Whole Genome Diversity, Population Structure, and Linkage Disequilibrium Analysis of Chickpea (Cicer arietinum L.) Genotypes Using Genome-Wide DArTseq-Based SNP Markers

 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and DNA Extraction
2.2. Genotyping by DArTseq Platform
2.3. Data Analysis
3. Results
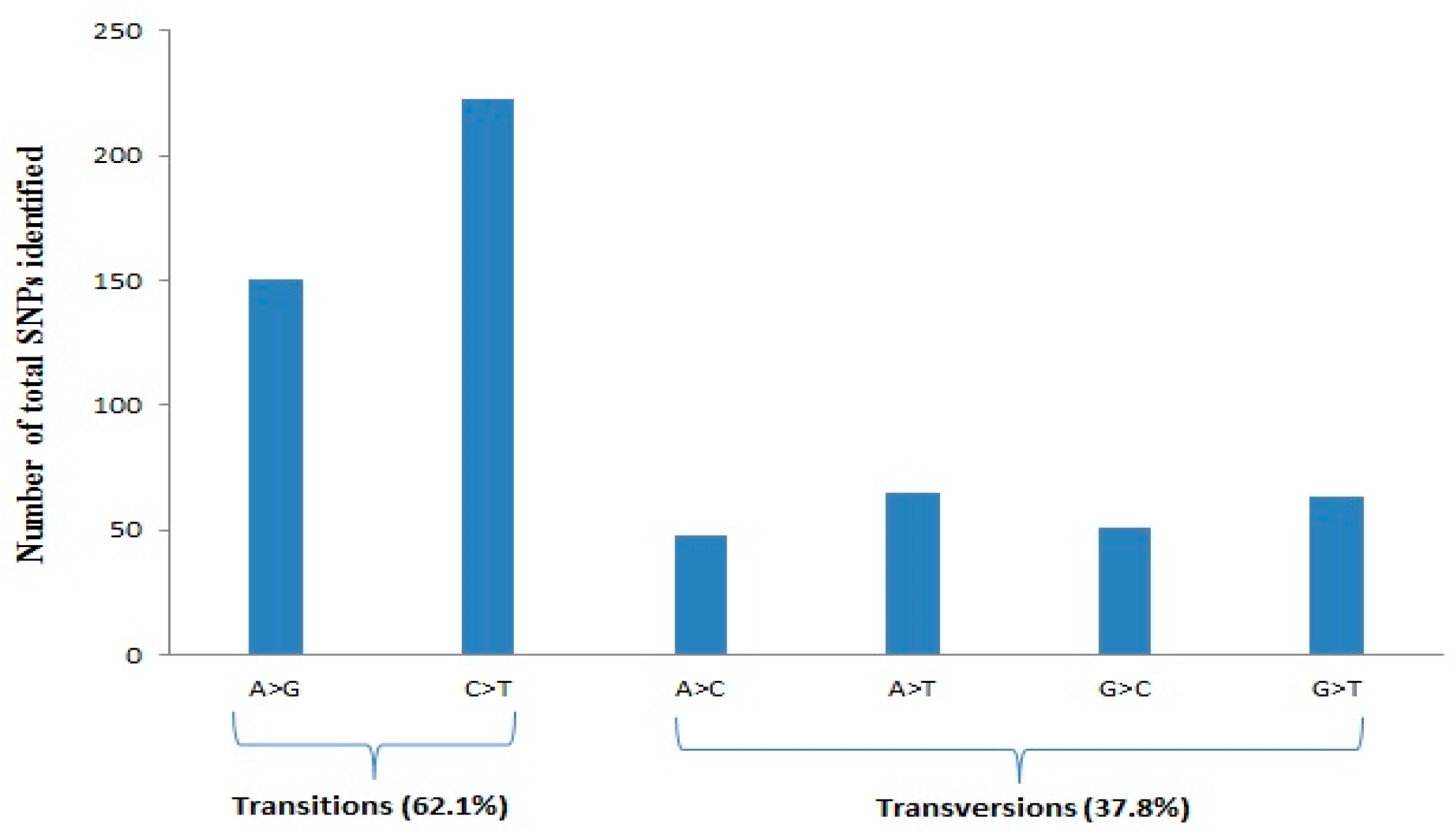
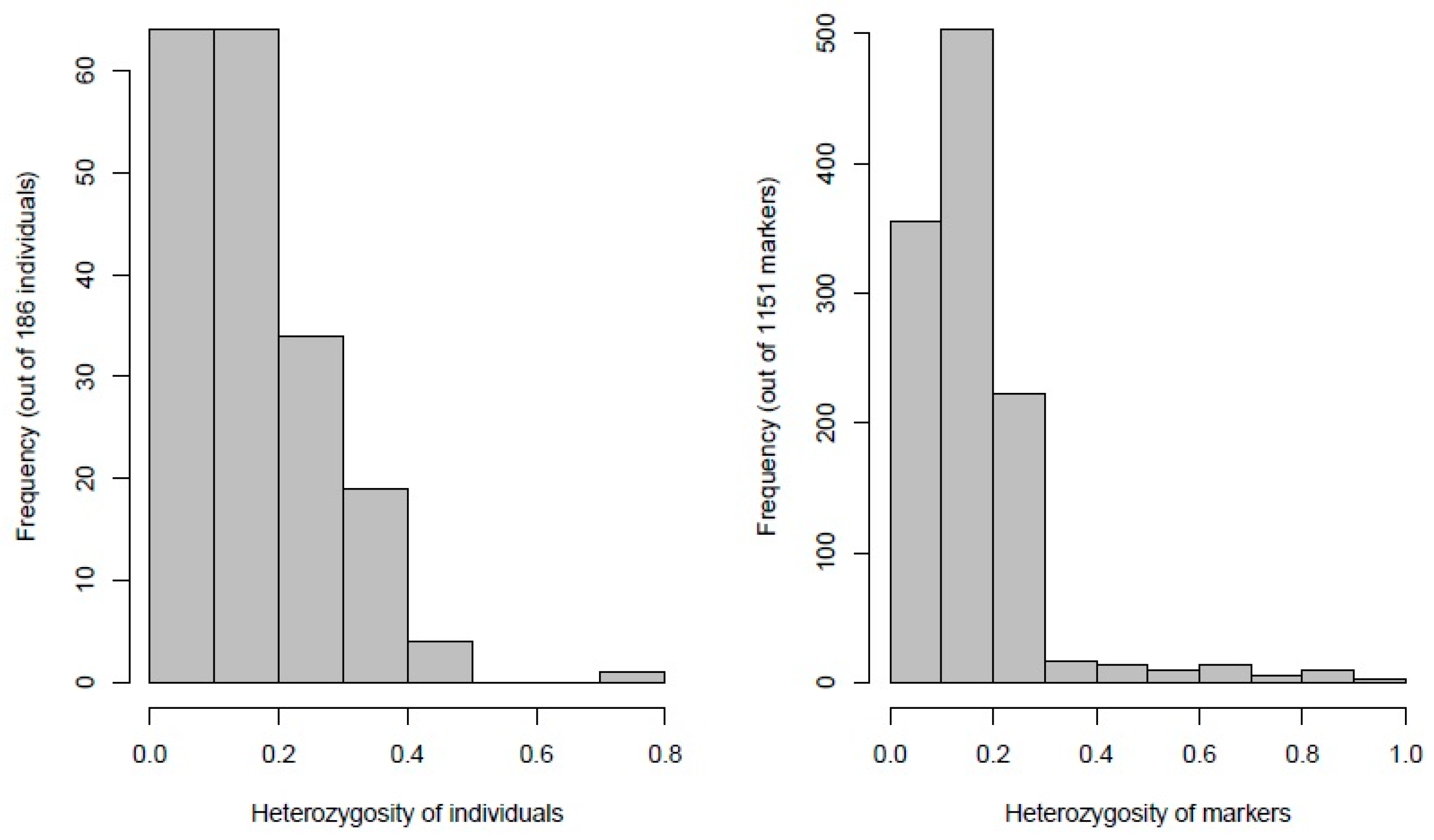
3.1. Quality, Diversity, and Functional Characterization of SNPs
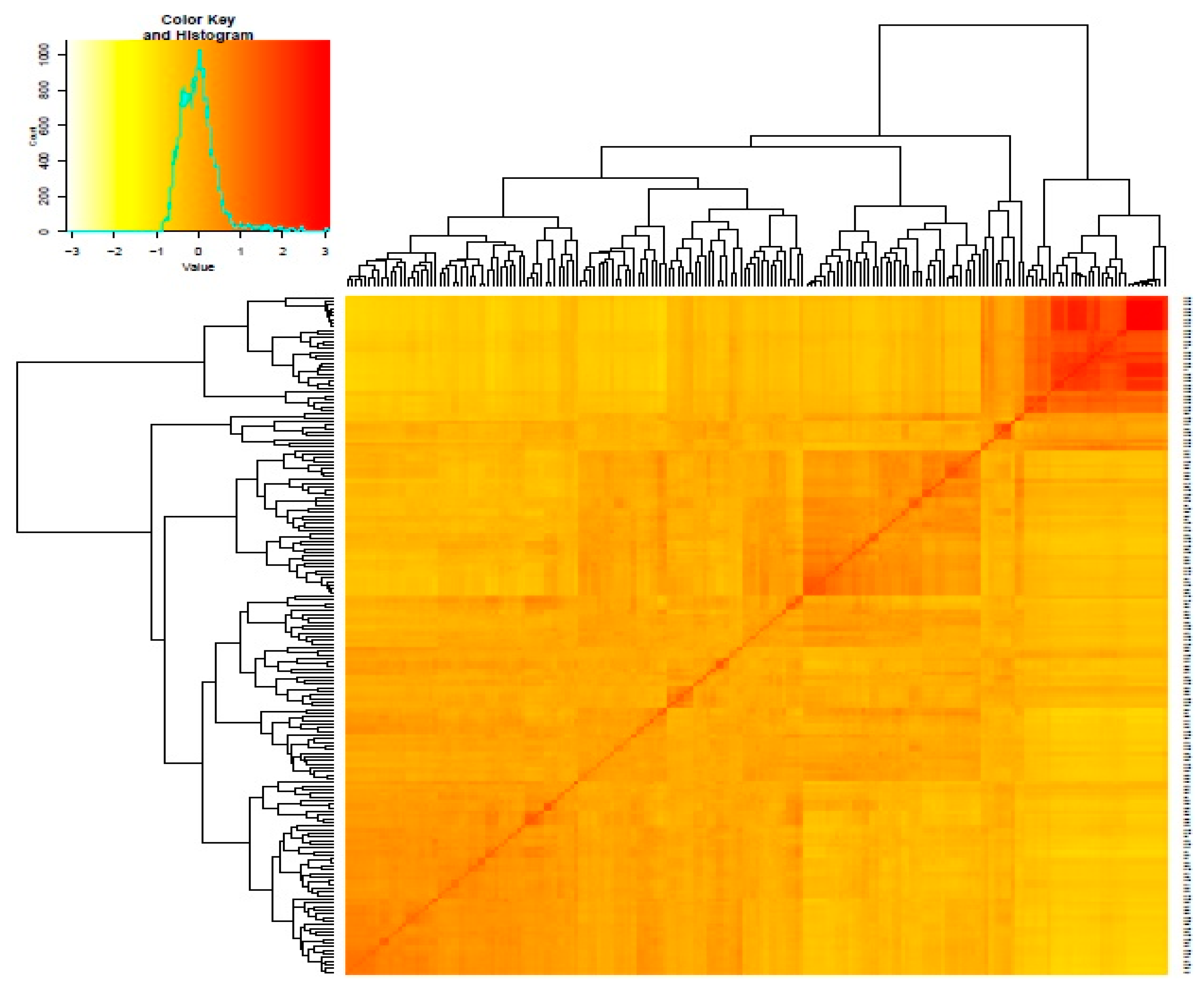
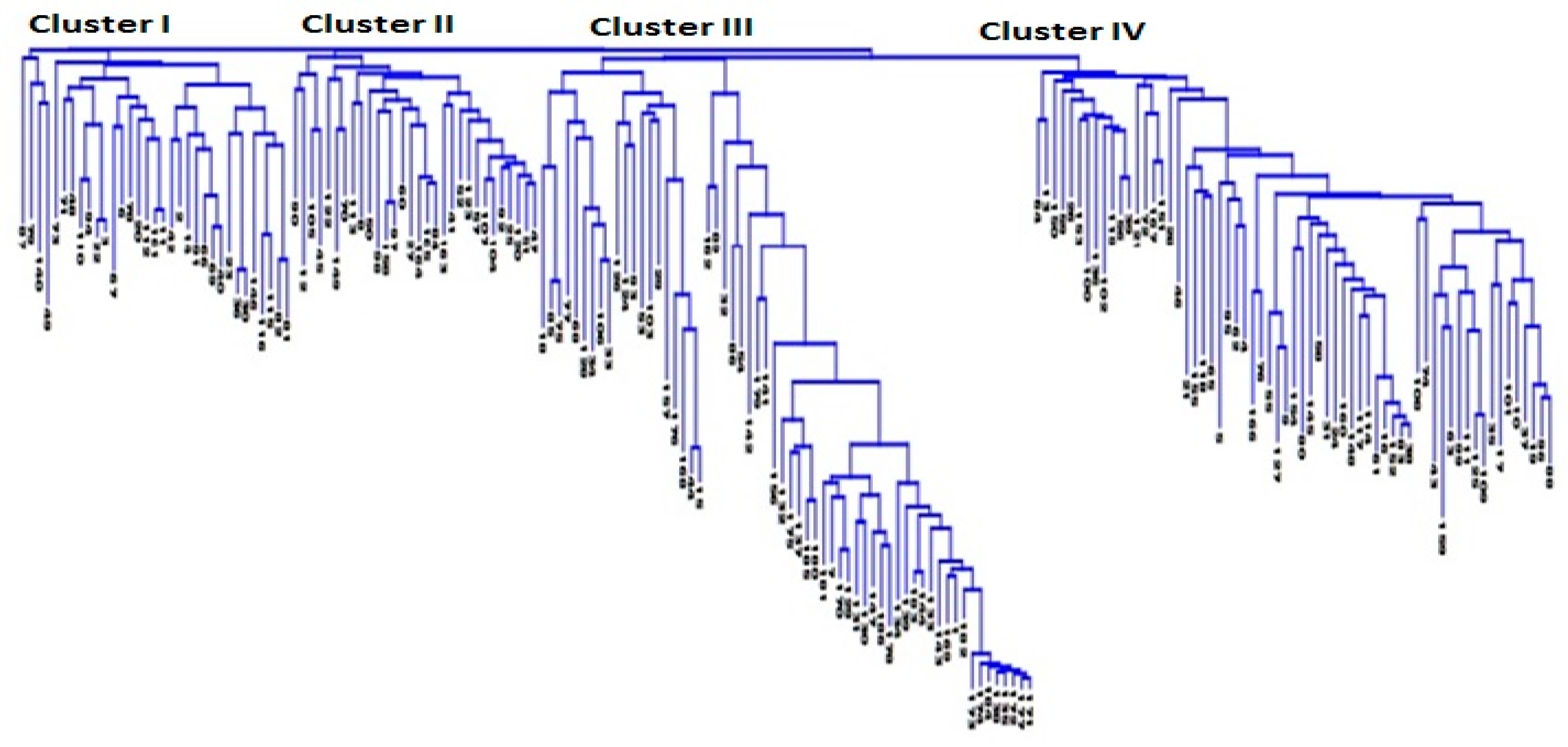
3.2. Genetic Distance and Relatedness Between Chickpea Genotypes
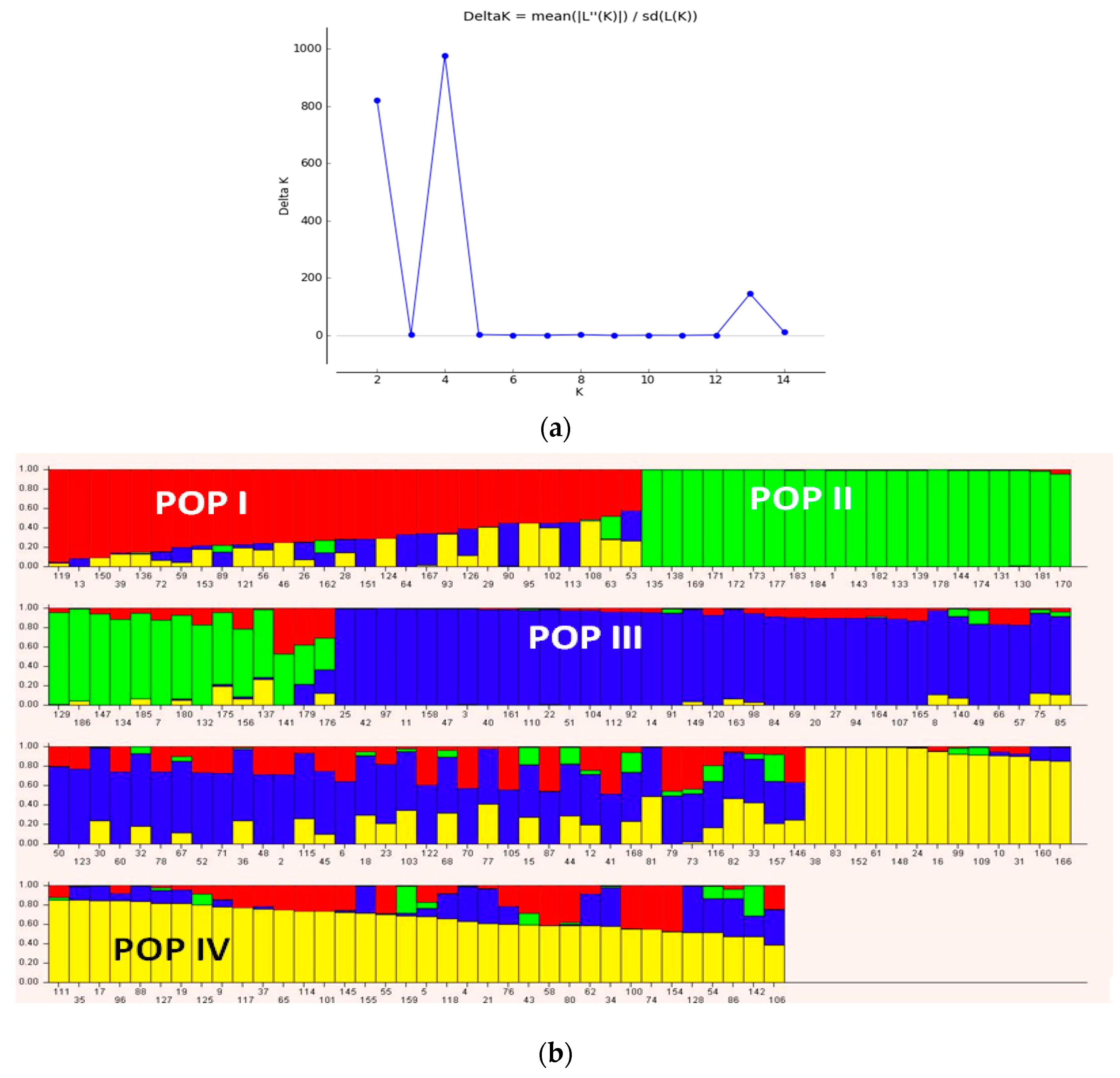
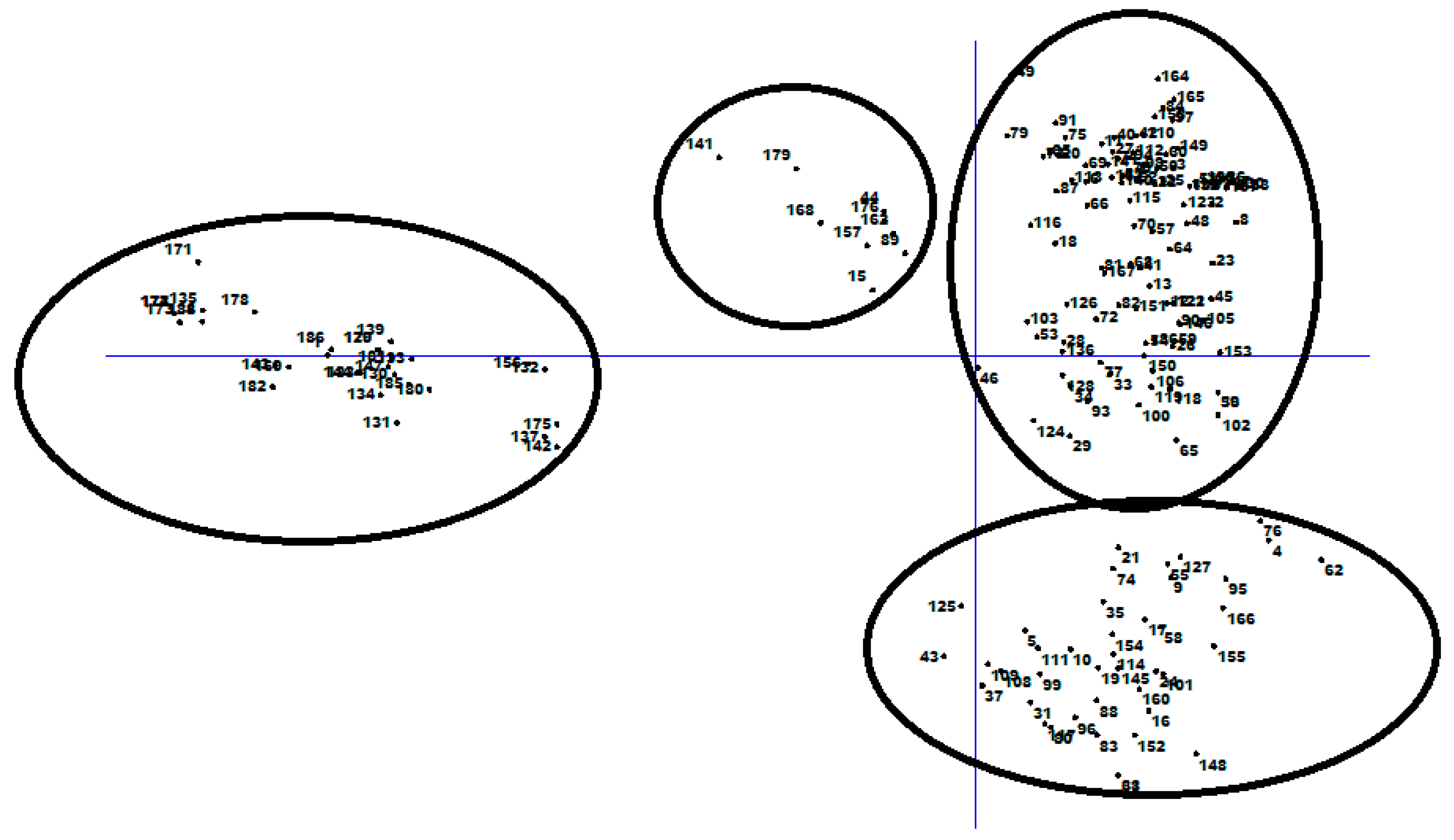
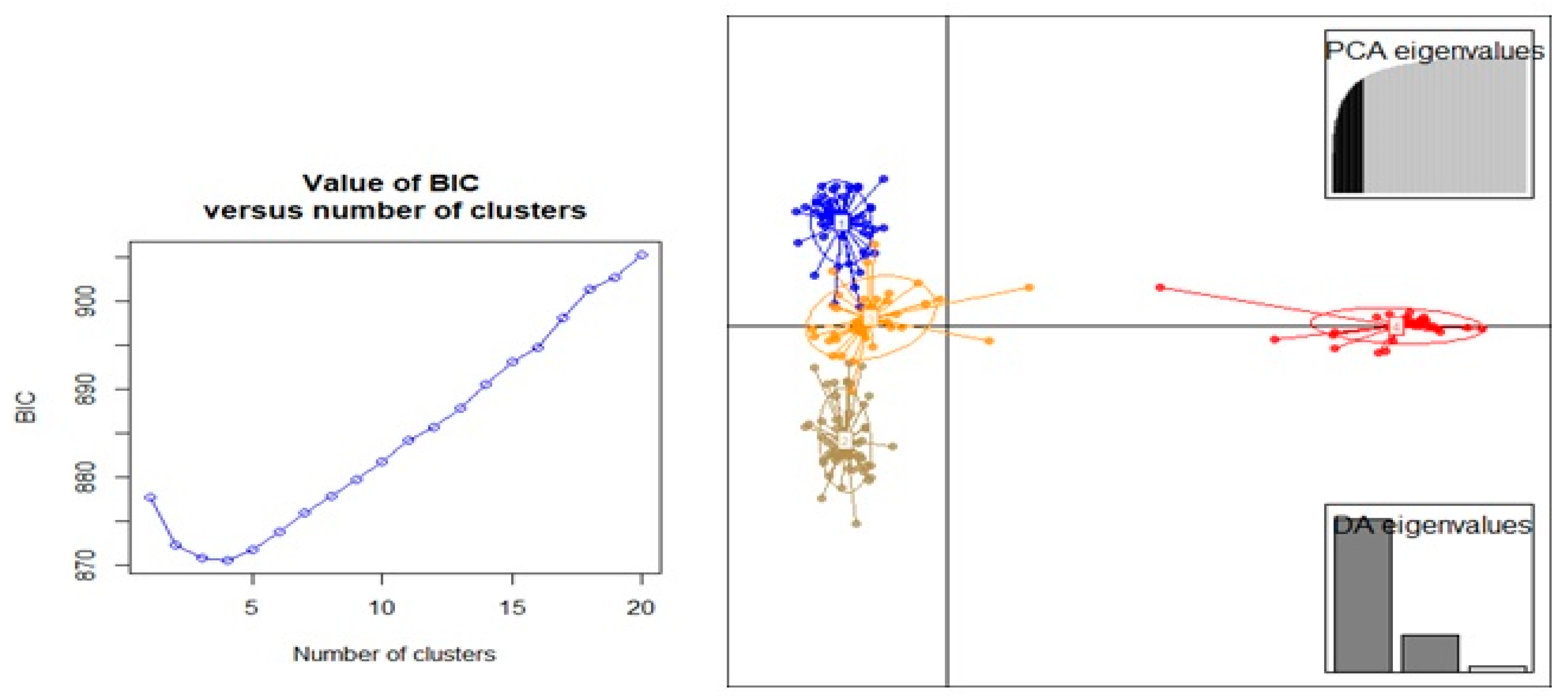
3.3. Population Structure and Discriminate Analysis of Principal Coordinate (DAPC)
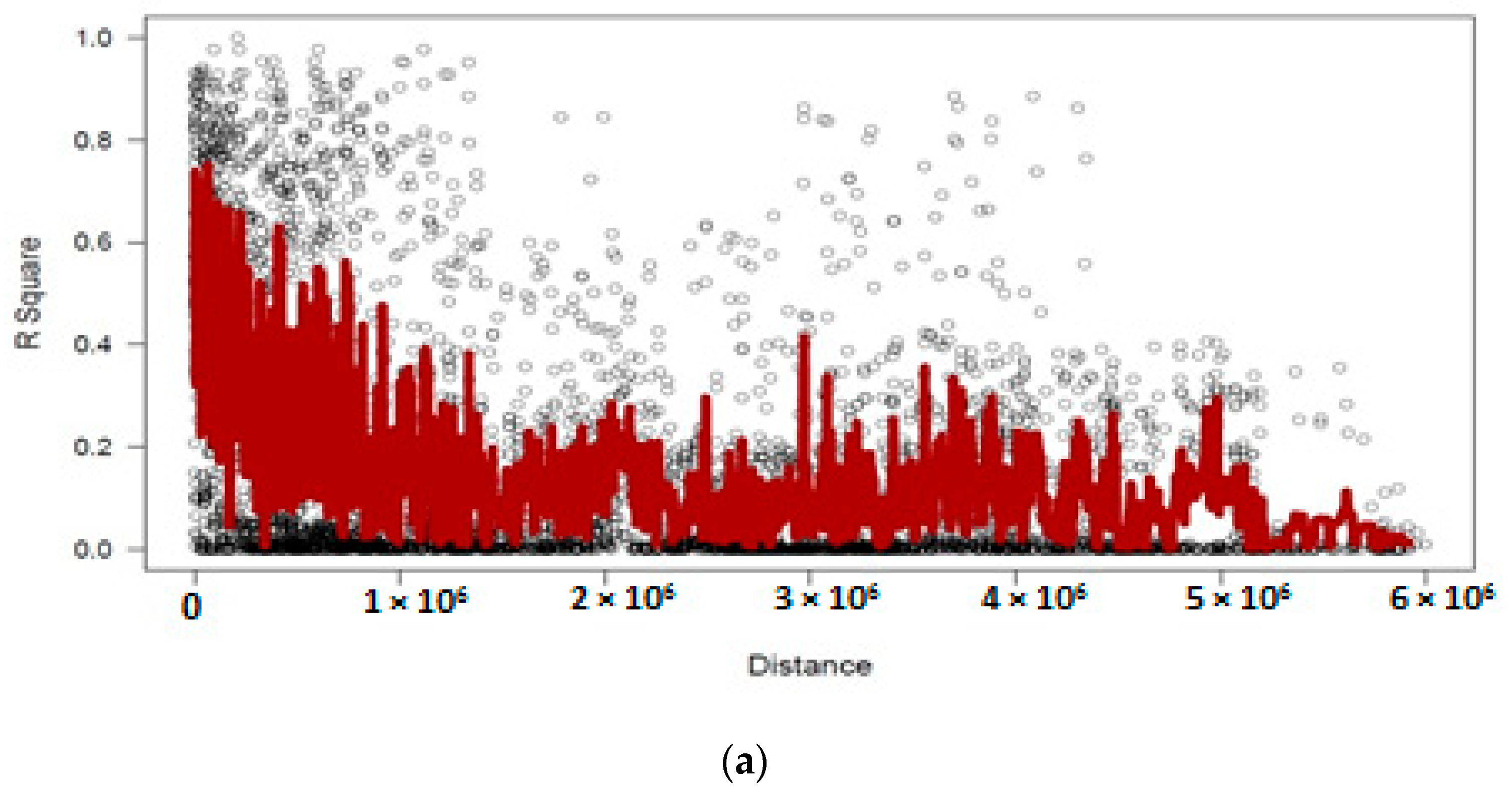
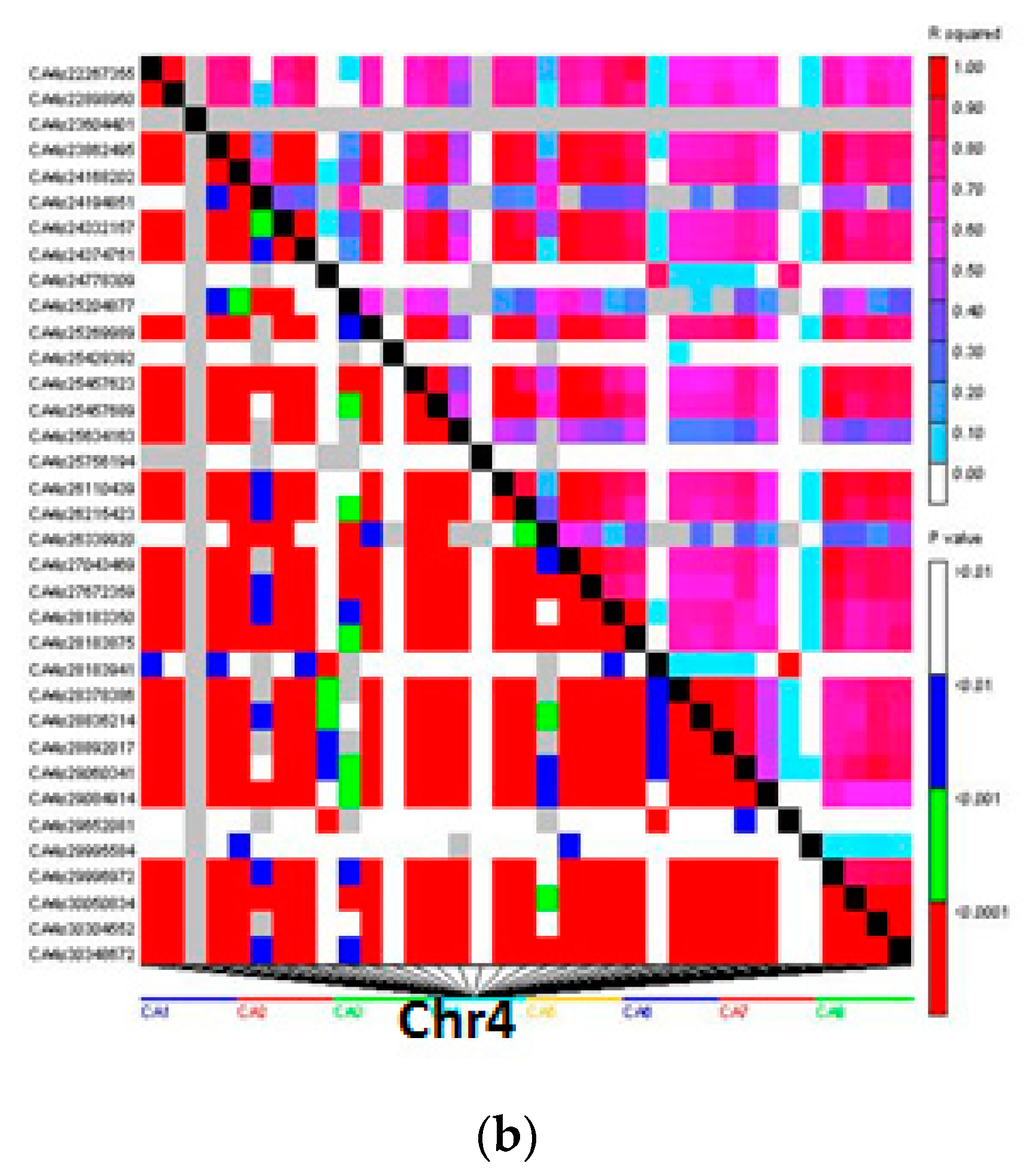
3.4. Linkage Disequilibrium Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Özer, S.; Karaköy, T.; Toklu, F.; Baloch, F.S.; Kilian, B.; Ozkan, H. Nutritional and physicochemical variation in Turkish kabuli chickpea (Cicer arietinum L.) landraces. Euphytica 2010, 175, 237–249. [Google Scholar] [CrossRef]
- Varshney, R.K.; Song, C.; Saxena, R.K.; Azam, S.; Yu, S.; Sharpe, A.G.; Cannon, S.; Baek, J.; Rosen, B.D.; Tar’An, B.; et al. Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat. Biotechnol. 2013, 31, 240–246. [Google Scholar] [CrossRef]
- Singh, R.; Singhal, V.; Randhawa, G.J. Molecular analysis of Chickpea (Cicer arietinum L.) cultivars using AFLP and STMS markers. J. Plant Biochem. Biotechnol. 2008, 17, 167–171. [Google Scholar] [CrossRef]
- Jain, M.; Misra, G.; Patel, R.K.; Priya, P.; Jhanwar, S.; Khan, A.W.; Shah, N.; Singh, V.K.; Garg, R.; Jeena, G.; et al. A draft genome sequence of the pulse crop chickpea (Cicer arietinum L.). Plant J. 2013, 74, 715–729. [Google Scholar] [CrossRef]
- Gupta, S.; Nawaz, K.; Parween, S.; Roy, R.; Sahu, K.; Pole, A.K.; Khandal, H.; Srivastava, R.; Parida, S.K.; Chattopadhyay, D. Draft genome sequence of Cicer reticulatum L., the wild progenitor of chickpea provides a resource for agronomic trait improvement. Curr. Neuropharmacol. 2016, 24, 1–10. [Google Scholar]
- Abbo, S.; Berger, J.; Turner, N.C. Viewpoint: Evolution of cultivated chickpea: Four bottlenecks limit diversity and constrain adaptation. Funct. Plant Biol. 2003, 30, 1081–1087. [Google Scholar] [CrossRef]
- Ahmad, F.; Gaur, P.M.; Croser, J.S. Chickpea (Cicer arietinum L.). In Genetic Resources, Chromosome Engineering, and Crop Improvement: Grain Legumes; Singh, R.J., Jauhar, P.P., Eds.; CRC Press: Boca Raton, FL, USA, 2005; pp. 187–217. [Google Scholar]
- Cui, C.; Mei, H.; Liu, Y.; Zhang, H.; Zheng, Y. Genetic diversity, population structure, and linkage disequilibrium of an association-mapping panel revealed by genome-wide SNP markers in sesame. Front. Plant Sci. 2017, 8, 1189. [Google Scholar] [CrossRef]
- Iruela, M.; Rubio, J.; Cubero, J.I.; Gil, J.; Millan, T. Phylogenetic analysis in the genus Cicer and cultivated chickpea using RAPD and ISSR markers. Theor. Appl. Genet. 2002, 104, 643–651. [Google Scholar] [CrossRef]
- Talebi, R.; Naji, A.; Fayaz, F. Geographical patterns of genetic diversity in cultivated chickpea (Cicer arietinum L.) characterized by amplified fragment length polymorphism. Plant Soil Environ. 2008, 54, 447–452. [Google Scholar] [CrossRef]
- Ahmad, F.; Khan, A.; Awan, F.; Sadia, B.; Sadaqat, H.; Bahadur, S.; Sadaqat, H. Genetic diversity of chickpea (Cicer arietinum L.) germplasm in Pakistan as revealed by RAPD analysis. Genet. Mol. Res. 2010, 9, 1414–1420. [Google Scholar] [CrossRef]
- Amirmoradi, B.; Talebi, R.; Karami, E. Comparative of genetic variation and differentiation among annual Cicer species using start codon targeted (SCoT) polymorphism, DAMD-PCR and ISSR markers. Plant Syst. Evol. 2010, 298, 1679–1688. [Google Scholar] [CrossRef]
- Aggarwal, H.; Kumar, A.R.A.; Singh, J.; Rana, J.S.; Naik, P.K.; Chhokar, V. Evaluation of genetic divergence and phylogenetic relationship using sequence-tagged microsatellite (STMS) sequences in Chickpea (Cicer arietinum L.) genotypes. Afr. J. Biotechnol. 2015, 14, 3051–3061. [Google Scholar]
- Talebi, R.; Fayaz, R.; Mardi, M.; Pirsyedi, S.M.; Naji, A.M. Genetic relationships among chickpea (Cicer arietinum) elite lines based on RAPD and agronomic markers. Int. J. Agric. Biol. 2008, 8, 1560–8530. [Google Scholar]
- Saeed, A.; Darvishzadeh, R. Genetic diversity in a minicore collection of Cicer accessions using amplified fragment length polymorphism (AFLP). Arch. Agron. Soil Sci. 2016, 62, 1711–1721. [Google Scholar] [CrossRef]
- Jannatabadi, A.A.; Talebi, R.; Armin, M.; Jamalabadi, J.; Baghebani, N. Genetic diversity of Iranian landrace chickpea (Cicer arietinum L.) accessions from different geographical origins as revealed by morphological and sequence tagged microsatellite markers. J. Plant. Biochem. Biotech. 2014, 23, 225–229. [Google Scholar] [CrossRef]
- Hajibarat, Z.; Saidi, A.; Hajibarat, Z.; Talebi, R. Characterization of genetic diversity in chickpea using SSR markers, start codon targeted polymorphism (SCoT) and conserved DNA-derived polymorphism (CDDP). Physiol. Mol. Boil. Plants 2015, 21, 365–373. [Google Scholar] [CrossRef]
- Gupta, P.K.; Rustgi, S. Molecular markers from the transcribed/expressed region of the genome in higher plants. Funct. Integr. Genom. 2004, 4, 139–162. [Google Scholar] [CrossRef]
- Kujur, A.; Bajaj, D.; Upadhyaya, H.D.; Das, S.; Ranjan, R.; Shree, T.; Saxena, M.S.; Badoni, S.; Kumar, V.; Tripathi, S.; et al. Employing genome-wide SNP discovery and genotyping strategy to extrapolate the natural allelic diversity and domestication patterns in chickpea. Front. Plant Sci. 2015, 6, 162. [Google Scholar] [CrossRef]
- Bajaj, D.; Das, S.; Badoni, S.; Kumar, V.; Singh, M.; Bansal, K.C.; Tyagi, A.K.; Parida, S.K. Genome-wide high-throughput SNP discovery and genotyping for understanding natural (functional) allelic diversity and domestication patterns in wild chickpea. Sci. Rep. 2015, 5, 12468. [Google Scholar] [CrossRef]
- Basu, U.; Srivastava, R.; Bajaj, D.; Thakro, V.; Daware, A.; Malik, N.; Upadhyaya, H.D.; Parida, S.K. Genome-wide generation and genotyping of informative SNPs to scan molecular signatures for seed yield in chickpea. Sci. Rep. 2018, 8, 13240. [Google Scholar] [CrossRef]
- Baloch, F.S.; Alsaleh, A.; Shahid, M.Q.; Çiftçi, V.; De Miera, L.E.S.; Aasim, M.; Nadeem, M.A.; Aktaş, H.; Özkan, H.; Hatipoğlu, R. A whole genome DArTseq and SNP analysis for genetic diversity assessment in durum wheat from central fertile crescent. PLoS ONE 2017, 12, e0167821. [Google Scholar] [CrossRef]
- Valdisser, P.A.M.R.; Pereira, W.J.; Filho, J.E.A.; Müller, B.S.F.; Coelho, G.R.C.; De Menezes, I.P.P.; Vianna, J.P.G.; Zucchi, M.I.; Lanna, A.C.; Coelho, A.S.G.; et al. In-depth genome characterization of a Brazilian common bean core collection using DArTseq high-density SNP genotyping. BMC Genom. 2017, 18, 423. [Google Scholar] [CrossRef]
- Ndjiondjop, M.N.; Semagn, K.; Gouda, A.C.; Kpeki, S.B.; Tia, D.D.; Sow, M.; Goungoulou, A.; Sie, M.; Perrier, X.; Ghesquiere, A.; et al. Genetic variation and population structure of Oryza glaberrima and development of a mini-core collection using DArTseq. Front. Plant Sci. 2017, 8, 1748. [Google Scholar] [CrossRef]
- Zaitoun, S.Y.A.; Jamous, R.M.; Shtaya, M.J.; Mallah, O.B.; Eid, I.S.; Ali-Shtayeh, M.S. Characterizing Palestinian snake melon (Cucumis melo var.flexuosus) germplasm diversity and structure using SNP and DArTseq markers. BMC Plant. Biol. 2018, 18, 246. [Google Scholar]
- Roorkiwal, M.; Rathore, A.; Das, R.R.; Singh, M.K.; Jain, A.; Srinivasan, S.; Gaur, P.M.; Chellapilla, B.; Tripathi, S.; Li, Y.; et al. Genome-enabled prediction models for yield related traits in chickpea. Front. Plant Sci. 2016, 7, 1666. [Google Scholar] [CrossRef]
- Murray, M.; Thompson, W. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980, 8, 4321–4326. [Google Scholar] [CrossRef]
- Sansaloni, C.; Petroli, C.; Jaccoud, D.; Carling, J.; Detering, F.; Grattapaglia, D.; Kilian, A. diversity arrays technology (DArT) and next-generation sequencing combined: Genome-wide, high throughput, highly informative genotyping for molecular breeding of Eucalyptus. BMC Proc. 2011, 5, 54. [Google Scholar] [CrossRef]
- Kilian, A.; Wenzl, P.; Huttner, E.; Carling, J.; Xia, L.; Blois, H.; Caig, V.; Heller-Uszynska, K.; Jaccoud, D.; Hopper, C.; et al. Diversity arrays technology: A generic genome profiling technology on open platforms. Methods Mol. Biol. 2012, 888, 67–89. [Google Scholar]
- Wenzl, P.; Carling, J.; Kudrna, D.; Jaccoud, D.; Huttner, E.; Kleinhofs, A.; Kilian, A. Diversity arrays technology (DArT) for whole-genome profiling of barley. Proc. Natl. Acad. Sci. USA 2004, 101, 9915–9920. [Google Scholar] [CrossRef]
- Liu, K.; Muse, S.V. PowerMarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Perrier, X.; Flori, A.; Bonnot, F. Data analysis methods. In Genetic Diversity of Cultivated Tropical Plants; Hamon, P., Seguin, M., Perrier, X., Glaszmann, J.C., Eds.; Science Publishers: Enfield, UK, 2003; pp. 43–76. [Google Scholar]
- Perrier, X.; Jacquemoud-Collet, J.P. DARwin Software. Available online: http://darwin.cirad.fr/darwin (accessed on 1 January 2006).
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Earl, D.A.; Vonholdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTU RE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Jombart, T.; Devillard, S.; Balloux, F. Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BMC Genet. 2010, 11, 94. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2018. [Google Scholar]
- Lipka, A.E.; Tian, F.; Wang, Q.; Peiffer, J.; Li, M.; Bradbury, P.J.; Gore, M.A.; Buckler, E.; Zhang, Z. GAPIT: Genome association and prediction integrated tool. Bioinformatics 2012, 28, 2397–2399. [Google Scholar] [CrossRef]
- Ghaffari, P.; Talebi, R.; Keshavarzi, F. Genetic diversity and geographical differentiation of Iranian landrace, cultivars, and exotic chickpea lines as revealed by morphological and microsatellite markers. Physiol. Mol. Boil. Plants 2014, 20, 225–233. [Google Scholar] [CrossRef][Green Version]
- Vavilov, N. Studies on the origin of cultivated plants. Inst. Appl. Bot. Plant. Breed. 1926. Leningrad. [Google Scholar]
- Nguyen, T.T.; Taylor, P.W.J.; Redden, R.J.; Ford, R. Genetic diversity estimates in Cicer using AFLP analysis. Plant Breed. 2004, 123, 173–179. [Google Scholar] [CrossRef]
- Nielsen, N.H.; Backes, G.; Stougaard, J.; Andersen, S.U.; Jahoor, A. Genetic diversity and population structure analysis of European hexaploid bread wheat (Triticum aestivum L.) varieties. PLoS ONE 2014, 9, e94000. [Google Scholar] [CrossRef]
- Ren, R.; Ray, R.; Li, P.; Xu, J.; Zhang, M.; Liu, G.; Yao, X.; Kilian, A.; Yang, X. Construction of a high-density DArTseq SNP-based genetic map and identification of genomic regions with segregation distortion in a genetic population derived from a cross between feral and cultivated-type watermelon. Mol. Genet. Genom. 2015, 290, 1457–1470. [Google Scholar] [CrossRef]
- Li, C.; Bai, G.; Chao, S.; Wang, Z. A high-density SNP and SSR consensus map reveals segregation distortion regions in wheat. BioMed Res. Int. 2015, 2015, 830618. [Google Scholar] [CrossRef]
- Alam, M.; Neal, J.; O’Connor, K.; Kilian, A.; Topp, B. Ultra-high-throughput DArTseq-based silicoDArT and SNP markers for genomic studies in macadamia. PLoS ONE 2018, 13, e0203465. [Google Scholar] [CrossRef]
- Thudi, M.; Upadhyaya, H.D.; Rathore, A.; Gaur, P.M.; Krishnamurthy, L.; Roorkiwal, M.; Nayak, S.N.; Chaturvedi, S.K.; Basu, P.S.; GangaRao, N.V.P.R.; et al. Genetic dissection of drought and heat tolerance in Chickpea through genome-wide and candidate gene-based association mapping approaches. PLoS ONE 2014, 9, e96758. [Google Scholar] [CrossRef]
- Monostori, I.; Szira, F.; Tondelli, A.; Árendás, T.; Gierczik, K.; Cattivelli, L.; Galiba, G.; Vágújfalvi, A. Genome-wide association study and genetic diversity analysis on nitrogen use efficiency in a Central European winter wheat (Triticum aestivum L.) collection. PLoS ONE 2017, 12, e0189265. [Google Scholar] [CrossRef]
- Dodds, K.G.; McEwan, J.C.; Brauning, R.; Anderson, R.M.; Van Stijn, T.C.; Kristjánsson, T.; Clarke, S.M. Construction of relatedness matrices using genotyping-by-sequencing data. BMC Genom. 2015, 16, 1047. [Google Scholar] [CrossRef]
- Upadhyaya, H.D.; Dwivedi, S.L.; Baum, M.; Varshney, R.K.; Udupa, S.M.; Gowda, C.L.; Hoisington, D.; Singh, S. Genetic structure, diversity, and allelic richness in composite collection and reference set in chickpea (Cicer arietinum L.). BMC Plant Boil. 2008, 8, 106. [Google Scholar] [CrossRef]
- Saxena, M.S.; Bajaj, D.; Kujur, A.; Das, S.; Badoni, S.; Kumar, V.; Singh, M.; Bansal, K.C.; Tyagi, A.K.; Parida, S.K. Natural allelic diversity, genetic structure and linkage disequilibrium pattern in wild chickpea. PLoS ONE 2014, 9, e107484. [Google Scholar] [CrossRef]
- De Giovanni, C.; Pavan, S.; Taranto, F.; Di Rienzo, V.; Miazzi, M.M.; Marcotrigiano, A.R.; Mangini, G.; Montemurro, C.; Ricciardi, L.; Lotti, C. Genetic variation of a global germplasm collection of chickpea (Cicer arietinum L.) including Italian accessions at risk of genetic erosion. Physiol. Mol. Biol. Plants 2017, 23, 197–205. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Linkage Group (Chromosome) | Number of SNPs | Chromosome Size (kbp) | Mean of SNPs per Mbp | PIC Range (Mean) | Call Rate | Average Reproducibility |
|---|---|---|---|---|---|---|
| LG1 | 192 | 44,634.56 | 4.20 | 0.05–0.49 (0.23) | 0.96 | 0.98 |
| LG2 | 89 | 36,915.99 | 2.41 | 0.05–0.49 (0.32) | 0.96 | 0.98 |
| LG3 | 105 | 61,351.17 | 1.71 | 0.05–0.49 (0.30) | 0.97 | 0.98 |
| LG4 | 378 | 57,562.47 | 6.56 | 0.05–0.50 (0.36) | 0.96 | 0.97 |
| LG5 | 74 | 67,923.99 | 1.08 | 0.09–0.48 (0.31) | 0.97 | 0.98 |
| LG6 | 141 | 63,087.8 | 2.34 | 0.05–0.50 (0.34) | 0.97 | 0.97 |
| LG7 | 121 | 54,252.93 | 2.23 | 0.05–0.50 (0.31) | 0.97 | 0.98 |
| LG8 | 52 | 16,236.36 | 3.20 | 0.06–0.49 (0.34) | 0.97 | 0.97 |
| Total | 1152 | 67,923.99 | 16.96 | 0.05–0.50 (0.32) | 0.97 | 0.98 |
| Population | Net Nucleotide Distance | Expected Heterozygosity | % of Membership | Mean Fixation Index (Fst) | ||
|---|---|---|---|---|---|---|
| POP2 | POP3 | POP4 | ||||
| POP1 | 0.44 | 0.25 | 0.26 | 0.28 | 0.20 | 0.56 |
| POP2 | 0.33 | 0.30 | 0.22 | 0.19 | 0.62 | |
| POP3 | 0.16 | 0.17 | 0.36 | 0.65 | ||
| POP4 | 0.19 | 0.26 | 0.63 | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farahani, S.; Maleki, M.; Mehrabi, R.; Kanouni, H.; Scheben, A.; Batley, J.; Talebi, R. Whole Genome Diversity, Population Structure, and Linkage Disequilibrium Analysis of Chickpea (Cicer arietinum L.) Genotypes Using Genome-Wide DArTseq-Based SNP Markers. Genes 2019, 10, 676. https://doi.org/10.3390/genes10090676
Farahani S, Maleki M, Mehrabi R, Kanouni H, Scheben A, Batley J, Talebi R. Whole Genome Diversity, Population Structure, and Linkage Disequilibrium Analysis of Chickpea (Cicer arietinum L.) Genotypes Using Genome-Wide DArTseq-Based SNP Markers. Genes. 2019; 10(9):676. https://doi.org/10.3390/genes10090676
Chicago/Turabian StyleFarahani, Somayeh, Mojdeh Maleki, Rahim Mehrabi, Homayoun Kanouni, Armin Scheben, Jacqueline Batley, and Reza Talebi. 2019. "Whole Genome Diversity, Population Structure, and Linkage Disequilibrium Analysis of Chickpea (Cicer arietinum L.) Genotypes Using Genome-Wide DArTseq-Based SNP Markers" Genes 10, no. 9: 676. https://doi.org/10.3390/genes10090676
APA StyleFarahani, S., Maleki, M., Mehrabi, R., Kanouni, H., Scheben, A., Batley, J., & Talebi, R. (2019). Whole Genome Diversity, Population Structure, and Linkage Disequilibrium Analysis of Chickpea (Cicer arietinum L.) Genotypes Using Genome-Wide DArTseq-Based SNP Markers. Genes, 10(9), 676. https://doi.org/10.3390/genes10090676