Genomic Prediction and Genome-Wide Association Studies of Flour Yield and Alveograph Quality Traits Using Advanced Winter Wheat Breeding Material

 , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Phenotyping
2.3. Genotyping
2.4. Statistical Analysis
3. Results
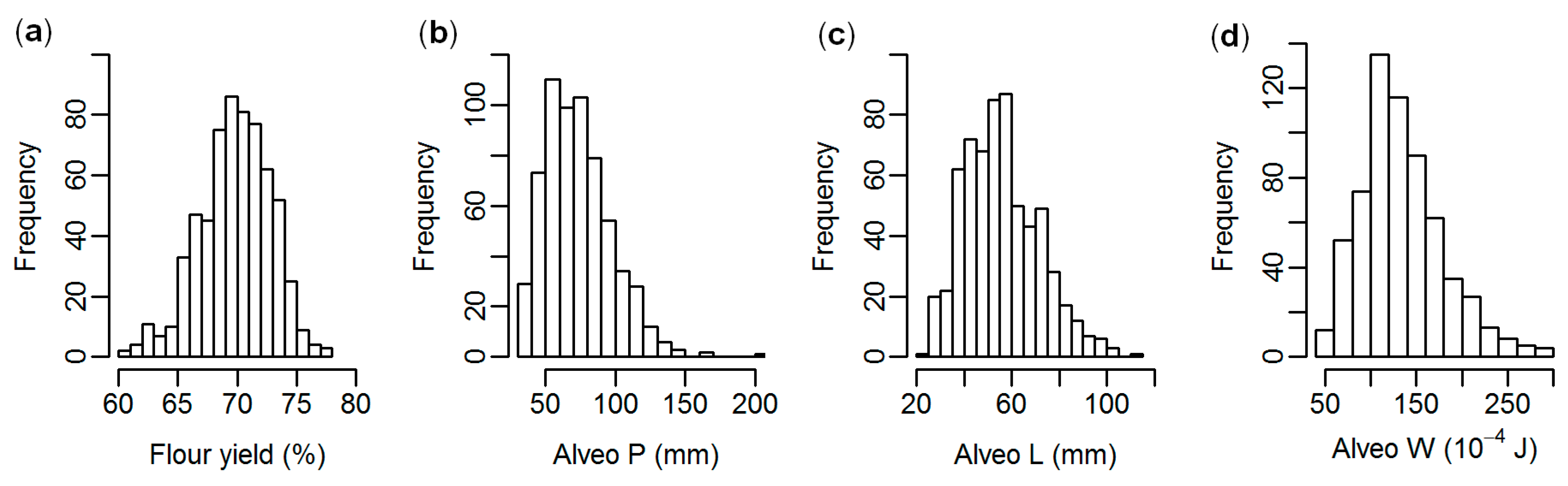
3.1. Phenotyping and Genotyping
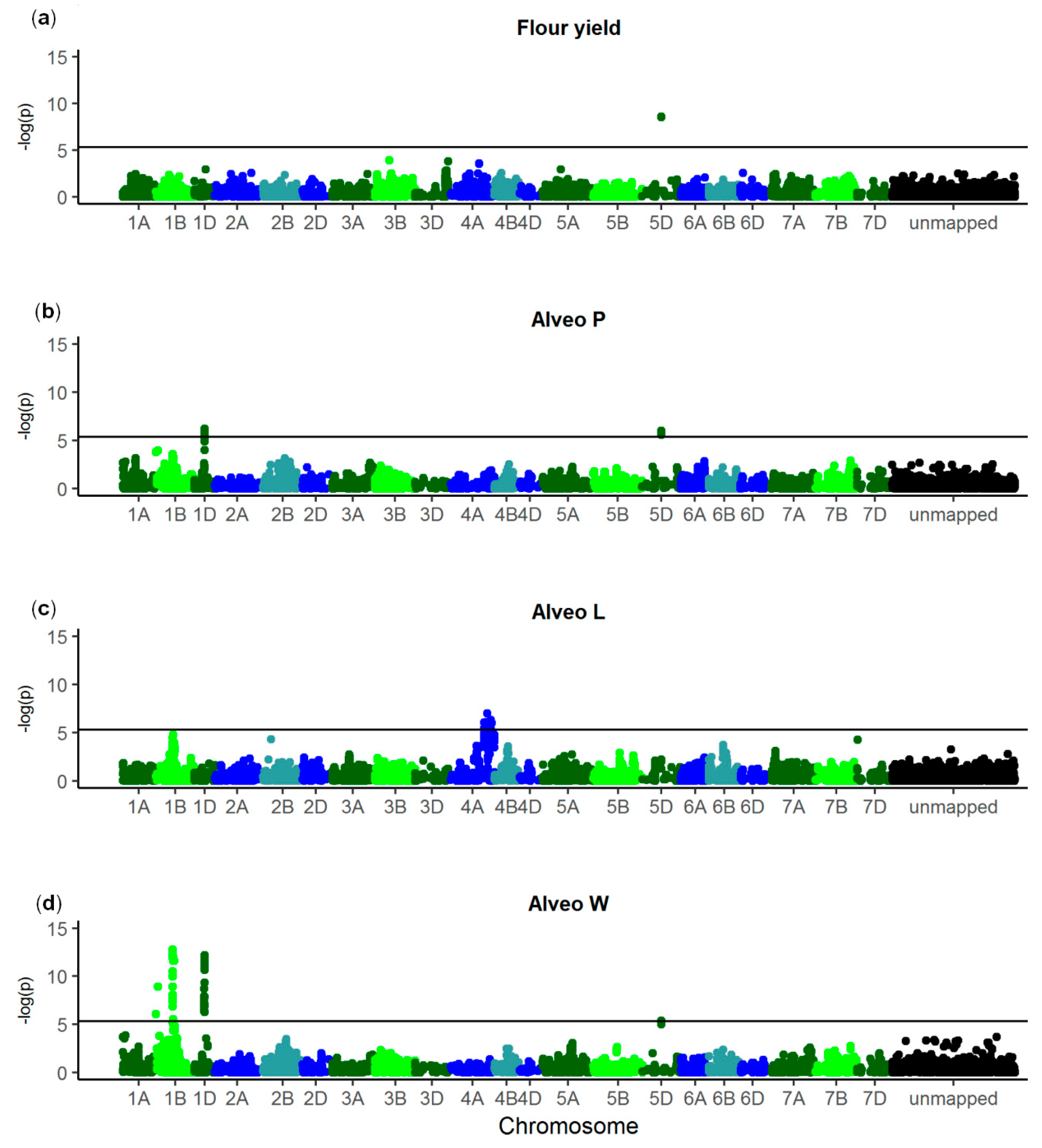
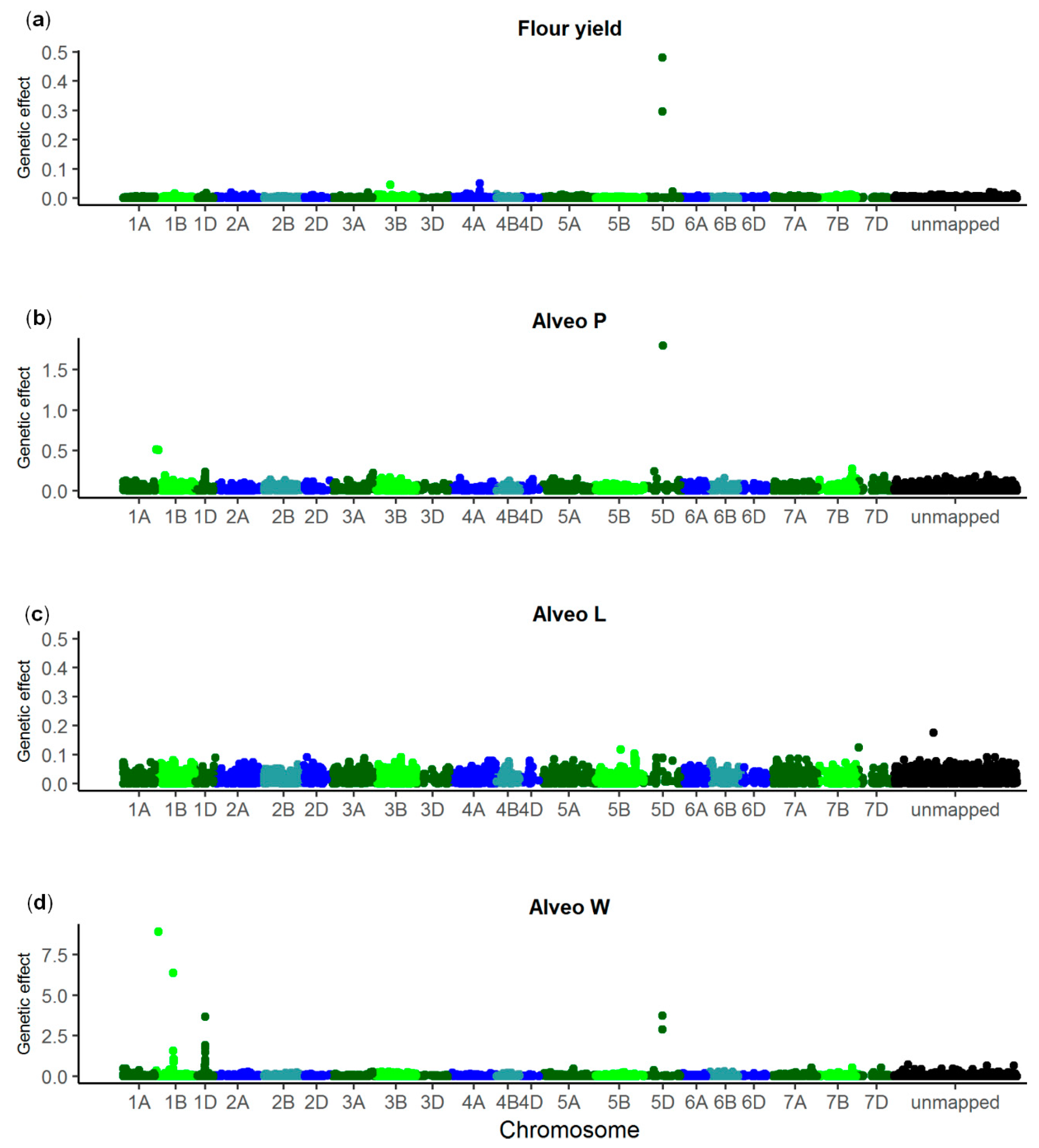
3.2. GWAS
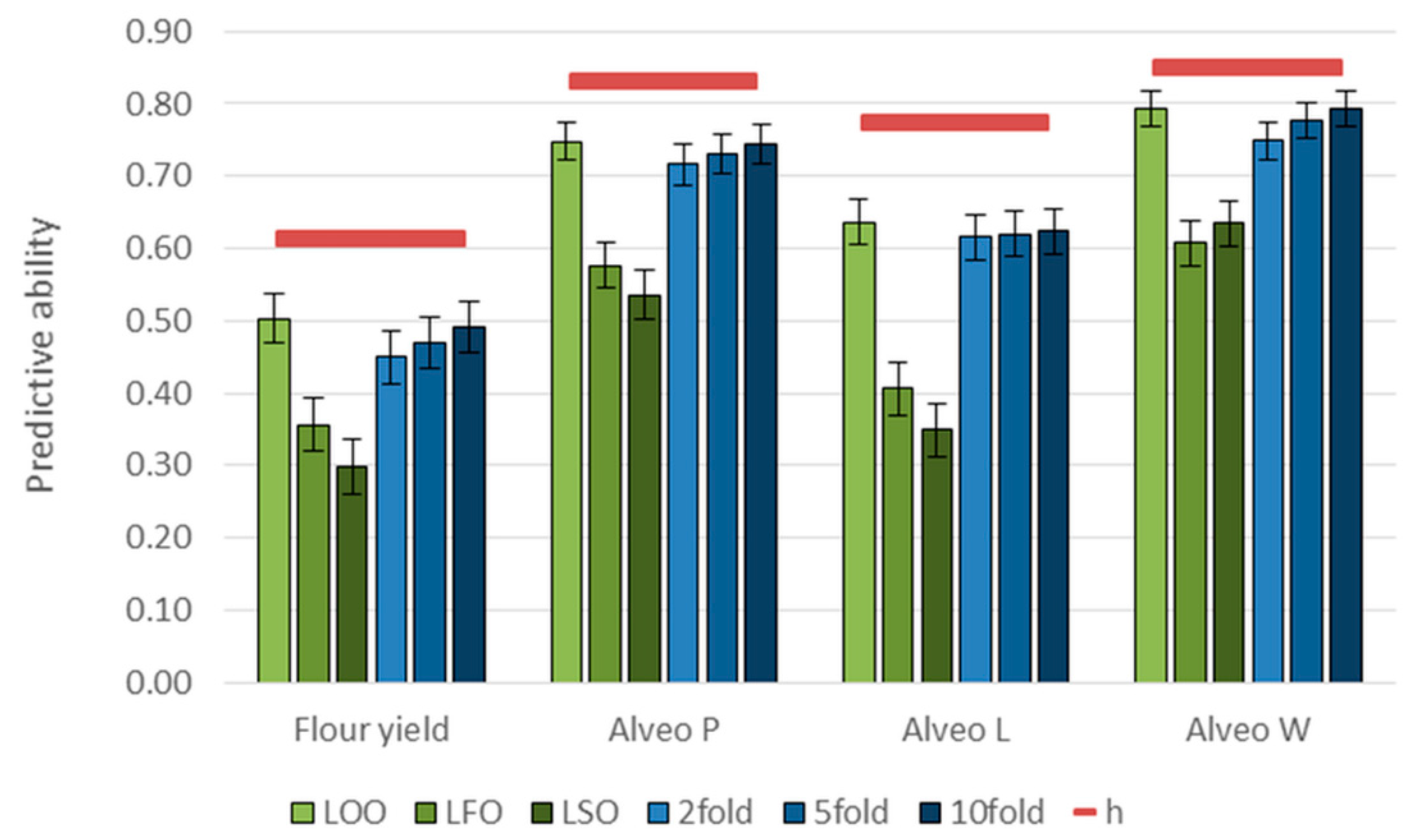
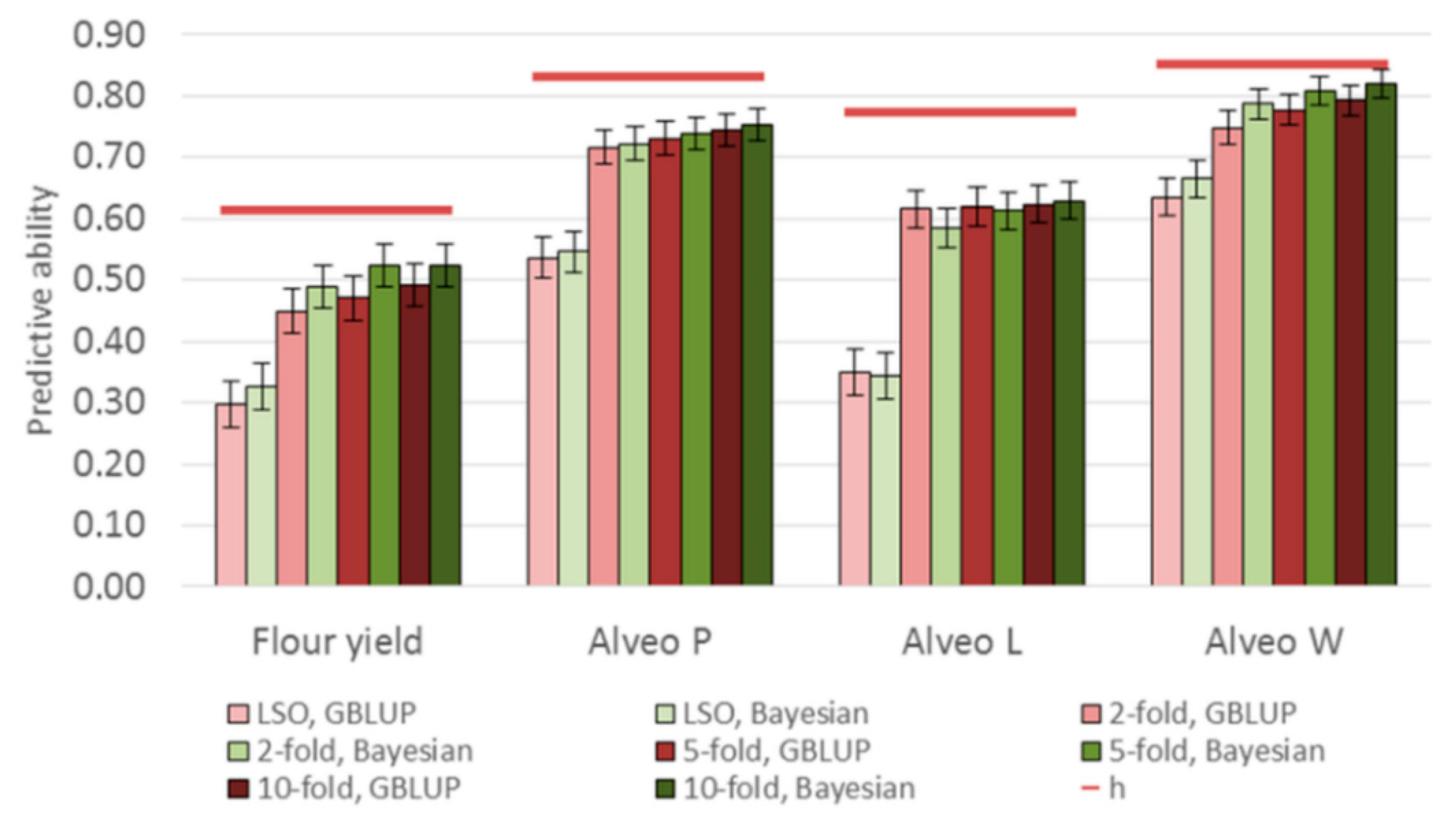
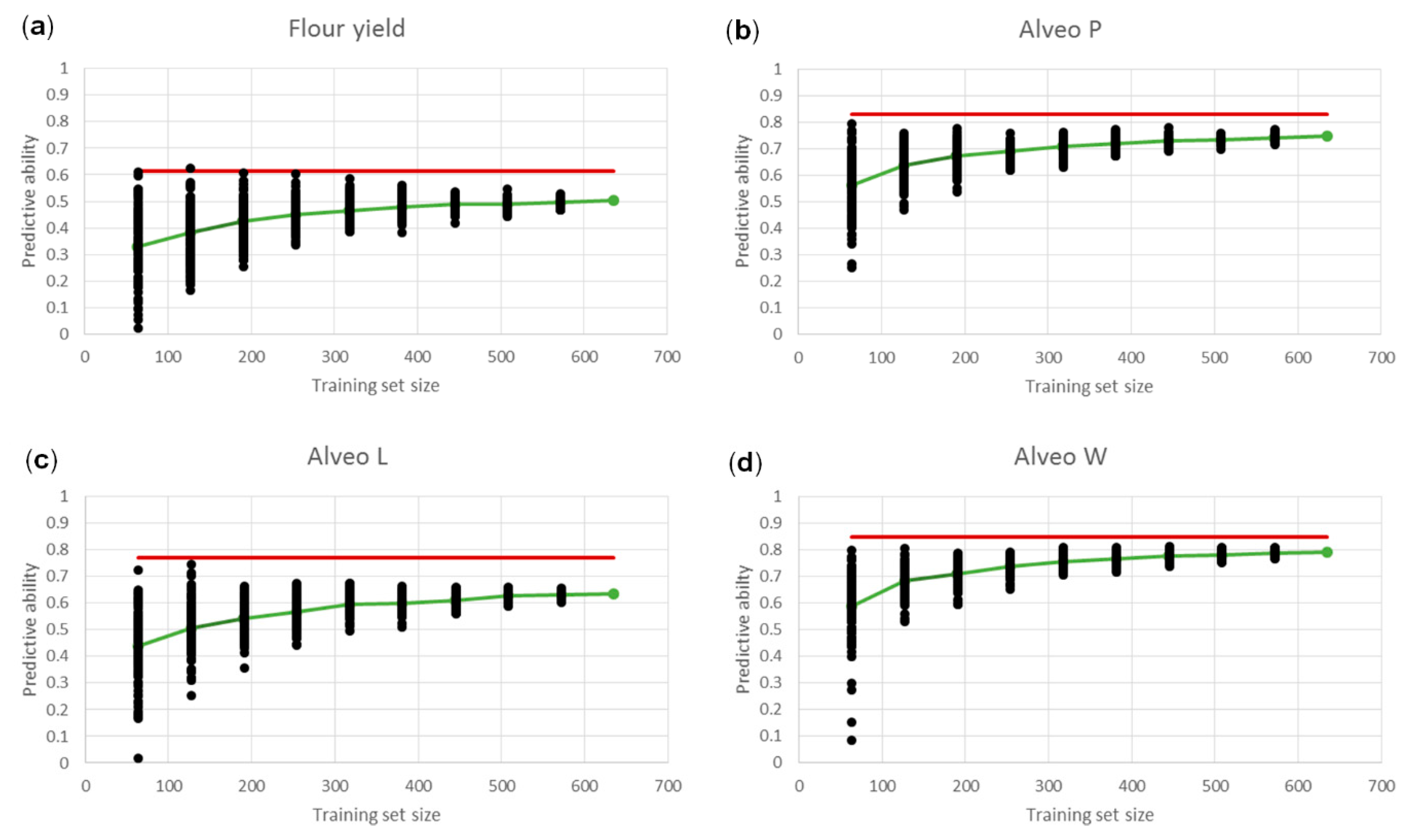
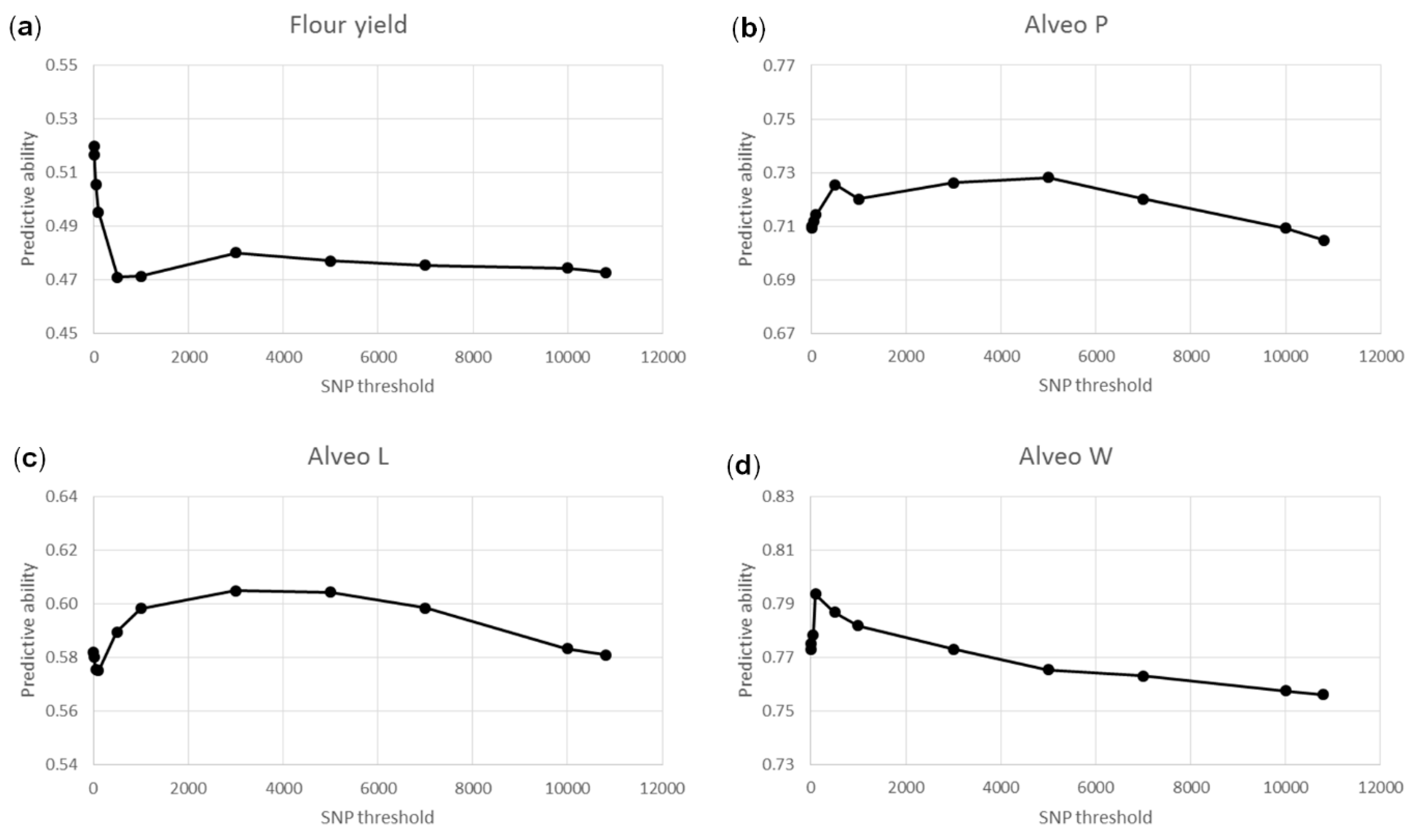
3.3. Genomic Predictions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bordes, J.; Ravel, C.; Le Gouis, J.; Lapierre, A.; Charmet, G.; Balfourier, F. Use of a global wheat core collection for association analysis of flour and dough quality traits. J. Cereal Sci. 2011, 54, 137–147. [Google Scholar] [CrossRef]
- Smith, N.; Guttieri, M.; Souza, E.; Shoots, J.; Sorrells, M.; Sneller, C. Identification and validation of QTL for grain quality traits in a cross of soft wheat cultivars pioneer brand 25R26 and Foster. Crop. Sci. 2011, 51, 1424–1436. [Google Scholar] [CrossRef]
- He, Z.H.; Liu, L.; Xia, X.C.; Liu, J.J.; Pena, R.J. Composition of HMW and LMW Glutenin Subunits and Their Effects on Dough Properties, Pan Bread, and Noodle Quality of Chinese Bread Wheats. Cereal Chem. 2005, 82, 345–350. [Google Scholar] [CrossRef]
- Payne, P.I.; Nightingale, M.A.; Krattiger, A.F.; Holt, L.M. The relationship between HMW glutenin subunit composition and the bread making quality of British grown wheat varieties. J. Sci. Food Agric. 1987, 40, 51–65. [Google Scholar] [CrossRef]
- Bhave, M.; Morris, C.F. Molecular genetics of puroindolines and related genes: Allelic diversity in wheat and other grasses. Plant. Mol. Biol. 2008, 66, 205–219. [Google Scholar] [CrossRef] [PubMed]
- Giroux, M.J.; Morris, C.F. A glycine to serine change in puroindoline b is associated with wheat grain hardness and low levels of starch-surface friabilin. Theor. Appl. Genet. 1997, 95, 857–864. [Google Scholar] [CrossRef]
- Graybosch, R.A.; Peterson, C.J.; Hareland, G.A.; Shelton, D.R.; Olewnik, M.C.; He, H.; Stearns, M.M. Relationships between small-scale wheat quality assays and commercial test bakes. Cereal Chem. 1999, 76, 428–433. [Google Scholar] [CrossRef]
- Guzmán, C.; Posadas-romano, G.; Hernández-Espinosa, N.; Morales-Dorantes, A.; Peña, R.J. A new standard water absorption criteria based on solvent retention capacity (SRC) to determine dough mixing properties, viscoelasticity, and bread-making quality. J. Cereal Sci. J. 2015, 66, 59–65. [Google Scholar] [CrossRef]
- Guzman, C.; Peña, R.J.; Singh, R.; Autrique, E.; Dreisigacker, S.; Crossa, J.; Rutkoski, J.; Poland, J.; Battenfield, S. Wheat quality improvement at CIMMYT and the use of genomic selection on it. Appl. Transl. Genomics. 2016, 11, 3–8. [Google Scholar] [CrossRef]
- Groos, C.; Bervas, E.; Charmet, G. Genetic analysis of grain protein content, grain hardness and dough rheology in a hard X hard bread wheat progeny. J. Cereal Sci. 2004, 40, 93–100. [Google Scholar] [CrossRef]
- Kerfal, S.; Giraldo, P.; Rodríguez-Quijano, M.; Vázquez, J.F.; Adams, K.; Lukow, O.M.; Röder, M.S.; Somers, D.J.; Carrillo, J.M. Mapping quantitative trait loci (QTLs) associated with dough quality in a soft×hard bread wheat progeny. J. Cereal Sci. 2010, 52, 46–52. [Google Scholar] [CrossRef]
- Nelson, J.C.; Andreescu, C.; Breseghello, F.; Finney, P.L.; Gualberto, D.G.; Bergman, C.J.; Peña, R.J.; Perretant, M.R.; Leroy, P.; Qualset, C.O.; et al. Quantitative trait locus analysis of wheat quality traits. Euphytica 2006, 149, 145–159. [Google Scholar] [CrossRef]
- Zanetti, S.; Winzeler, M.; Feuilet, C.; Keller, B.; Messmer, M. Genetic analysis of bread-making quality in wheat and spelt. Plant. Breed. 2001, 120, 13–19. [Google Scholar] [CrossRef]
- Tadesse, W.; Ogbonnaya, F.C.; Jighly, A.; Sohail, Q.; Rajaram, S. Genome-Wide Association Mapping of Yield and Grain Quality Traits in Winter Wheat Genotypes. PLoS ONE 2015, 10, e0141339. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, A.; Guttieri, M.; Smith, N.; Souza, E.; Sturbaum, A.; Hua, D.; Griffey, C.; Barnett, M.; Murphy, P.; Ohm, H.; et al. Identification of milling and baking quality QTL in multiple soft wheat mapping populations. Theor. Appl. Genet. 2015, 128, 2227–2242. [Google Scholar] [CrossRef] [PubMed]
- Fox, G.P.; Martin, A.; Kelly, A.M.; Sutherland, M.W.; Martin, D.; Banks, P.M.; Sheppard, J. QTLs for water absorption and flour yield identified in the doubled haploid wheat population Lang/QT8766. Euphytica 2013, 192, 453–462. [Google Scholar] [CrossRef]
- Ishikawa, G.; Nakamura, K.; Ito, H.; Saito, M.; Sato, M.; Jinno, H.; Yoshimura, Y.; Nishimura, T.; Maejima, H.; Uehara, Y.; et al. Association mapping and validation of QTLs for flour yield in the soft winter wheat variety Kitahonami. PLoS ONE 2014, 9, e111337. [Google Scholar] [CrossRef] [PubMed]
- McCartney, C.A.; Somers, D.J.; Lukow, O.; Ames, N.; Noll, J.; Cloutier, S.; Humphreys, D.G.; McCallum, B.D. QTL analysis of quality traits in the spring wheat cross RL4452 x “AC domain”. Plant. Breed. 2006, 125, 565–575. [Google Scholar] [CrossRef]
- Sherman, J.D.; Nash, D.; Lanning, S.P.; Martin, J.M.; Blake, N.K.; Morris, C.F.; Talbert, L.E. Genetics of end-use quality differences between a modern and historical spring wheat. Crop. Sci. 2014, 54, 1972–1980. [Google Scholar] [CrossRef]
- Tsilo, T.J.; Hareland, G.A.; Chao, S.; Anderson, J.A. Genetic mapping and QTL analysis of flour color and milling yield related traits using recombinant inbred lines in hard red spring wheat. Crop. Sci. 2011, 51, 237–246. [Google Scholar] [CrossRef]
- Bagningsanalyser af sorter af Vinterhvede ved TystofteFonden. Available online: https://www.tystofte.dk/wp-content/uploads/2018/06/broedhvedekriterier-til-www.pdf (accessed on 10 October 2018).
- Brødhvede. Available online: https://lbst.dk/landbrug/goedning/kvaelstofregulering/broedhvede/#c51695 (accessed on 10 October 2018).
- Bernardo, R. Molecular markers and selection for complex traits in plants: Learning from the last 20 years. Crop. Sci. 2008, 48, 1649–1664. [Google Scholar] [CrossRef]
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [PubMed]
- Heffner, E.L.; Jannink, J.L.; Iwata, H.; Souza, E.; Sorrells, M.E. Genomic selection accuracy for grain quality traits in biparental wheat populations. Crop. Sci. 2011, 51, 2597–2606. [Google Scholar] [CrossRef]
- Zhao, Y.; Mette, M.F.; Gowda, M.; Longin, C.F.H.; Reif, J.C. Bridging the gap between marker-assisted and genomic selection of heading time and plant height in hybrid wheat. Heredity (Edinb) 2014, 112, 638–645. [Google Scholar] [CrossRef] [PubMed]
- VanRaden, P.M. Efficient methods to compute genomic predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef]
- Meuwissen, T.H.E.; Hayes, B.; Goddard, M. Genomic selection: A paradigm shift in animal breeding. Anim. Front. 2016, 6, 6. [Google Scholar] [CrossRef]
- Cericola, F.; Jahoor, A.; Orabi, J.; Andersen, J.R.; Janss, L.L.; Jensen, J. Optimizing Training Population Size and Genotyping Strategy for Genomic Prediction Using Association Study Results and Pedigree Information. A Case of Study in Advanced Wheat Breeding Lines. PLoS ONE 2017, 12, e0169606. [Google Scholar] [CrossRef] [PubMed]
- Crossa, J.; Pérez-Rodríguez, P.; Cuevas, J.; Montesinos-López, O.; Jarquín, D.; de los Campos, G.; Burgueño, J.; González-Camacho, J.M.; Pérez-Elizalde, S.; Beyene, Y.; et al. Genomic Selection in Plant Breeding: Methods, Models, and Perspectives. Trends Plant. Sci. 2017, 22, 961–975. [Google Scholar] [CrossRef]
- Marulanda, J.J.; Mi, X.; Melchinger, A.E.; Xu, J.L.; Würschum, T.; Longin, C.F.H. Optimum breeding strategies using genomic selection for hybrid breeding in wheat, maize, rye, barley, rice and triticale. Theor. Appl. Genet. 2016, 129, 1901–1913. [Google Scholar] [CrossRef]
- Nielsen, N.H.; Jahoor, A.; Jensen, J.D.; Orabi, J.; Cericola, F.; Edriss, V.; Jensen, J. Genomic prediction of seed quality traits using advanced barley breeding lines. PLoS ONE 2016, 11, e0164494. [Google Scholar] [CrossRef]
- Heffner, E.L.; Jannink, J.; Sorrells, M.E. Genomic Selection Accuracy using Multifamily Prediction Models in a Wheat Breeding Program. Plant Genome 2011, 4, 65–75. [Google Scholar] [CrossRef]
- Arruda, M.P.; Lipka, A.E.; Brown, P.J.; Krill, A.M.; Thurber, C.; Brown-Guedira, G.; Dong, Y.; Foresman, B.J.; Kolb, F.L. Comparing genomic selection and marker-assisted selection for Fusarium head blight resistance in wheat (Triticum aestivum L.). Mol. Breed. 2016, 36, 1–11. [Google Scholar] [CrossRef]
- Charmet, G.; Storlie, E.; Oury, F.X.; Laurent, V.; Beghin, D.; Chevarin, L.; Lapierre, A.; Perretant, M.R.; Rolland, B.; Heumez, E.; et al. Genome-wide prediction of three important traits in bread wheat. Mol. Breed. 2014, 34, 1843–1852. [Google Scholar] [CrossRef] [PubMed]
- Battenfield, S.D.; Guzmán, C.; Gaynor, R.C.; Singh, R.P.; Peña, R.J.; Dreisigacker, S.; Fritz, A.K.; Poland, J.A. Genomic Selection for Processing and End-Use Quality Traits in the CIMMYT Spring Bread Wheat Breeding Program. Plant Genome 2016, 9. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, P.S.; Jahoor, A.; Andersen, J.R.; Cericola, F.; Orabi, J.; Janss, L.; Jensen, J. Genome-Wide Association Studies and Comparison of Models and Cross-Validation Strategies for Genomic Prediction of Quality Traits in Advanced Winter Wheat Breeding Lines. Front. Plant. Sci. 2018, 9, 69. [Google Scholar] [CrossRef] [PubMed]
- Michel, S.; Kummer, C.; Gallee, M.; Hellinger, J.; Ametz, C.; Akgöl, B.; Epure, D.; Löschenberger, F.; Buerstmayr, H. Improving the baking quality of bread wheat by genomic selection in early generations. Theor. Appl. Genet. 2018, 131, 477–493. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, R. Genomewide selection when major genes are known. Crop. Sci. 2014, 54, 68–75. [Google Scholar] [CrossRef]
- Moore, J.K.; Manmathan, H.K.; Anderson, V.A.; Poland, J.A.; Morris, C.F.; Haley, S.D. Improving genomic prediction for pre-harvest sprouting tolerance in wheat by weighting large-effect quantitative trait loci. Crop. Sci. 2017, 57, 1315–1324. [Google Scholar] [CrossRef]
- Norman, A.; Taylor, J.; Edwards, J.; Kuchel, H. Optimising Genomic Selection in Wheat: Effect of Marker Density, Population Size and Population Structure on Prediction Accuracy. G3-Genes Genomes Genetics 2018, 8, 2889–2899. [Google Scholar] [CrossRef]
- Thorwarth, P.; Liu, G.; Ebmeyer, E.; Schacht, J.; Schachschneider, R.; Kazman, E.; Reif, J.C.; Würschum, T.; Longin, C.F.H. Dissecting the genetics underlying the relationship between protein content and grain yield in a large hybrid wheat population. Theor. Appl. Genet. 2019, 132, 489–500. [Google Scholar] [CrossRef]
- American Association of Cereal Chemists. Approved Methods of Analysis, 11th ed.; AACC International: St. Paul, MN, USA, 2000. [Google Scholar]
- Rogers, S.O.; Bendich, A.J. Extraction of DNA from milligram amounts of fresh, herbarium and mummified plant tissues. Plant. Mol. Biol. 1985, 5, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Madsen, P.; Jensen, J. DMU: A User’s Guide. A Package for Analysing Multivariate Mixed Models. 2013. Available online: http://dmu.agrsci.dk (accessed on 8 August 2019).
- Hinrichs, A.L.; Larkin, E.K.; Suarez, B.K. Population stratification and patterns of linkage disequilibrium. Genet. Epidemiol. 2009, 33, S88–S92. [Google Scholar] [CrossRef] [PubMed]
- IWGSC (The International Wheat Genome Sequencing Consortium). Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361, 7191. [Google Scholar] [CrossRef] [PubMed]
- Kersey, P.J.; Allen, J.E.; Allot, A.; Barba, M.; Boddu, S.; Bolt, B.J.; Carvalho-Silva, D.; Christensen, M.; Davis, P.; Grabmueller, C.; et al. Ensembl Genomes 2018: An integrated omics infrastructure for non-vertebrate species. Nucleic Acids Res. 2018, 46, D802–D808. [Google Scholar] [CrossRef] [PubMed]
- Janss, L.L. Bayz Manual. Available online: http://bayz.biz (accessed on 8 August 2019).
- Gao, H.; Su, G.; Janss, L.; Zhang, Y.; Lund, M.S. Model comparison on genomic predictions using high-density markers for different groups of bulls in the Nordic Holstein population. J. Dairy Sci. 2013, 96, 4678–4687. [Google Scholar] [CrossRef] [PubMed]
- Spiegelhalter, D.J.; Best, N.G.; Carlin, B.P.; Van Der Linde, A. Bayesian measures of model complexity and fit. J. R Statist. Soc. B 2002, 64, 583–616. [Google Scholar] [CrossRef]
- Plummer, M.; Best, N.; Cowles, K.; Vines, K. CODA: Convergence Diagnosis and Output Analysis for MCMC. R News. 2006, 6, 7–11. Available online: https://www.r-project.org/doc/Rnews/Rnews_2006-1.pdf (accessed on 8 August 2019).
- Rohde, P.D.; Gaertner, B.; Ward, K.; Sørensen, P.; Mackay, T.F.C. Genomic analysis of genotype by social environment interaction for Drosophila aggressive behavior. Genetics 2017, 206, 1969–1984. [Google Scholar] [CrossRef]
- Sarup, P.; Jensen, J.; Ostersen, T.; Henryon, M.; Sørensen, P. Increased prediction accuracy using a genomic feature model including prior information on quantitative trait locus regions in purebred Danish Duroc pigs. BMC Genet. 2016, 17, 1–17. [Google Scholar] [CrossRef]
- Deng, Z.; Tian, J.; Chen, F.; Li, W.; Zheng, F.; Chen, J.; Shi, C.; Sun, C.; Wang, S.; Zhang, Y. Genetic dissection on wheat flour quality traits in two related populations. Euphytica 2015, 203, 221–235. [Google Scholar] [CrossRef]
- Jin, H.; Wen, W.; Liu, J.; Zhai, S.; Zhang, Y.; Yan, J.; Liu, Z.; Xia, X.; He, Z. Genome-Wide QTL Mapping for Wheat Processing Quality Parameters in a Gaocheng 8901/Zhoumai 16 Recombinant Inbred Line Population. Front. Plant. Sci. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Maphosa, L.; Langridge, P.; Taylor, H.; Emebiri, L.C.; Mather, D.E. Genetic control of grain protein, dough rheology traits and loaf traits in a bread wheat population grown in three environments. J. Cereal Sci. 2015, 64, 147–152. [Google Scholar] [CrossRef]
- Mohler, V.; Schmolke, M.; Paladey, E.; Seling, S.; Hartl, L. Association analysis of Puroindoline-D1 and Puroindoline b-2 loci with 13 quality traits in European winter wheat (Triticum aestivum L.). J. Cereal Sci. 2012, 56, 623–628. [Google Scholar] [CrossRef]
- Morris, C.F. Puroindolines: The molecular basis of wheat grain hardness. Plant Mol Biol Puroindolines: The molecular genetic basis of wheat grain hardness. Plant. Mol. Biol. 2002, 48, 633–647. [Google Scholar] [CrossRef] [PubMed]
- Vagndorf, N.; Kristensen, P.S.; Andersen, J.R.; Jahoor, A.; Orabi, J. Marker-assisted breeding in eheat. In Next Generation Plant Breeding; IntechOpen: London, UK, 2018; pp. 3–22. [Google Scholar] [CrossRef]
- Beavis, W.D. QTL analyses: power, precision, and accuracy. In Molecular Dissection of Complex Traits; Paterson, A.H., Ed.; CRC Press: New York, NY, USA, 1998; pp. 145–162. [Google Scholar]
- Xu, S. Theoretical Basis of the Beavis Effect. Genetics 2003, 165, 2259–2268. [Google Scholar] [PubMed]
- Jarquín, D.; Crossa, J.; Lacaze, X.; Du Cheyron, P.; Daucourt, J.; Lorgeou, J.; Piraux, F.; Guerreiro, L.; Pérez, P.; Calus, M.; et al. A reaction norm model for genomic selection using high-dimensional genomic and environmental data. Theor. Appl. Genet. 2014, 127, 595–607. [Google Scholar] [CrossRef]
- Michel, S.; Ametz, C.; Gungor, H.; Epure, D.; Grausgruber, H.; Löschenberger, F.; Buerstmayr, H. Genomic selection across multiple breeding cycles in applied bread wheat breeding. Theor. Appl. Genet. 2016, 129, 1179–1189. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Mean | Range | Coefficient of Variation (%) |
|---|---|---|---|
| Flour yield (%) | 69.9 | 60.8–77.3 | 4.3 |
| Alveo P (mm) | 73.8 | 31–201 | 32.5 |
| Alveo L (mm) | 56.1 | 21–114 | 28.0 |
| Alveo W (10−4 J) | 134.2 | 40–293 | 33.4 |
| Trait | Varg | Vare | h2 |
|---|---|---|---|
| Flour yield | 2.8 ± 0.30 | 4.6 ± 0.36 | 0.38 ± 0.046 |
| Alveo P | 327.6 ± 23.7 | 146.8 ± 15.3 | 0.69 ± 0.053 |
| Alveo L | 138.6 ± 11.3 | 94.1 ± 8.9 | 0.60 ± 0.054 |
| Alveo W | 1108.3 ± 74.4 | 429.0 ± 45.3 | 0.72 ± 0.050 |
| Trait | SNP 1 | Chromo-some | p Value | Allele Frequency (%) 2 | Genetic Effect | Explained Genetic Variance (%) | Gene ID | Annotation |
|---|---|---|---|---|---|---|---|---|
| Flour yield | NOS_WW_ SNP_688 | 5D | 2.44 × 10−9 | 28 | 0.96 ± 0.14 | 13.3 | TraesCS5D02G004300 | Nontranslating coding sequence |
| Alveo P | NOS_WW_ SNP_5054 | 1D | 6.22 × 10−7 | 15 | 7.99 ± 1.29 | 5.0 | TraesCS1D02G322300 | Uncharacterized protein |
| NOS_WW_ SNP_688 | 5D | 1.02 × 10−6 | 28 | 6.01 ± 0.99 | 4.4 | TraesCS5D02G004300 | Nontranslating coding sequence | |
| Alveo L | NOS_WW_ SNP_2731 | 4A | 9.59 × 10−8 | 37 | 4.87 ± 0.82 | 8.0 | TraesCS4A02G447000 | Glycosyl-transferase |
| Alveo W | NOS_WW_ SNP_11809 | 1B | 1.77 × 10−13 | 64 | 14.58 ± 1.66 | 8.8 | TraesCS1B02G329600 | Uncharacterized protein |
| NOS_WW_ SNP_6663 | 1B | 1.21 × 10−9 | 64 | 12.04 ± 1.67 | 6.0 | TraesCS1B02G016400 | Uncharacterized protein | |
| NOS_WW_ SNP_3056 | 1D | 6.30 × 10−13 | 13 | 20.18 ± 2.35 | 8.3 | TraesCS1D02G317100 | Histone deacetylase | |
| NOS_WW_ SNP_688 | 5D | 4.16 × 10−6 | 28 | 9.41 ± 1.74 | 3.2 | TraesCS5D02G004300 | Nontranslating coding sequence |
| NOS_WW_ SNP_5054 (Chr. 1D) | NOS_WW_ SNP_688 (Chr. 5D) | Alveo P (mm) | Number of Lines |
|---|---|---|---|
| G | C | 64.0 | 386 |
| G | T | 84.2 | 125 |
| A | C | 100.7 | 43 |
| A | T | 104.7 | 36 |
| NOS_WW_ SNP_3056 (Chr. 1D) | NOS_WW_ SNP_11809 (Chr. 1B) | NOS_WW_ SNP_6663 (Chr. 1B) | NOS_WW_ SNP_688 (Chr. 5D) | Alveo W (10−4 J) | Number of Lines |
|---|---|---|---|---|---|
| C | G | G | C | 88.9 | 75 |
| C | G | G | T | 83.8 | 11 |
| C | G | A | C | 127.4 | 86 |
| C | A | G | T | 132.9 | 26 |
| C | A | A | C | 137.0 | 144 |
| C | A | A | T | 164.7 | 74 |
| T | G | G | C | 131.8 | 5 |
| T | G | G | T | 127.0 | 3 |
| T | G | A | C | 160.2 | 5 |
| T | G | A | T | 167.3 | 3 |
| T | A | G | C | 145.5 | 6 |
| T | A | G | T | 170.5 | 2 |
| T | A | A | C | 204.7 | 14 |
| T | A | A | T | 214.5 | 26 |
| Trait | LOO 1 | LFO | LSO | 2-Fold | 5-Fold | 10-Fold |
|---|---|---|---|---|---|---|
| Flour yield | 1.00 ± 0.07 | 0.89 ± 0.09 | 0.71 ± 0.09 | 0.95 ± 0.08 | 0.97 ± 0.07 | 0.99 ± 0.07 |
| Alveo P | 1.00 ± 0.04 | 0.98 ± 0.06 | 0.90 ± 0.06 | 1.02 ± 0.04 | 0.99 ± 0.04 | 1.00 ± 0.04 |
| Alveo L | 0.99 ± 0.05 | 0.94 ± 0.08 | 0.81 ± 0.09 | 1.03 ± 0.05 | 0.97 ± 0.05 | 0.98 ± 0.05 |
| Alveo W | 1.02 ± 0.03 | 0.95 ± 0.05 | 0.96 ± 0.05 | 1.03 ± 0.04 | 1.02 ± 0.03 | 1.03 ± 0.03 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kristensen, P.S.; Jensen, J.; Andersen, J.R.; Guzmán, C.; Orabi, J.; Jahoor, A. Genomic Prediction and Genome-Wide Association Studies of Flour Yield and Alveograph Quality Traits Using Advanced Winter Wheat Breeding Material. Genes 2019, 10, 669. https://doi.org/10.3390/genes10090669
Kristensen PS, Jensen J, Andersen JR, Guzmán C, Orabi J, Jahoor A. Genomic Prediction and Genome-Wide Association Studies of Flour Yield and Alveograph Quality Traits Using Advanced Winter Wheat Breeding Material. Genes. 2019; 10(9):669. https://doi.org/10.3390/genes10090669
Chicago/Turabian StyleKristensen, Peter S., Just Jensen, Jeppe R. Andersen, Carlos Guzmán, Jihad Orabi, and Ahmed Jahoor. 2019. "Genomic Prediction and Genome-Wide Association Studies of Flour Yield and Alveograph Quality Traits Using Advanced Winter Wheat Breeding Material" Genes 10, no. 9: 669. https://doi.org/10.3390/genes10090669
APA StyleKristensen, P. S., Jensen, J., Andersen, J. R., Guzmán, C., Orabi, J., & Jahoor, A. (2019). Genomic Prediction and Genome-Wide Association Studies of Flour Yield and Alveograph Quality Traits Using Advanced Winter Wheat Breeding Material. Genes, 10(9), 669. https://doi.org/10.3390/genes10090669