Eukaryote Genes Are More Likely than Prokaryote Genes to Be Composites


Abstract
1. Introduction
2. Materials and Methods
2.1. Dataset Construction and BLAST Analysis
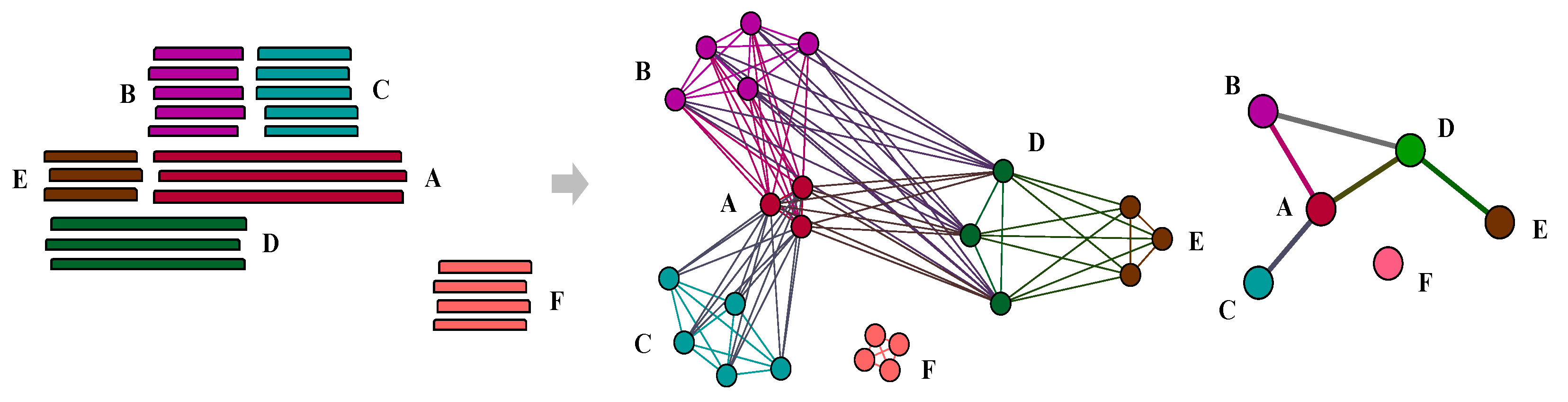
2.2. Composite Gene Identification
2.3. Functional Annotations
2.4. Statistical Analysis
3. Results
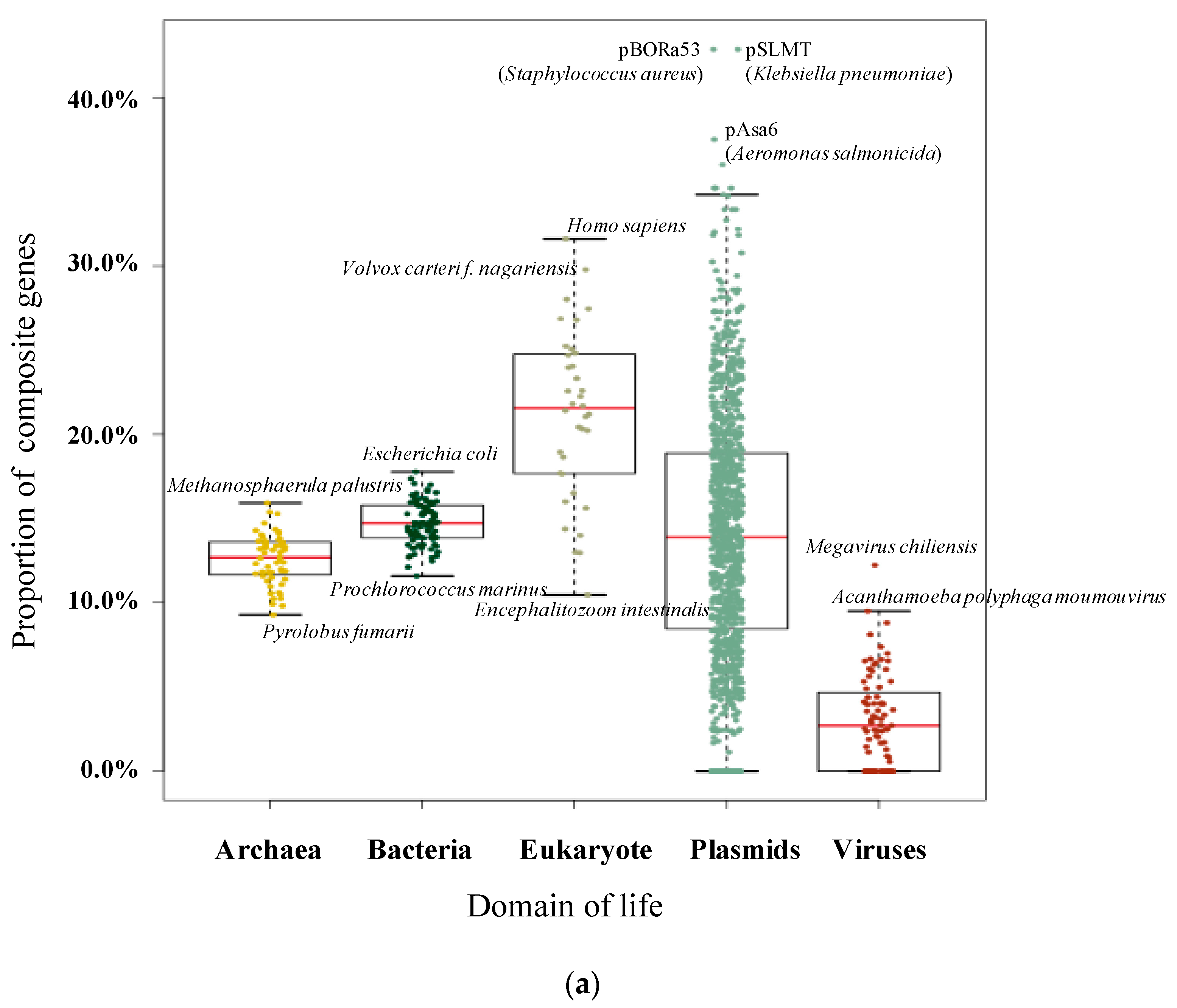
3.1. Pervasive Existence of Composite Genes across All of Life
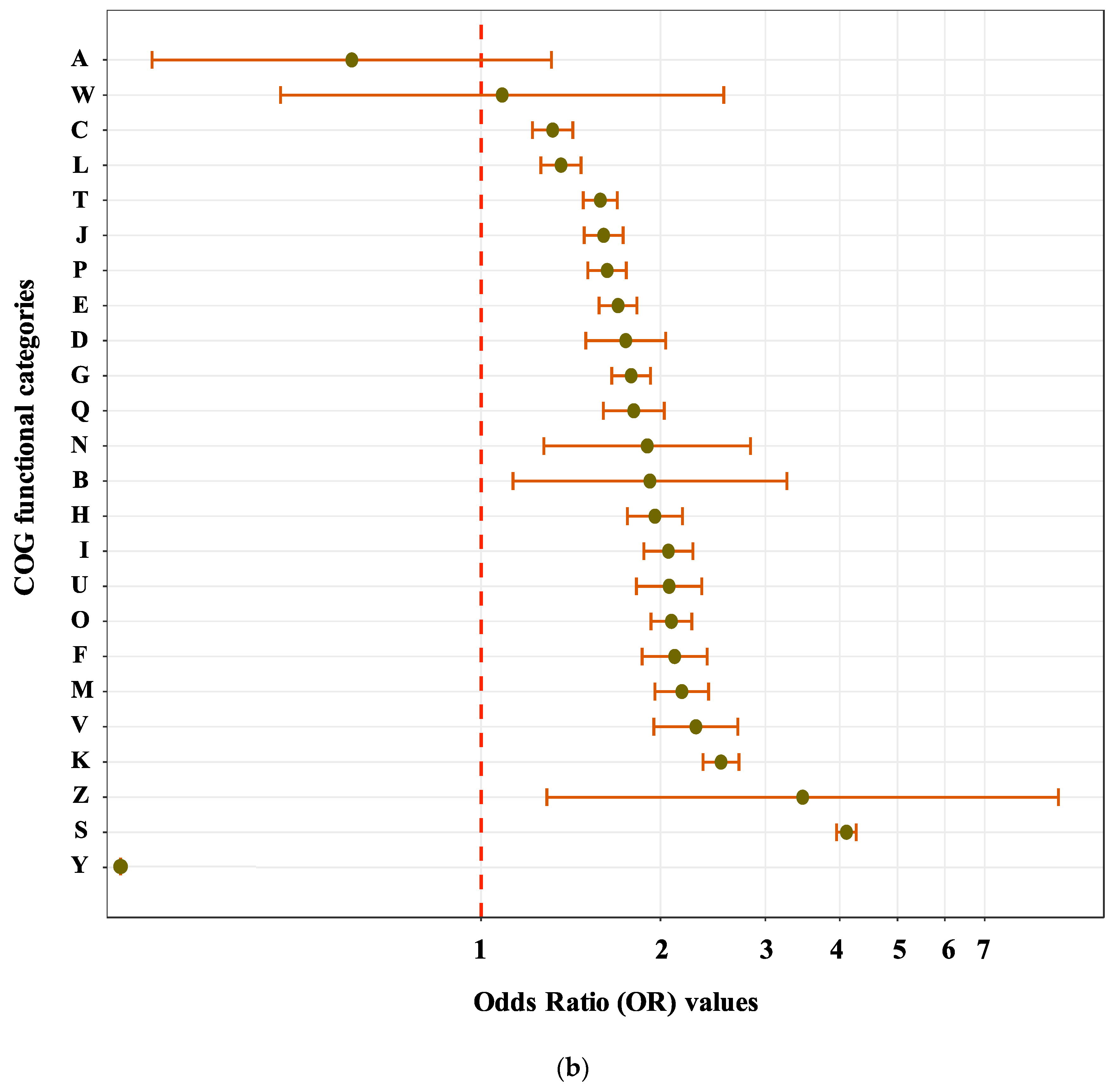
3.2. Sequence Functional Annotations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nelson-Sathi, S.; Dagan, T.; Landan, G.; Janssen, A.; Steel, M.; McInerney, J.O.; Deppenmeier, U.; Martin, W.F. Acquisition of 1000 eubacterial genes physiologically transformed a methanogen at the origin of Haloarchaea. Proc. Natl. Acad. Sci. USA 2012, 109, 20537–20542. [Google Scholar] [CrossRef]
- Oakley, T.H. Furcation and fusion: The phylogenetics of evolutionary novelty. Dev. Biol. 2017, 431, 69–76. [Google Scholar] [CrossRef]
- Linder, C.R.; Moret, B.M.E.; Nakhleh, L.; Warnow, T. Network (reticulate) evolution: Biology, models, and algorithms. In Proceedings of the Ninth Pacific Symposium on Biocomputing (PSB), Big Island, HI, USA, 6–10 January 2004. [Google Scholar]
- Corel, E.; Lopez, P.; Méheust, R.; Bapteste, E. Network-Thinking: Graphs to Analyze Microbial Complexity and Evolution. Trends Microbiol. 2016, 24, 224–237. [Google Scholar] [CrossRef]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An Open Source Software for Exploring and Manipulating Networks. In Proceedings of the Third International AAAI Conference on Weblogs and Social Media, San Jose, CA, USA, 17–20 May 2009. [Google Scholar]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Haggerty, L.S.; Jachiet, P.A.; Hanage, W.P.; Fitzpatrick, D.A.; Lopez, P.; O’Connell, M.J.; Pisani, D.; Wilkinson, M.; Bapteste, E.; McInerney, J.O. A pluralistic account of homology: Adapting the models to the data. Mol. Biol. Evol. 2014, 31, 501–516. [Google Scholar] [CrossRef]
- Coleman, O.; Hogan, R.; McGoldrick, N.; Rudden, N.; McInerney, J. Evolution by Pervasive Gene Fusion in Antibiotic Resistance and Antibiotic Synthesizing Genes. Computation 2015, 3, 114–127. [Google Scholar] [CrossRef]
- Enright, A.J.; Iliopoulos, I.; Kyrpides, N.C.; Ouzounis, C.A. Protein interaction maps for complete genomes based on gene fusion events. Nature 1999, 402, 86. [Google Scholar] [CrossRef]
- Jachiet, P.A.; Pogorelcnik, R.; Berry, A.; Lopez, P.; Bapteste, E. MosaicFinder: Identification of fused gene families in sequence similarity networks. Bioinformatics 2013, 29, 837–844. [Google Scholar] [CrossRef]
- Jachiet, P.A.; Colson, P.; Lopez, P.; Bapteste, E. Extensive gene remodeling in the viral world: New evidence for nongradual evolution in the mobilome network. Genome Biol. Evol. 2014, 6, 2195–2205. [Google Scholar] [CrossRef]
- Méheust, R.; Zelzion, E.; Bhattacharya, D.; Lopez, P.; Bapteste, E. Protein networks identify novel symbiogenetic genes resulting from plastid endosymbiosis. Proc. Natl. Acad. Sci. USA 2016, 113, 3579–3584. [Google Scholar] [CrossRef]
- Ocaña-Pallarès, E.; Najle, S.R.; Scazzocchio, C.; Ruiz-Trillo, I. Reticulate evolution in eukaryotes: Origin and evolution of the nitrate assimilation pathway. PLoS Genet. 2019, 15, e1007986. [Google Scholar] [CrossRef]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI reference sequences (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2006, 35, D61–D65. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Pathmanathan, J.S.; Lopez, P.; Lapointe, F.-J.; Bapteste, E. CompositeSearch: A generalized network approach for composite gene families detection. Mol. Biol. Evol. 2017, 35, 252–255. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2015, 44, D286–D293. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef]
- Consortium, G.O. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004, 32, D258–D261. [Google Scholar] [CrossRef]
- Szumilas, M. Explaining odds ratios. J. Can. Acad. child Adolesc. Psychiatry 2010, 19, 227. [Google Scholar]
- Sedgwick, P. Multiple significance tests: The Bonferroni correction. BMJ 2012, 344, e509. [Google Scholar] [CrossRef]
- Alvarez-Ponce, D.; Lopez, P.; Bapteste, E.; McInerney, J.O. Gene similarity networks provide tools for understanding eukaryote origins and evolution. Proc. Natl. Acad. Sci. USA 2013, 110, E1594–E1603. [Google Scholar] [CrossRef] [PubMed]
- McInerney, J.O.; O’Connell, M.J.; Pisani, D. The hybrid nature of the Eukaryota and a consilient view of life on Earth. Nat. Rev. Microbiol. 2014, 12, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Halary, S.; McInerney, J.O.; Lopez, P.; Bapteste, E. EGN: A wizard for construction of gene and genome similarity networks. BMC Evol. Biol. 2013, 13, 146. [Google Scholar] [CrossRef] [PubMed]
- McInerney, J.O.; Pisani, D.; Bapteste, E.; O’Connell, M.J. The public goods hypothesis for the evolution of life on Earth. Biol. Direct 2011, 6, 41. [Google Scholar] [CrossRef] [PubMed]
- Barbour, A.G.; Dai, Q.; Restrepo, B.I.; Stoenner, H.G.; Frank, S.A. Pathogen escape from host immunity by a genome program for antigenic variation. Proc. Natl. Acad. Sci. USA 2006, 103, 18290–18295. [Google Scholar] [CrossRef]
- Chaconas, G.; Kobryn, K. Structure, function, and evolution of linear replicons in Borrelia. Annu. Rev. Microbiol. 2010, 64, 185–202. [Google Scholar] [CrossRef]
- Corel, E.; Méheust, R.; Watson, A.K.; Mcinerney, J.O.; Lopez, P.; Bapteste, E. Bipartite network analysis of gene sharings in the microbial world. Mol. Biol. Evol. 2018, 35, 899–913. [Google Scholar] [CrossRef]
- Sibbald, S.J.; Hopkins, J.F.; Filloramo, G.V.; Archibald, J.M. Ubiquitin fusion proteins in algae: Implications for cell biology and the spread of photosynthesis. BMC Genomics 2019, 20, 1–13. [Google Scholar] [CrossRef]
- AM, M.; Hyland, E.M.; Cormican, P.; Moran, R.J.; Webb, A.E.; Lee, K.D.; Hernandez, J.; Prado-Martinez, J.; Creevey, C.J.; Aspden, J.L. Gene Fusions derived by transcriptional readthrough are Driven by Segmental Duplication in Human. Genome Biol. Evol. 2019. [Google Scholar] [CrossRef]
- Méheust, R.; Watson, A.K.; Lapointe, F.J.; Papke, R.T.; Lopez, P.; Bapteste, E. Hundreds of novel composite genes and chimeric genes with bacterial origins contributed to haloarchaeal evolution. Genome Biol. 2018, 19, 1–12. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
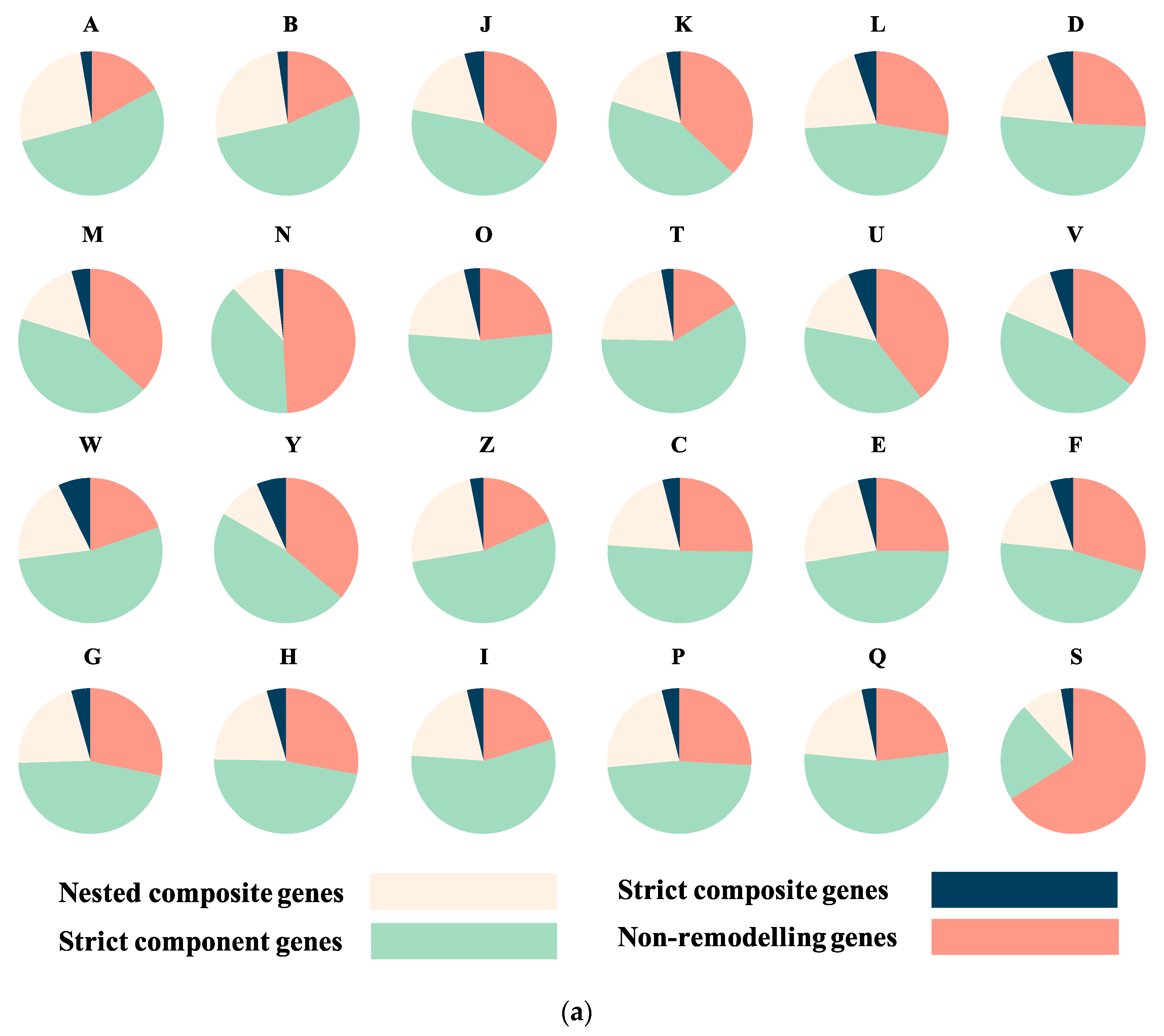{kind=link}
{kind=link}
| Species | Total Number of Genes | Number of Composite Genes | Proportion | Species | Total Number of Genes | Number of Composite Genes | Proportion |
|---|---|---|---|---|---|---|---|
| Homo sapiens | 109,018 | 34,455 | 31.60% | Fervidicoccus fontis | 1385 | 152 | 10.97% |
| Volvox carteri f. nagariensis | 14,436 | 4298 | 29.77% | Thermoproteus uzoniensis | 2112 | 224 | 10.61% |
| Aureococcus anophagefferens | 11,520 | 3227 | 28.01% | Nanoarchaeum equitans | 540 | 57 | 10.56% |
| Capsaspora owczarzaki | 8792 | 2413 | 27.45% | Staphylothermus marinus | 1598 | 168 | 10.51% |
| Chlorella variabilis | 9780 | 2626 | 26.85% | Encephalitozoon intestinalis | 1939 | 203 | 10.47% |
| Polysphondylium pallidum | 12,367 | 3313 | 26.79% | Ignisphaera aggregans | 1930 | 198 | 10.26% |
| Monosiga brevicollis | 9203 | 2322 | 25.23% | Methanopyrus kandleri | 1687 | 173 | 10.25% |
| Salpingoeca rosetta | 11,731 | 2939 | 25.05% | Pyrobaculum neutrophilum | 1966 | 195 | 9.92% |
| Allomyces macrogynus | 19,446 | 4829 | 24.83% | Hyperthermus butylicus | 1681 | 165 | 9.82% |
| Tetrahymena thermophila | 10,626 | 2625 | 24.70% | Pyrolobus fumarii | 1885 | 175 | 9.28% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ou, Y.; McInerney, J.O. Eukaryote Genes Are More Likely than Prokaryote Genes to Be Composites. Genes 2019, 10, 648. https://doi.org/10.3390/genes10090648
Ou Y, McInerney JO. Eukaryote Genes Are More Likely than Prokaryote Genes to Be Composites. Genes. 2019; 10(9):648. https://doi.org/10.3390/genes10090648
Chicago/Turabian StyleOu, Yaqing, and James O. McInerney. 2019. "Eukaryote Genes Are More Likely than Prokaryote Genes to Be Composites" Genes 10, no. 9: 648. https://doi.org/10.3390/genes10090648
APA StyleOu, Y., & McInerney, J. O. (2019). Eukaryote Genes Are More Likely than Prokaryote Genes to Be Composites. Genes, 10(9), 648. https://doi.org/10.3390/genes10090648