Origin and Evolution of Deleterious Mutations in Horses


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
3. Results
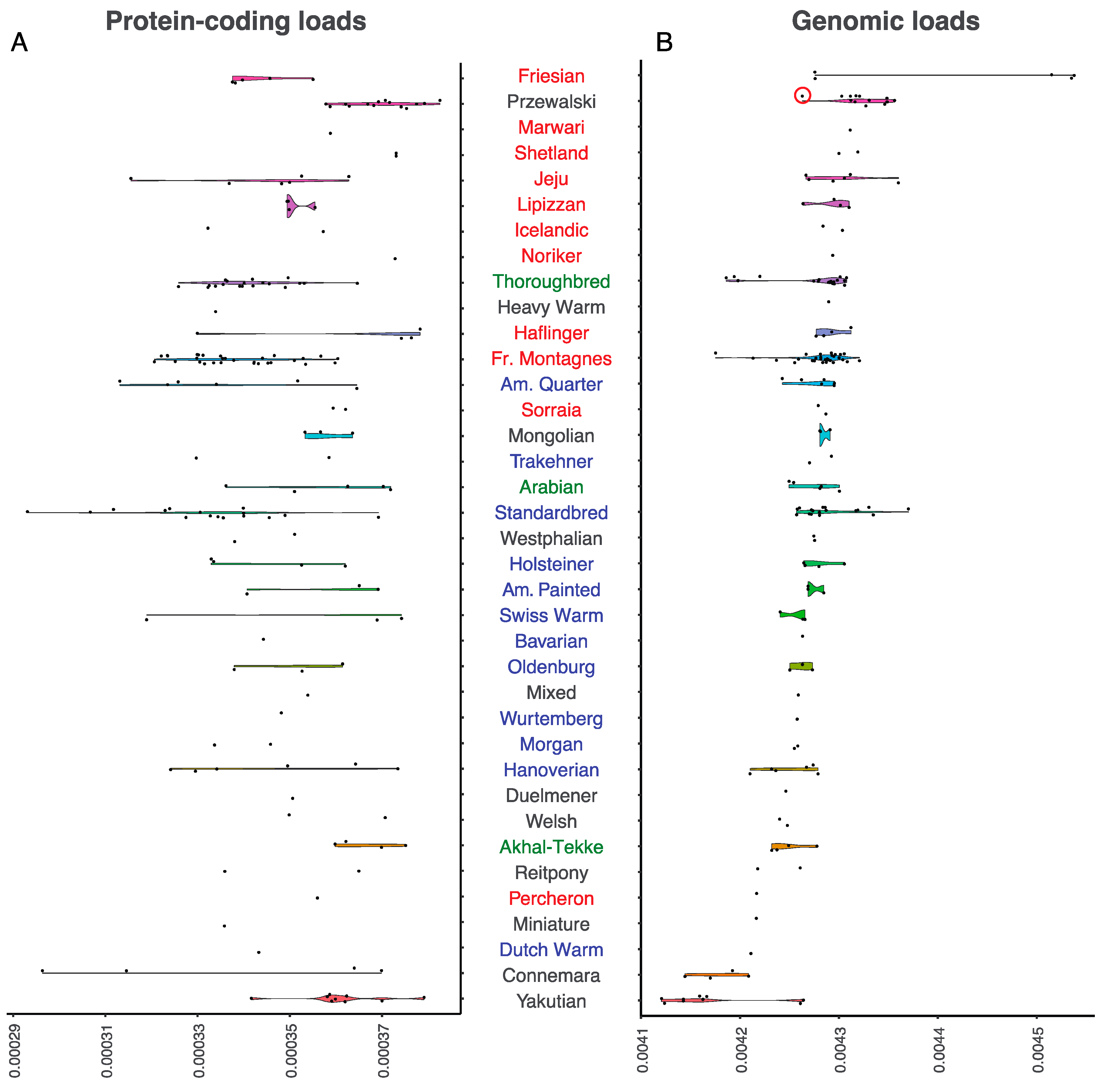
3.1. Levels and Patterns of Mutational Loads, Across Site Categories and Breed Types
3.2. Phylogenetic Reconstruction Supports Recent Population Decays in Working Breeds
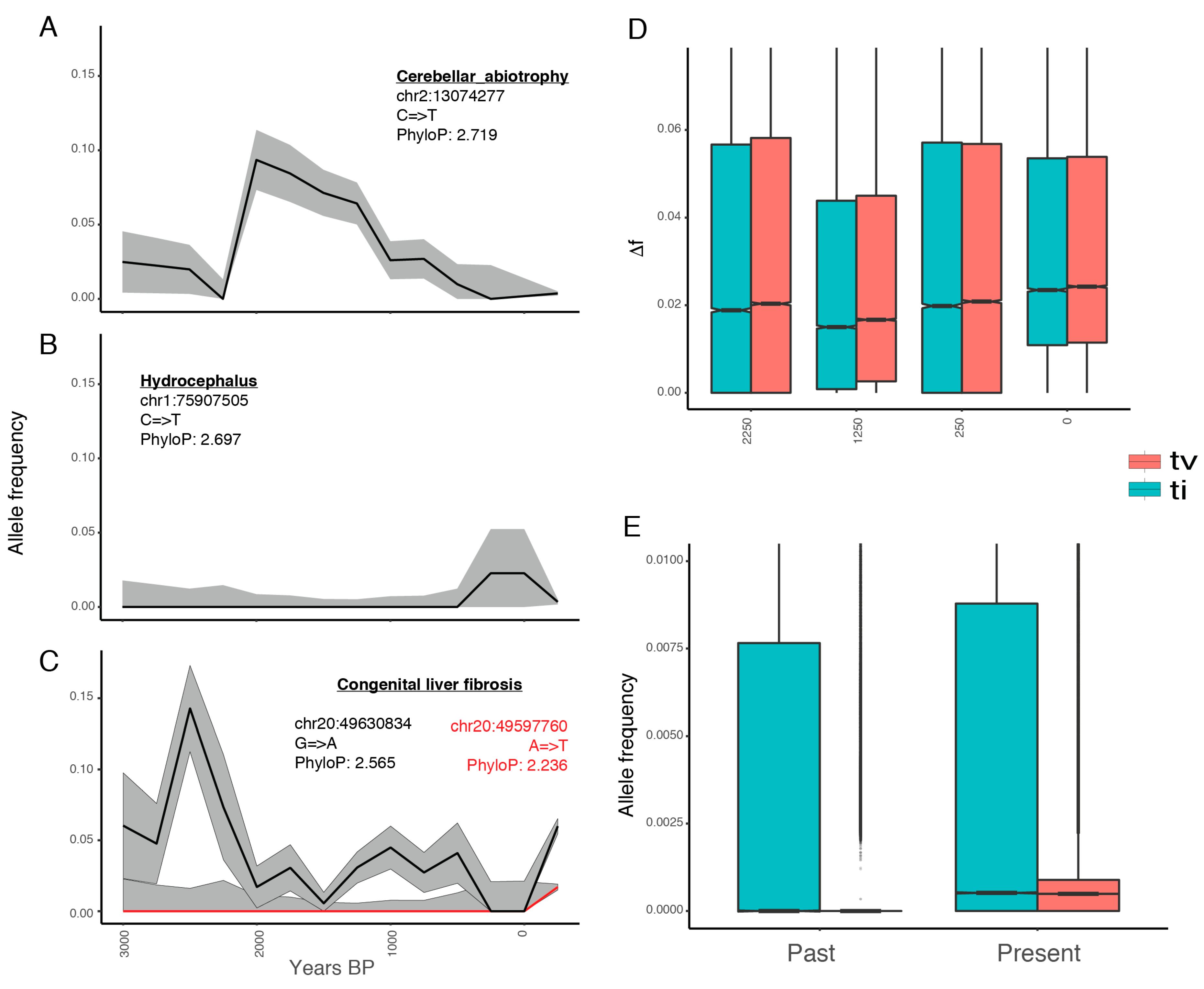
3.3. Deleterious Mutations Segregated at Low Frequencies Until the Last ~250 Years
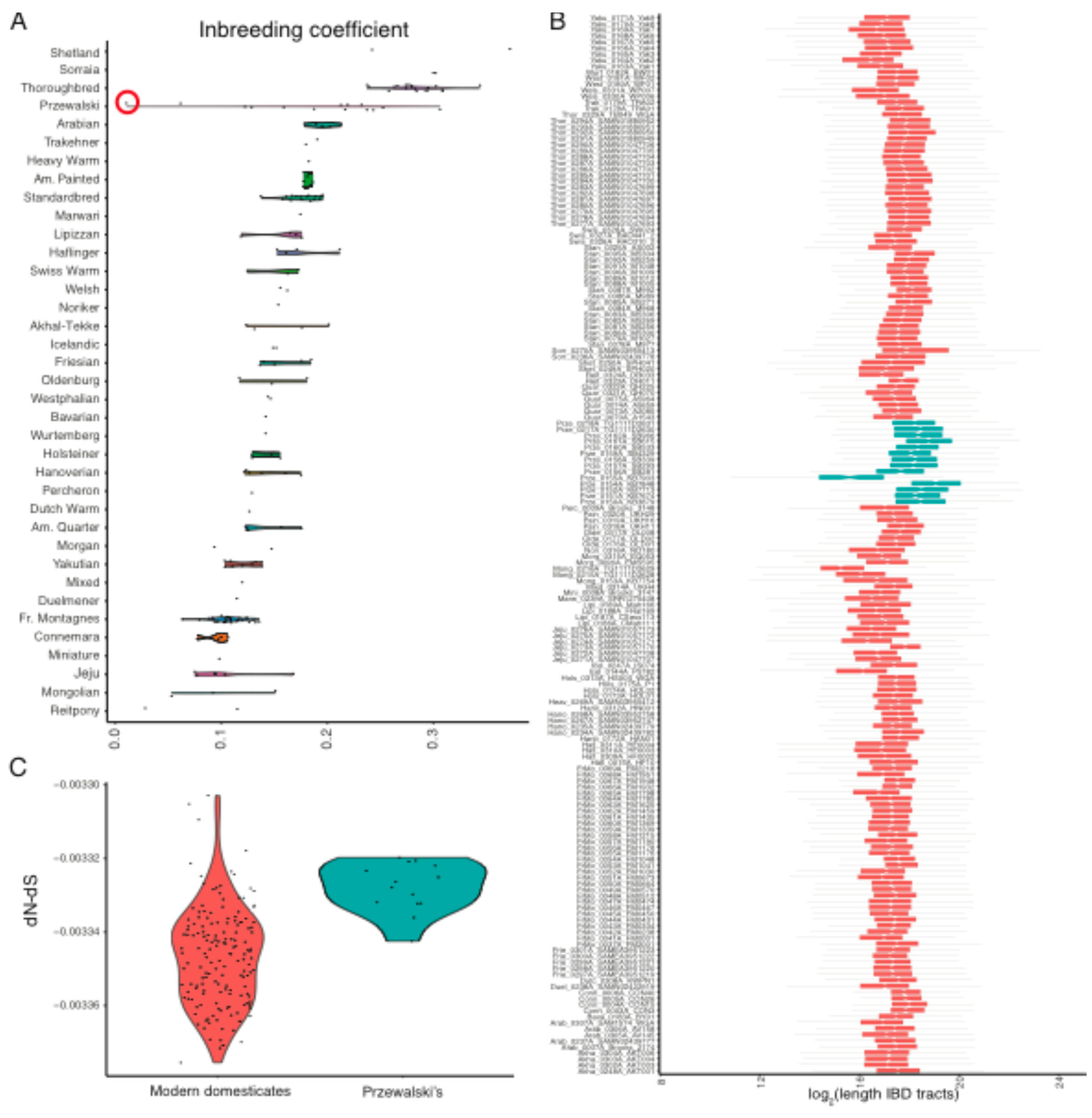
3.4. Inbreeding Depression and Genetic Purging Shaped Loads in Modern Domesticates
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Anthony, D.W. The Horse, the Wheel, and Language: How Bronze-Age Riders from the Eurasian Steppes Shaped the Modern World, Reprint ed.; Princeton University Press: Princeton, NJ, USA, 2010; ISBN 978-0-691-14818-2. [Google Scholar]
- Kelekna, P. The Horse in Human History; Cambridge University Press: Cambridge, UK, 2009. [Google Scholar]
- Equine Industry Statistics Overview l Equine Business Association. Available online: https://www.equinebusinessassociation.com/equine-industry-statistics/ (accessed on 7 June 2019).
- Librado, P.; Fages, A.; Gaunitz, C.; Leonardi, M.; Wagner, S.; Khan, N.; Hanghøj, K.; Alquraishi, S.A.; Alfarhan, A.H.; Al-Rasheid, K.A.; et al. The Evolutionary Origin and Genetic Makeup of Domestic Horses. Genetics 2016, 204, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Fages, A.; Hanghøj, K.; Khan, N.; Gaunitz, C.; Seguin-Orlando, A.; Leonardi, M.; McCrory Constantz, C.; Gamba, C.; Al-Rasheid, K.A.S.; Albizuri, S.; et al. Tracking Five Millennia of Horse Management with Extensive Ancient Genome Time Series. Cell 2019, 177, 1419–1435.e31. [Google Scholar] [CrossRef] [PubMed]
- Wallner, B.; Vogl, C.; Shukla, P.; Burgstaller, J.P.; Druml, T.; Brem, G. Identification of Genetic Variation on the Horse Y Chromosome and the Tracing of Male Founder Lineages in Modern Breeds. PLoS ONE 2013, 8, e60015. [Google Scholar] [CrossRef] [PubMed]
- Felkel, S.; Vogl, C.; Rigler, D.; Dobretsberger, V.; Chowdhary, B.P.; Distl, O.; Fries, R.; Jagannathan, V.; Janečka, J.E.; Leeb, T.; et al. The horse Y chromosome as an informative marker for tracing sire lines. Sci. Rep. 2019, 9, 6095. [Google Scholar] [CrossRef] [PubMed]
- Bosse, M.; Megens, H.; Derks, M.F.L.; de Cara, Á.M.R.; Groenen, M.A.M. Deleterious alleles in the context of domestication, inbreeding, and selection. Evol. Appl. 2018, 12, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Szpiech, Z.A.; Xu, J.; Pemberton, T.J.; Peng, W.; Zöllner, S.; Rosenberg, N.A.; Li, J.Z. Long Runs of Homozygosity Are Enriched for Deleterious Variation. Am. J. Hum. Genet. 2013, 93, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, F.W. Online Mendelian Inheritance in Animals (OMIA): A comparative knowledgebase of genetic disorders and other familial traits in non-laboratory animals. Nucleic Acids Res. 2003, 31, 275–277. [Google Scholar] [CrossRef]
- Korte, A.; Farlow, A. The advantages and limitations of trait analysis with GWAS: A review. Plant. Methods 2013, 9, 29. [Google Scholar] [CrossRef]
- Charlesworth, B. Effective population size and patterns of molecular evolution and variation. Nat. Rev. Genet. 2009, 10, 195–205. [Google Scholar] [CrossRef]
- Charlesworth, D.; Willis, J.H. The genetics of inbreeding depression. Nat. Rev. Genet. 2009, 10, 783–796. [Google Scholar] [CrossRef]
- Lynch, M.; Conery, J.; Burger, R. Mutation Accumulation and the Extinction of Small Populations. Am. Nat. 1995, 146, 489–518. [Google Scholar] [CrossRef]
- Understanding and Predicting the Fitness Decline of Shrunk Populations: Inbreeding, Purging, Mutation, and Standard Selection|Genetics. Available online: https://www.genetics.org/content/190/4/1461.long (accessed on 20 August 2019).
- Schubert, M.; Ermini, L.; Der Sarkissian, C.; Jónsson, H.; Ginolhac, A.; Schaefer, R.; Martin, M.D.; Fernández, R.; Kircher, M.; McCue, M.; et al. Characterization of ancient and modern genomes by SNP detection and phylogenomic and metagenomic analysis using PALEOMIX. Nat. Protoc. 2014, 9, 1056–1082. [Google Scholar] [CrossRef] [PubMed]
- Siepel, A.; Pollard, K.S.; Haussler, D. New Methods for Detecting Lineage-Specific Selection. In Proceedings of the Research in Computational Molecular Biology; Apostolico, A., Guerra, C., Istrail, S., Pevzner, P.A., Waterman, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; pp. 190–205. [Google Scholar]
- Pollard, K.S.; Hubisz, M.J.; Rosenbloom, K.R.; Siepel, A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010, 20, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Wade, C.M.; Giulotto, E.; Sigurdsson, S.; Zoli, M.; Gnerre, S.; Imsland, F.; Lear, T.L.; Adelson, D.L.; Bailey, E.; Bellone, R.R.; et al. Genome sequence, comparative analysis, and population genetics of the domestic horse. Science 2009, 326, 865–867. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Gamba, C.; Gaunitz, C.; Sarkissian, C.D.; Pruvost, M.; Albrechtsen, A.; Fages, A.; Khan, N.; Schubert, M.; Jagannathan, V.; et al. Ancient genomic changes associated with domestication of the horse. Science 2017, 356, 442–445. [Google Scholar] [CrossRef] [PubMed]
- Korneliussen, T.S.; Albrechtsen, A.; Nielsen, R. ANGSD: Analysis of Next Generation Sequencing Data. BMC Bioinform. 2014, 15, 356. [Google Scholar] [CrossRef] [PubMed]
- Dabney, J.; Meyer, M.; Pääbo, S. Ancient DNA Damage. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Lefort, V.; Desper, R.; Gascuel, O. FastME 2.0: A Comprehensive, Accurate, and Fast Distance-Based Phylogeny Inference Program. Mol. Biol. Evol. 2015, 32, 2798–2800. [Google Scholar] [CrossRef]
- Hedrick, P.W.; Garcia-Dorado, A. Understanding Inbreeding Depression, Purging, and Genetic Rescue. Trends Ecol. Evol. 2016, 31, 940–952. [Google Scholar] [CrossRef]
- Hill, W.G.; Robertson, A. Linkage disequilibrium in finite populations. Theor. Appl. Genet. 1968, 38, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Fox, E.A.; Wright, A.E.; Fumagalli, M.; Vieira, F.G. ngsLD: Evaluating linkage disequilibrium using genotype likelihoods. Bioinformatics 2019. [Google Scholar] [CrossRef] [PubMed]
- Van Dongen, S. Graph Clustering Via a Discrete Uncoupling Process. SIAM J. Matrix Anal. Appl. 2008, 30, 121–141. [Google Scholar] [CrossRef]
- Vieira, F.G.; Albrechtsen, A.; Nielsen, R. Estimating IBD tracts from low coverage NGS data. Bioinformatics 2016, 32, 2096–2102. [Google Scholar] [CrossRef] [PubMed]
- The IUCN Red List of Threatened Species. Available online: https://www.iucnredlist.org/en (accessed on 7 June 2019).
- Schubert, M.; Jónsson, H.; Chang, D.; Der Sarkissian, C.; Ermini, L.; Ginolhac, A.; Albrechtsen, A.; Dupanloup, I.; Foucal, A.; Petersen, B.; et al. Prehistoric genomes reveal the genetic foundation and cost of horse domestication. Proc. Natl. Acad. Sci. USA 2014, 111, E5661–E5669. [Google Scholar] [CrossRef] [PubMed]
- Gaunitz, C.; Fages, A.; Hanghøj, K.; Albrechtsen, A.; Khan, N.; Schubert, M.; Seguin-Orlando, A.; Owens, I.J.; Felkel, S.; Bignon-Lau, O.; et al. Ancient genomes revisit the ancestry of domestic and Przewalski’s horses. Science 2018, 360, 111–114. [Google Scholar] [CrossRef]
- Shetland Ponies, about Shetland Ponies—The Breed and Stud-Book. Available online: http://www.shetlandponystudbooksociety.co.uk/about-the-breed (accessed on 7 June 2019).
- Luís, C.; Cothran, E.G.; Oom, M.d.M. Inbreeding and Genetic Structure in the Endangered Sorraia Horse Breed: Implications for its Conservation and Management. J. Hered. 2007, 98, 232–237. [Google Scholar] [CrossRef]
- Horses—Breeds of Livestock, Department of Animal Science. Available online: http://afs.okstate.edu/breeds/horses/horses-w.html#r (accessed on 9 June 2019).
- Librado, P.; Sarkissian, C.D.; Ermini, L.; Schubert, M.; Jónsson, H.; Albrechtsen, A.; Fumagalli, M.; Yang, M.A.; Gamba, C.; Seguin-Orlando, A.; et al. Tracking the origins of Yakutian horses and the genetic basis for their fast adaptation to subarctic environments. Proc. Natl. Acad. Sci. USA 2015, 112, E6889–E6897. [Google Scholar] [CrossRef]
- Achilli, A.; Olivieri, A.; Soares, P.; Lancioni, H.; Hooshiar Kashani, B.; Perego, U.A.; Nergadze, S.G.; Carossa, V.; Santagostino, M.; Capomaccio, S.; et al. Mitochondrial genomes from modern horses reveal the major haplogroups that underwent domestication. Proc. Natl. Acad. Sci. USA 2012, 109, 2449–2454. [Google Scholar] [CrossRef]
- Jansen, T.; Forster, P.; Levine, M.A.; Oelke, H.; Hurles, M.; Renfrew, C.; Weber, J.; Olek, K. Mitochondrial DNA and the origins of the domestic horse. Proc. Natl. Acad. Sci. USA 2002, 99, 10905–10910. [Google Scholar] [CrossRef]
- Drögemüller, M.; Jagannathan, V.; Welle, M.M.; Graubner, C.; Straub, R.; Gerber, V.; Burger, D.; Signer-Hasler, H.; Poncet, P.-A.; Klopfenstein, S.; et al. Congenital Hepatic Fibrosis in the Franches-Montagnes Horse Is Associated with the Polycystic Kidney and Hepatic Disease 1 (PKHD1) Gene. PLoS ONE 2014, 9, e110125. [Google Scholar] [CrossRef] [PubMed]
- Brault, L.S.; Cooper, C.A.; Famula, T.R.; Murray, J.D.; Penedo, M.C.T. Mapping of equine cerebellar abiotrophy to ECA2 and identification of a potential causative mutation affecting expression of MUTYH. Genomics 2011, 97, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Ducro, B.J.; Schurink, A.; Bastiaansen, J.W.M.; Boegheim, I.J.M.; van Steenbeek, F.G.; Vos-Loohuis, M.; Nijman, I.J.; Monroe, G.R.; Hellinga, I.; Dibbits, B.W.; et al. A nonsense mutation in B3GALNT2 is concordant with hydrocephalus in Friesian horses. BMC Genomics 2015, 16, 761. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J. Rates of conservative and radical nonsynonymous nucleotide substitutions in mammalian nuclear genes. J. Mol. Evol. 2000, 50, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Lyons, D.M.; Lauring, A.S. Evidence for the Selective Basis of Transition-to-Transversion Substitution Bias in Two RNA Viruses. Mol. Biol. Evol. 2017, 34, 3205–3215. [Google Scholar] [CrossRef] [PubMed]
- O’Grady, J.J.; Brook, B.W.; Reed, D.H.; Ballou, J.D.; Tonkyn, D.W.; Frankham, R. Realistic levels of inbreeding depression strongly affect extinction risk in wild populations. Biol. Conserv. 2006, 133, 42–51. [Google Scholar] [CrossRef]
- Ebert, D.; Haag, C.; Kirkpatrick, M.; Riek, M.; Hottinger, J.W.; Pajunen, V.I. A selective advantage to immigrant genes in a Daphnia metapopulation. Science 2002, 295, 485–488. [Google Scholar] [CrossRef]
- Der Sarkissian, C.; Ermini, L.; Schubert, M.; Yang, M.A.; Librado, P.; Fumagalli, M.; Jónsson, H.; Bar-Gal, G.K.; Albrechtsen, A.; Vieira, F.G.; et al. Evolutionary Genomics and Conservation of the Endangered Przewalski’s Horse. Curr. Biol. 2015, 25, 2577–2583. [Google Scholar] [CrossRef]
- Genes|Free Full-Text|The Genomic Makeup of Nine Horse Populations Sampled in the Netherlands. Available online: https://www.mdpi.com/2073-4425/10/6/480 (accessed on 2 July 2019).
- Orlando, L.; Ginolhac, A.; Zhang, G.; Froese, D.; Albrechtsen, A.; Stiller, M.; Schubert, M.; Cappellini, E.; Petersen, B.; Moltke, I.; et al. Recalibrating Equus evolution using the genome sequence of an early Middle Pleistocene horse. Nature 2013, 499, 74–78. [Google Scholar] [CrossRef]
- Data export|Domestic Animal Diversity Information System (DAD-IS)|Food and Agriculture Organization of the United Nations. Available online: http://www.fao.org/dad-is/dataexport/en/ (accessed on 11 June 2019).
- Mischka, J. The Percheron Horse in America; Mischka Press/Heart Prairie: Walworth, WI, USA, 1991; ISBN 978-0-9622663-5-5. [Google Scholar]
- Montgomery, E.S. The Thoroughbred; Arco Pub: New York, NY, USA, 1972; ISBN 978-0-668-02824-0. [Google Scholar]
- Outram, A.K.; Stear, N.A.; Bendrey, R.; Olsen, S.; Kasparov, A.; Zaibert, V.; Thorpe, N.; Evershed, R.P. The Earliest Horse Harnessing and Milking. Science 2009, 323, 1332–1335. [Google Scholar] [CrossRef]
- Ludwig, A.; Pruvost, M.; Reissmann, M.; Benecke, N.; Brockmann, G.A.; Castaños, P.; Cieslak, M.; Lippold, S.; Llorente, L.; Malaspinas, A.-S.; et al. Coat Color Variation at the Beginning of Horse Domestication. Science 2009, 324, 485. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Tang, T.; Tang, H.; Huang, J.; Shi, S.; Wu, C.-I. The accumulation of deleterious mutations in rice genomes: A hypothesis on the cost of domestication. Trends Genet. 2006, 22, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Marsden, C.D.; Vecchyo, D.O.-D.; O’Brien, D.P.; Taylor, J.F.; Ramirez, O.; Vilà, C.; Marques-Bonet, T.; Schnabel, R.D.; Wayne, R.K.; Lohmueller, K.E. Bottlenecks and selective sweeps during domestication have increased deleterious genetic variation in dogs. Proc. Natl. Acad. Sci. USA 2016, 113, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Koenig, D.; Jiménez-Gómez, J.M.; Kimura, S.; Fulop, D.; Chitwood, D.H.; Headland, L.R.; Kumar, R.; Covington, M.F.; Devisetty, U.K.; Tat, A.V.; et al. Comparative transcriptomics reveals patterns of selection in domesticated and wild tomato. Proc. Natl. Acad. Sci. USA 2013, 110, E2655–E2662. [Google Scholar] [CrossRef] [PubMed]
- Moyers, B.T.; Morrell, P.L.; McKay, J.K. Genetic Costs of Domestication and Improvement. J. Hered. 2018, 109, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Allaby, R.G.; Ware, R.L.; Kistler, L. A re-evaluation of the domestication bottleneck from archaeogenomic evidence. Evol. Appl. 2018, 12, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hill, W.G.; Charlesworth, D.; Charlesworth, B. Dynamics of inbreeding depression due to deleterious mutations in small populations: Mutation parameters and inbreeding rate. Genet. Res. 1999, 74, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, B. Mutational load, inbreeding depression and heterosis in subdivided populations. Mol. Ecol. 2018, 27, 4991–5003. [Google Scholar] [CrossRef] [PubMed]
- Caballero, A.; Bravo, I.; Wang, J. Inbreeding load and purging: Implications for the short-term survival and the conservation management of small populations. Heredity 2017, 118, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Todd, E.T.; Ho, S.Y.W.; Thomson, P.C.; Ang, R.A.; Velie, B.D.; Hamilton, N.A. Founder-specific inbreeding depression affects racing performance in Thoroughbred horses. Sci. Rep. 2018, 8, 6167. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orlando, L.; Librado, P. Origin and Evolution of Deleterious Mutations in Horses. Genes 2019, 10, 649. https://doi.org/10.3390/genes10090649
Orlando L, Librado P. Origin and Evolution of Deleterious Mutations in Horses. Genes. 2019; 10(9):649. https://doi.org/10.3390/genes10090649
Chicago/Turabian StyleOrlando, Ludovic, and Pablo Librado. 2019. "Origin and Evolution of Deleterious Mutations in Horses" Genes 10, no. 9: 649. https://doi.org/10.3390/genes10090649
APA StyleOrlando, L., & Librado, P. (2019). Origin and Evolution of Deleterious Mutations in Horses. Genes, 10(9), 649. https://doi.org/10.3390/genes10090649