DNA Barcoding Reveals High Levels of Divergence among Mitochondrial Lineages of Brycon (Characiformes, Bryconidae)


Abstract
1. Introduction
2. Materials and Methods
2.1. Study Area and Sample Collection
2.2. Extraction of the DNA, Amplification, Purification, and Sequencing
2.3. Data Analysis
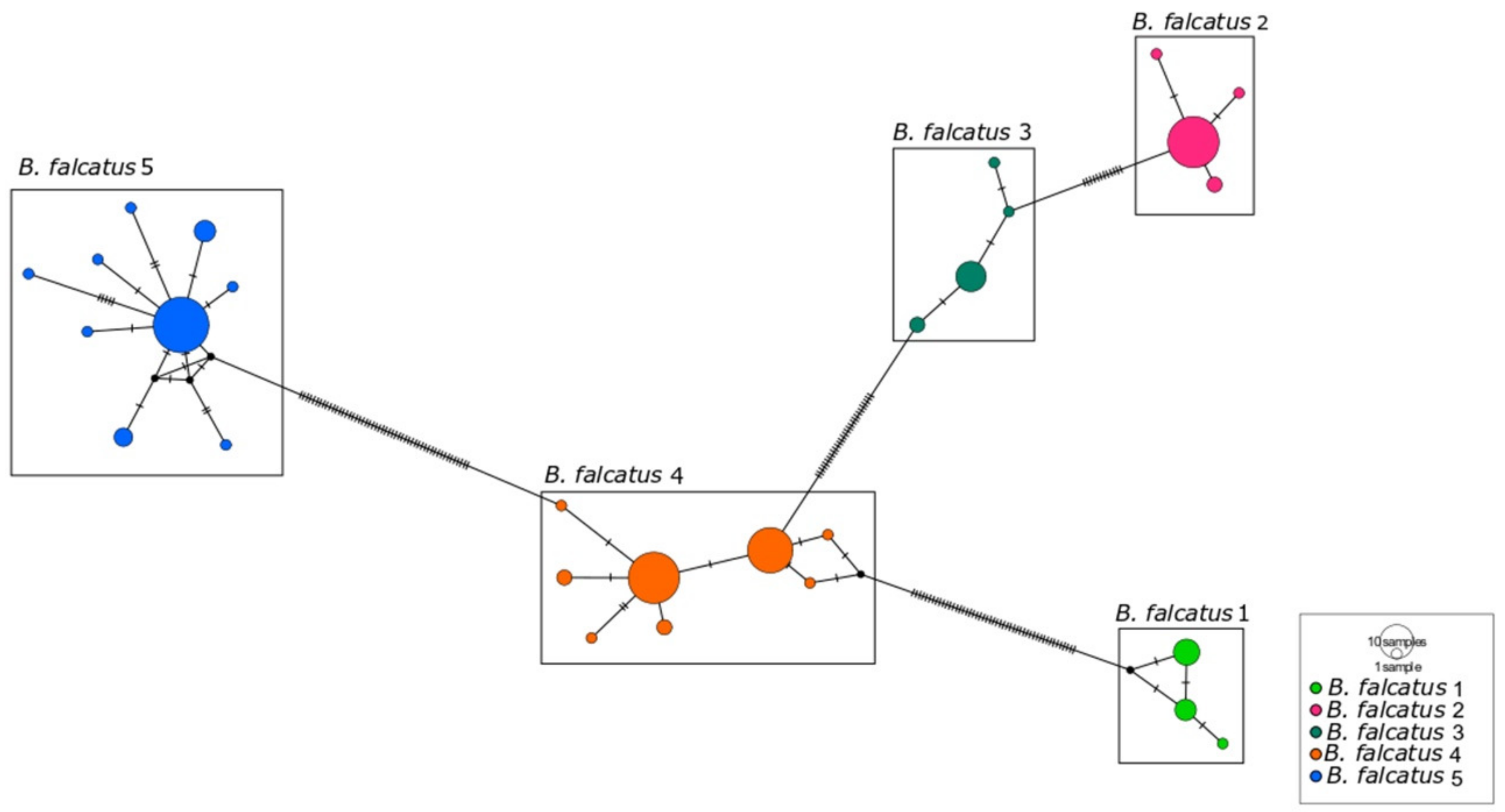
3. Results
3.1. Species Delimitation
3.2. Diversity Genetic of the B. falcatus, B. pesu, and B. amazonicus Lineages
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lima, F.C.T. A revision of the cis-andean species of the genus Brycon Müller & Troschel (Characiformes: Characidae). Zootaxa 2017, 4222, 1–189. [Google Scholar]
- Lima, F.C.T. Subfamily Bryconinae (Characins, Tetras). In Checklist of the Freshwater Fishes of South and Central America, 1st ed.; Reis, R.E., Kullander, S.O., Ferraris, C.J., Jr., Eds.; EDIPUCRS: Porto Alegre, Brazil, 2003; pp. 174–181. [Google Scholar]
- Lima, F.C.T.; Castro, R.M.C. Brycon vermelha, a new species of characid fish from the Rio Mucuri, a coastal river of eastern Brazil (Ostariophysi: Characiformes). Ichthyol. Explor. Freshwaters. 2000, 11, 155–162. [Google Scholar]
- Rodriguez-Rodriguez, M.P.; Lopera-Barrero, N.M.; Ribeiro, R.P.; Povh, J.A.; Vargas, L.; Sirol, R.N.; Jacometo, C.B. Diversidad genética de piracanjuba usada en programas de repoblación con marcadores microsatélites. Pesq. Agropec. Bras. 2010, 45, 56–63. [Google Scholar] [CrossRef]
- Goulding, M. The Fishes and the Forest. Explorations in Amazonian Natural History; University of California Press: Berkeley, CA, USA, 1980; p. 280. [Google Scholar]
- Gomiero, L.M.; Briani, D.C.; Giasson, L.O.M. Vertebrados consumidos por Brycon opalinus (Pisces, Characidae) em rios do Parque Estadual da Serra do Mar, SP. Biota Neotrop. 2006, 6, 1–5. [Google Scholar] [CrossRef]
- Gomiero, L.M.; Manzatto, A.G.B.; Braga, F.M.S. The role of riverine forests for food supply for the omnivorous fish Brycon opalinus Cuvier, 1819 (Characidae) in the Serra do Mar, Southeast Brazil. Braz. J. Biol. 2008, 68, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Hebert, P.D.N.; Cywinska, A.; Ball, S.L.; Dewaard, J.R. Biological identifications through DNA barcodes. Proc. R. Soc. Lond. 2003, 270, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.D.; Zemlak, T.S.; Innes, B.H.; Last, P.R.; Hebert, P.D.N. Dna barcoding Australia’s fish species. Philos. Trans. R. Soc. B 2005, 360, 1847–1857. [Google Scholar] [CrossRef] [PubMed]
- Mabragaña, E.; Díaz de Astarloa, J.M.; Hanner, R.; Zhang, J.; Castro, M.G. DNA Barcoding Identifies Argentine Fishes from Marine and Brackish Waters. PLoS ONE 2011, 6, e28655. [Google Scholar] [CrossRef]
- Hubert, N.; Hanner, R.; Holm, E.; Mandrak, N.E.; Taylor, E.; Burridge, M.; Watkinson, D.; Dumont, P.; Curry, A.; Bentzen, P.; et al. Identifying Canadian freshwater fishes through DNA barcodes. PLoS ONE 2008, 3, e2490. [Google Scholar] [CrossRef]
- Costa-Silva, G.J.; Rodriguez, M.S.; Roxo, F.F.; Foresti, F.; Oliveira, C. Using Different Methods to Access the Difficult Task of Delimiting Species in a Complex Neotropical Hyperdiverse Group. PLoS ONE 2015, 10, e0135075. [Google Scholar] [CrossRef]
- Roxo, F.F.; Ochoa, L.E.; Costa-Silva, G.J.; Oliveira, C. Species delimitation in Neoplecostomus (Siluriformes: Loricariidae) using morphologic and genetic approaches. DNA Barcodes. 2015, 3, 110–117. [Google Scholar] [CrossRef]
- Gomes, L.C.; Pessali, T.C.; Sales, N.G.; Pompeu, P.S.; Carvalho, D.C. Integrative taxonomy detects cryptic and overlooked fish species in a Neotropical river basin. Genetica 2015, 143, 581–588. [Google Scholar] [CrossRef]
- Shen, Y.; Guan, L.; Wang, D.; Gan, X. DNA barcoding and evaluation of genetic diversity in Cyprinidae fish in the midstream of the Yangtze River. Ecol. Evol. 2016, 6, 2702–2713. [Google Scholar] [CrossRef]
- Machado, C.D.B.; Ishizuka, T.K.; Freitas, P.D.D.; Valiati, V.H.; Galetti, P.M., Jr. DNA barcoding reveals taxonomic uncertainty in Salminus (Characiformes). Syst. Biodivers. 2016, 1–11. [Google Scholar] [CrossRef]
- Ribolli, J.; Scaranto, B.M.; Shibatta, O.A.; Bombardelli, R.Q.; Zaniboni-Filho, E. DNA barcoding confirms the occurrence of Rhamdia branneri and Rhamdia voulezi (Siluriformes: Heptapteridae) in the Iguaçu River Basin. Neotrop. Ichthyol. 2017, 15, e160147. [Google Scholar] [CrossRef]
- Laskar, B.A.; Kumar, V.; Kundu, S.; Darshan, A.; Tyagi, K.; Chandra, K. DNA barcoding of fishes from River Diphlu within Kaziranga National Park in northeast India. Mitochondrial DNA Part A 2019, 30, 126–134. [Google Scholar] [CrossRef]
- Aljanabi, S.M.; Martinez, L. Universal and rapid salt-extraction of high quality genomic DNA for PCR-based techniques. Nuleic. Acids. Research. 1997, 25, 4692–4693. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA 6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 406–425. [Google Scholar]
- Brown, S.D.J.; Collins, R.A.; Boyer, S.; Lefort, M.C.; Malumbres-Olarte, J.; Vink, C.J.; Cruickshank, R.H. Spider: An R package for the analysis of species identity and evolution, with particular reference to DNA barcoding. Mol. Ecol. Res. 2012, 12, 562–565. [Google Scholar] [CrossRef]
- Pons, J.; Barraclough, T.G.; Gomez-Zurita, J.; Cardoso, A.; Duran, D.P.; Hazell, S.; Kamoun, S.; Sumlin, W.D.; Vogler, A.P. Sequence-based species delimitation for the DNA taxonomy of undescribed insects. Syst. Biol. 2006, 55, 595–609. [Google Scholar] [CrossRef]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.-H.; Xie, D. Drummond AJ. BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS. Comput. Biol. 2014, 10, 1–6. [Google Scholar] [CrossRef]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUTi and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef]
- Rambaut, A.; Suchard, M.A.; Xie, D.; Drummond, A.J. Tracer v1.6. 2013. Available online: http://tree.bio.ed.ac.uk/software/tracer/ (accessed on 2 February 2019).
- Puillandre, N.; Lambert, A.; Brouillet, S.; Achaz, G. ABGD, Automatic Barcode Gap Discovery for primary species delimitation. Mol. Ecol. 2012, 21, 1864–1877. [Google Scholar] [CrossRef]
- Ronquist, F.; Huelsenbeck, J.F. MrBayes: Bayesian phylogenetic inferenceunder mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef]
- Zwickl, D.J. Genetic Algorithm Approaches for the Phylogenetic Analysis of Large Biological Sequence Datasets Under the Maximum Likelihood Criterion. Ph.D. Thesis, The University of Texas at Austin, Austin, TX, USA, 2006. [Google Scholar]
- Rambaut, A. FigTree v1.4.3. 2016. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 2 February 2019).
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Bandelt, H.J.; Forster, P.; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Leigh, J.W.; Bryant, D. POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Res. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Costa, F.O.; Landi, M.; Martins, R.; Costa, M.H.; Costa, M.E.; Carneiro, M.; Alves, M.J.; Steinke, D.; Carvalho, G.R. A Ranking System for Reference Libraries of DNA Barcodes: Application to Marine Fish Species from Portugal. PLoS ONE 2012, 7, 1–9. [Google Scholar] [CrossRef]
- Pereira, L.H.G.; Hanner, R.; Foresti, F.; Oliveira, C. Can DNA barcoding accurately discriminate megadiverse Neotropical freshwater fish fauna? BMC Genet. 2013, 14, 1–4. [Google Scholar] [CrossRef]
- Bellafronte, E.; Mariguela, T.C.; Pereira, L.H.G.; Oliveira, C.; Moreira-Filho, O. DNA barcode of Parodontidae species from the La Plata river basin—applying new data to clarify taxonomic problems. Neotrop. Ichthyol. 2013, 11, 497–506. [Google Scholar] [CrossRef]
- Knebelsberger, T.; Landi, M.; Neumann, H.; Kloppmann, K.; Sell, A.F.; Campbell, P.D.; Laakmann, S.; Raupach, M.J.; Carvalho, G.R.; Costa, F.O. A reliable DNA barcode reference library for the identification of the North European shelf fish fauna. Mol. Ecol. Res. 2014, 14, 1060–1071. [Google Scholar] [CrossRef]
- Landi, M.; Dimech, M.; Arculeo, M.; Biondo, G.; Martins, R.; Carneiro, M.; Carvalho, G.R.; Lo Brutto, S.; Costa, F.O. DNA Barcoding for Species Assignment: The Case of Mediterranean Marine Fishes. PLoS ONE 2014, 9, e106135. [Google Scholar] [CrossRef]
- Díaz, J.; Villanova, G.V.; Brancolini, F.; Del Pazo, F.; Posner, V.N.; Grimberg, A.; Arranz, E.S. First DNA barcode reference library for the identification of South American freshwater fish from the lower Paraná River. PLoS ONE 2016, 11, e0157419. [Google Scholar] [CrossRef]
- Serrano, E.A.; Melo, B.F.; Freitas-Souza, D.; Oliveira, M.L.M.; Utsunomia, R.; Oliveira, C.; Foresti, F. Species delimitation in neotropical fishes of the genus Characidium (Teleostei, Characiformes). Zoo Scr. 2018, 48, 1–12. [Google Scholar] [CrossRef]
- Mateussi, N.T.B.; Melo, B.F.; Forest, F.; Oliveira, C. Molecular Data Reveal Multiple Lineages in Piranhas of the Genus Pygocentrus (Teleostei, Characiformes). Genes 2019, 10, 371. [Google Scholar] [CrossRef]
- Ward, R.D. DNA barcode divergence among species and genera of birds and fishes. Mol. Ecol. Res. 2009, 9, 1077–1085. [Google Scholar] [CrossRef]
- Chen, W.; Ma, X.; Shen, Y.; Mao, Y.; He, S. The fish diversity in the upper reaches of the Salween River, Nujiang River, revealed by DNA barcoding. Sci. Rep. 2015, 5, 1–12. [Google Scholar] [CrossRef]
- Silva-Santos, R.; Ramirez, J.L.; Galetti, P.M., Jr.; Freitas, P.D. Molecular Evidences of a Hidden Complex Scenario in Leporinus cf. Friderici Front. Genet. 2018, 9, 1–9. [Google Scholar] [CrossRef]
- Abe, K.T.; Mariguela, T.C.; Avelino, G.S.; Foresti, F.; Oliveira, C. Systematic and historical biogeography of the Bryconidae (Ostariophysi: Characiformes) suggesting a new rearrangement of its genera and an old origin of Mesoamerican ichthyofauna. BMC Evol. Biol. 2014, 14, 1–15. [Google Scholar] [CrossRef]
- Travenzoli, N.M.; Silva, P.C.; Santos, U.; Zanuncio, J.C.; Oliveira, C.; Dergam, J.A. Cytogenetic and Molecular Data Demonstrate that the Bryconinae (Ostariophysi, Bryconidae) Species from Southeastern Brazil Form a Phylogenetic and Phylogeographic Unit. PLoS ONE. 2015, 10, e0137843. [Google Scholar] [CrossRef]
- Excoffier, L.; Smouse, P.E. Quattro JM. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source of the Variation | d.f. | Sum of Squares | Components of the Variance | % Variation |
|---|---|---|---|---|
| Among groups | 4 | 2626.838 | 25.06425 Va | 96.72 |
| Among populations within groups | 3 | 7.016 | 0.45465 Vb | 1.75 |
| Within populations | 131 | 51.779 | 0.39526 Vc | 1.53 |
| Source of the Variation | d.f. | Sum of Squares | Components of the Variance | % Variation |
|---|---|---|---|---|
| Among groups | 6 | 1271.181 | 20.05707 Va | 91.96 |
| Among populations within groups | 3 | 20.335 | 1.21388 Vb | 5.57 |
| Within populations | 76 | 41.055 | 0.54019 Vc | 2.48 |
| Source of the Variation | d.f. | Sum of Squares | Components of the Variance | % Variation |
|---|---|---|---|---|
| Among groups | 1 | 48.125 | 5.97295 Va | 97.52 |
| Among populations within groups | 2 | 0.417 | 0.04094 Vb | 0.67 |
| Within populations | 12 | 1.333 | 0.11111 Vc | 1.81 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arruda, P.S.S.; Ferreira, D.C.; Oliveira, C.; Venere, P.C. DNA Barcoding Reveals High Levels of Divergence among Mitochondrial Lineages of Brycon (Characiformes, Bryconidae). Genes 2019, 10, 639. https://doi.org/10.3390/genes10090639
Arruda PSS, Ferreira DC, Oliveira C, Venere PC. DNA Barcoding Reveals High Levels of Divergence among Mitochondrial Lineages of Brycon (Characiformes, Bryconidae). Genes. 2019; 10(9):639. https://doi.org/10.3390/genes10090639
Chicago/Turabian StyleArruda, Pábila S. S., Daniela C. Ferreira, Claudio Oliveira, and Paulo C. Venere. 2019. "DNA Barcoding Reveals High Levels of Divergence among Mitochondrial Lineages of Brycon (Characiformes, Bryconidae)" Genes 10, no. 9: 639. https://doi.org/10.3390/genes10090639
APA StyleArruda, P. S. S., Ferreira, D. C., Oliveira, C., & Venere, P. C. (2019). DNA Barcoding Reveals High Levels of Divergence among Mitochondrial Lineages of Brycon (Characiformes, Bryconidae). Genes, 10(9), 639. https://doi.org/10.3390/genes10090639