecular Characterization of the Maize Rp-1 Rust Resistance Haplotype and Its Mutants. Plant Cell 1999, 11, 1365. [Google Scholar] [CrossRef] [PubMed]
- Baumgarten, A.; Cannon, S.; Spangler, R.; May, G. Genome-level evolution of resistance genes in Arabidopsis thaliana. Genetics 2003, 165, 309–319. [Google Scholar] [PubMed]
- Wijnker, E.; Velikkakam James, G.; Ding, J.; Becker, F.; Klasen, J.R.; Rawat, V.; Rowan, B.A.; de Jong, D.F.; de Snoo, C.B.; Zapata, L.; et al. The genomic landscape of meiotic crossovers and gene conversions in Arabidopsis thaliana. Elife 2013, 2, e01426. [Google Scholar] [CrossRef] [PubMed]
- Meagher, R.B.; Berry-Lowe, S.; Rice, K. Molecular evolution of the small subunit of ribulose bisphosphate carboxylase: Nucleotide substitution and gene conversion. Genetics 1989, 123, 845–863. [Google Scholar] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| All Coding Sites (2766 bp) | LRR (888 bp) | |||||
|---|---|---|---|---|---|---|
| Segregating Sites (S) 1 | Average Number of SNPs/Allele 1 | Number of Unique Haplotypes | Segregating Sites (S) 1 | Average Number of SNPs/Allele 1 | Number of Unique Haplotypes | |
| P1 | 358 | 108.0 | 15/15 | 254 | 61.2 | 20/21 |
| S | 276 | 109.3 | 8/8 | 153 | 54.2 | 11/11 |
| D1 | 246 | 103.4 | 7/7 | 212 | 68.0 | 9/10 |
| D2 | 174 | 80.5 | 7/7 | 162 | 51.8 | 17/19 |
| P3 | 302 | 110.8 | 9/9 | 179 | 63.7 | 9/10 |
| all | 470 | 118.0 | 31/31 | 327 | 60.4 | 45/50 |
| In X, Not in Y 1 | S | D1 | D2 | P3 |
|---|---|---|---|---|
| S | - | 0 | 27 | 0 |
| D1 | 0 | - | 14 | 0 |
| D2 | 4 | 0 | - | 3 |
| P3 | 4 | 0 | 27 | - |
| In X and Y 1 | S | D1 | D2 | P3 |
|---|---|---|---|---|
| S | - | 316 | 183 | 326 |
| D1 | - | - | 161 | 307 |
| D2 | - | - | - | 162 |
| Coding Region | LRR | |||||||
|---|---|---|---|---|---|---|---|---|
| πs | πa | πa/πs | Ks | Ka | Ka/Ks | πa/πs | Ka/Ks | |
| Genome Average 1 | 0.005 (0.004–0.006) | 0.0014 | 0.23 | 0.13 (0.02, 0.24) | 0.025 (0, 0.12) | 0.19 (0, 0.07) | n/a | n/a |
| P1 | 0.0429 | 0.0355 | 0.829 | 0.143 | 0.0789 | 0.527 | 1.56 | 0.746 |
| P2 | 0.0341 | 0.0279 | 0.815 | 0.130 | 0.0823 | 0.612 | 2.27 | 0.965 |
| P3 | 0.0459 | 0.0383 | 0.830 | 0.144 | 0.0814 | 0.538 | 1.06 | 0.751 |
| LRR a | 1 b | 2 | 3 | 4 c | 5d | 6 | 7 | 8 e | 9 | 10 f | 11 | 12 | 13 | 14 | Average Fraction Derived (~Ka) g | Fold Increase over Genomic Ka (C.I.) h | Distinct Amino Acids (C.I.) i |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| l | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0.007 +/− 0.002 | 0.3 | 1.1 (1, 1.5) |
| x | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 4 | 0 | 0 | 0.014 +/− 0.003 | 0.6 | 1.2 (1, 2) |
| x | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0.014 +/− 0.002 | 0.6 | 1.2 (1, 2) |
| l | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 7 | 0 | 0 | 1 | 0 | 0.019 +/− 0.004 | 0.8 | 1.1 (1, 2) |
| x | 0 | 0 | 0 | 0 | 6 | 0 | 4 | 0 | 0 | 0 | 4 | 14 | 0 | 0 | 0.067 +/− 0.01 | 2.7 | 1.4 (1, 2) |
| x | 0 | 0 | 0 | 1 | 7 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0.024 +/− 0.004 | 1 | 1.3 (1, 2) |
| l | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0.002 +/− 0.001 | 0.1 | 1.1 (1, 1.5) |
| X | 21 | 0 | 0 | 0 | 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0.06 +/− 0.013 | 2.4 | 1.4 (1, 2) |
| X | 11 | 1 | 1 | 4 | 1 | 7 | 26 | 5 | 12 | 16 | 0 | 1 | 1 | 0 | 0.205 * +/− 0.018 | 8.2* | 2.9* (1.5, 4.5) |
| L | 8 | 0 | 0 | 1 | 2 | 0 | 2 | 0 | 1 | 6 | 0 | 1 | 0 | 0 | 0.05 +/− 0.006 | 2 | 1.6 (1, 2.5) |
| X | 25 | 4 | 10 | 10 | 9 | 22 | 17 | 28 | 2 | 3 | 13 | 22 | 0 | 3 | 0.4 * +/− 0.022 | 16* | 3.5* (2, 5.5) |
| L | 2 | 0 | 0 | 4 | 5 | 6 | 1 | 18 | 1 | 21 | 1 | 0 | 0 | 0 | 0.14 * +/− 0.016 | 5.6* | 1.9 (1, 3) |
| X | 25 | 2 | 12 | 12 | 0 | 0 | 3 | 7 | 11 | 3 | 18 | 0 | 8 | 12 | 0.269 * +/− 0.018 | 10.8* | 2.9 (1, 5) |
| X | 0 | 3 | 25 | 3 | 11 | 0 | 0 | 6 | 3 | 18 | 18 | 10 | 2 | 5 | 0.248 * +/− 0.019 | 9.9* | 3.3* (1.5, 5.5) |
| X | 21 | 0 | 0 | 10 | 18 | 0 | 1 | 7 | 1 | 18 | 18 | 13 | 0 | 0 | 0.255 * +/− 0.02 | 10.2* | 2.8 (1, 5) |
| X | 4 | 0 | 11 | 6 | 2 | 0 | 0 | 18 | 0 | 0 | 0 | 6 | 14 | 0 | 0.145 * +/− 0.014 | 5.8* | 1.9 (0.5, 3.5) |
| x | 0 | 0 | 1 | 9 | 19 | 2 | 0 | 9 | 0 | 15 | 0 | 0 | 0 | 0 | 0.131 +/− 0.015 | 5.2 | 1.7 (1, 3) |
| x | 0 | 0 | 0 | 8 | 0 | 12 | 4 | 11 | 0 | 0 | 7 | 0 | 0 | 1 | 0.102 +/− 0.011 | 4.1 | 1.6 (1, 3) |
| x | 0 | 0 | 0 | 7 | 0 | 0 | 1 | 10 | 0 | 3 | 0 | 0 | 0 | 0 | 0.05 +/− 0.007 | 2 | 1.6 (0.5, 3) |
| x | 0 | 0 | 0 | 2 | 3 | 2 | 1 | 7 | 0 | 2 | 13 | 3 | 0 | 0 | 0.079 +/− 0.009 | 3.2 | 1.6 (1.5, 2.5) |
| x | 0 | 0 | 0 | 0 | 0 | 0 | 4 | 3 | 0 | 0 | 0 | 0 | 0 | 1 | 0.019 +/− 0.003 | 0.8 | 1.2 (1, 2) |
| x | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 3 | 0 | 1 | 0 | 0 | 0 | 0.015 +/− 0.002 | 0.6 | 1.3 (1, 2) | |
| x | 0 | 0 | 0 | 1 | 0 | 18 | 0 | 0 | 0 | 0 | 0 | 0.058 +/− 0.016 | 2.3 | 1.2 (1, 2) | |||
| x | 8 | 0 | 1 | 3 | 0 | 0 | 0 | 1 | 0 | 0.048 +/− 0.01 | 1.9 | 1.4 (1, 2) | |||||
| x | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0.01 +/− 0.002 | 0.4 | 1.1 (1, 2) | |||||||
| x | 0 | 0 | 1 | 0.011 +/− 0.006 | 0.4 | 1.3 (1, 2) |
| X/Y | S | D1 | D2 | P3 |
|---|---|---|---|---|
| S | 0.0477 ± 0.0100 | 0.0449 ± 0.0079 | 0.0537 ± 0.0067 | 0.0526 ± 0.0130 |
| D1 | - | 0.0392 ± 0.0110 | 0.0559 ± 0.0076 | 0.0476 ± 0.0138 |
| D2 | - | - | 0.0333 ± 0.0156 | 0.0597 ± 0.0140 |
| P3 | - | - | - | 0.0451 ± 0.0195 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MacQueen, A.; Tian, D.; Chang, W.; Holub, E.; Kreitman, M.; Bergelson, J. Population Genetics of the Highly Polymorphic RPP8 Gene Family. Genes 2019, 10, 691. https://doi.org/10.3390/genes10090691
MacQueen A, Tian D, Chang W, Holub E, Kreitman M, Bergelson J. Population Genetics of the Highly Polymorphic RPP8 Gene Family. Genes. 2019; 10(9):691. https://doi.org/10.3390/genes10090691
Chicago/Turabian StyleMacQueen, Alice, Dacheng Tian, Wenhan Chang, Eric Holub, Martin Kreitman, and Joy Bergelson. 2019. "Population Genetics of the Highly Polymorphic RPP8 Gene Family" Genes 10, no. 9: 691. https://doi.org/10.3390/genes10090691
APA StyleMacQueen, A., Tian, D., Chang, W., Holub, E., Kreitman, M., & Bergelson, J. (2019). Population Genetics of the Highly Polymorphic RPP8 Gene Family. Genes, 10(9), 691. https://doi.org/10.3390/genes10090691