Transcriptome Landscape Variation in the Genus Thymus



Abstract
1. Introduction
2. Material and Methods
2.1. Ethics Statement
2.2. Plant Material
2.3. Genome Size Measures
2.4. RNA Extraction, Library Preparation and Sequencing
2.5. Data Processing, Transcriptome Assembly and Annotation
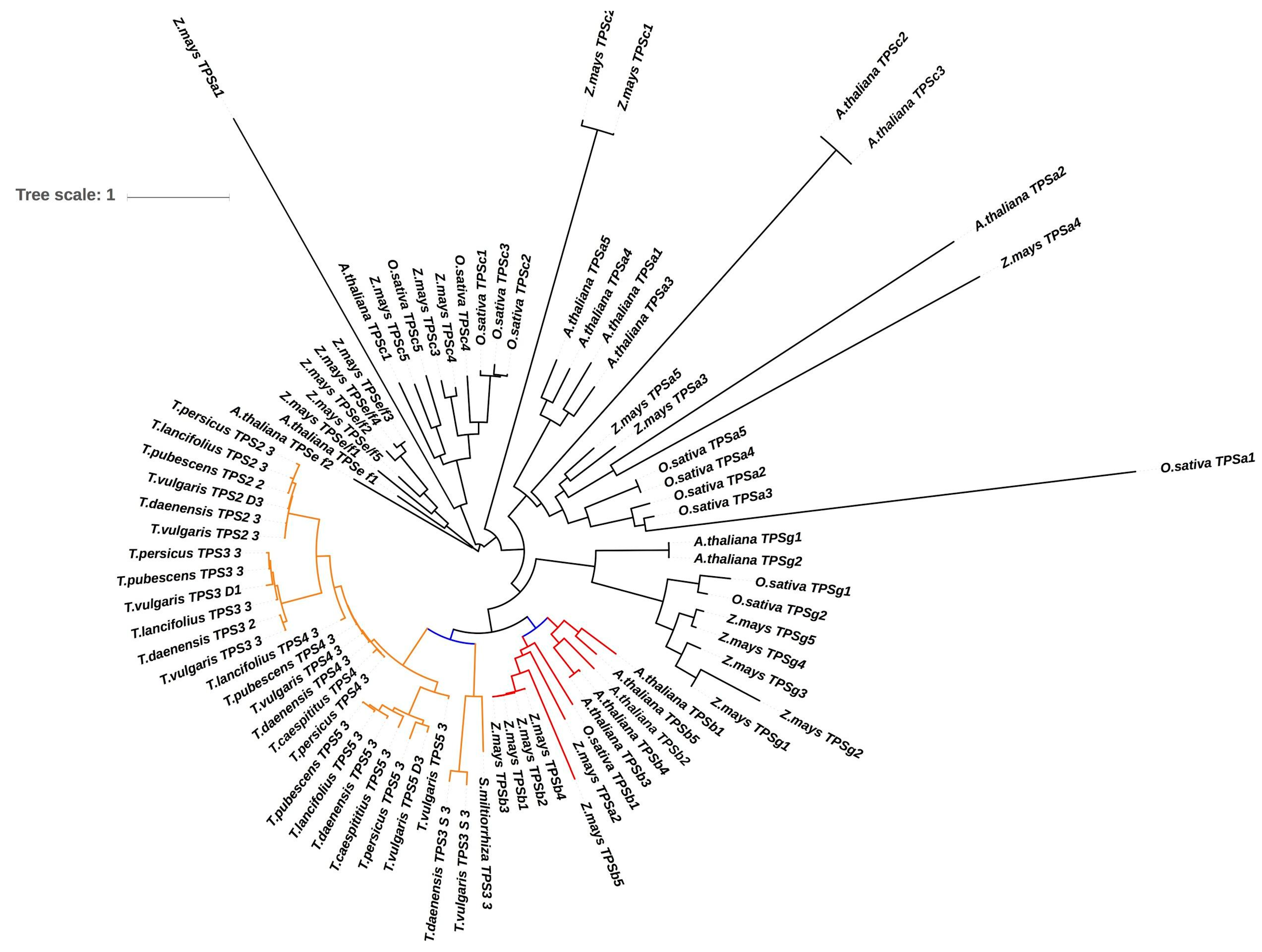
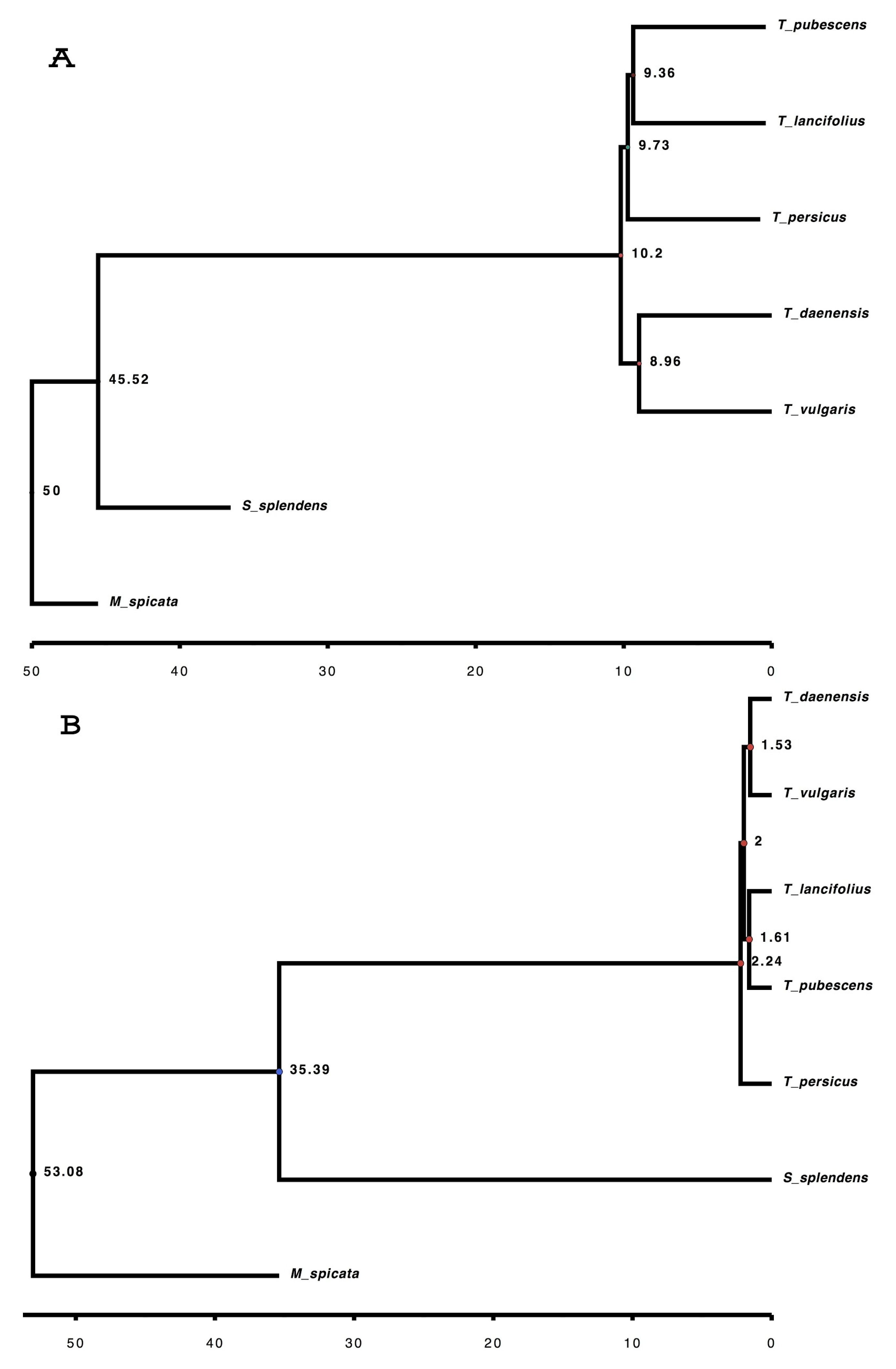
2.6. Identification of Orthologous Genes and Comparative Phylogenetic Analysis
2.7. Positive Selection Analysis
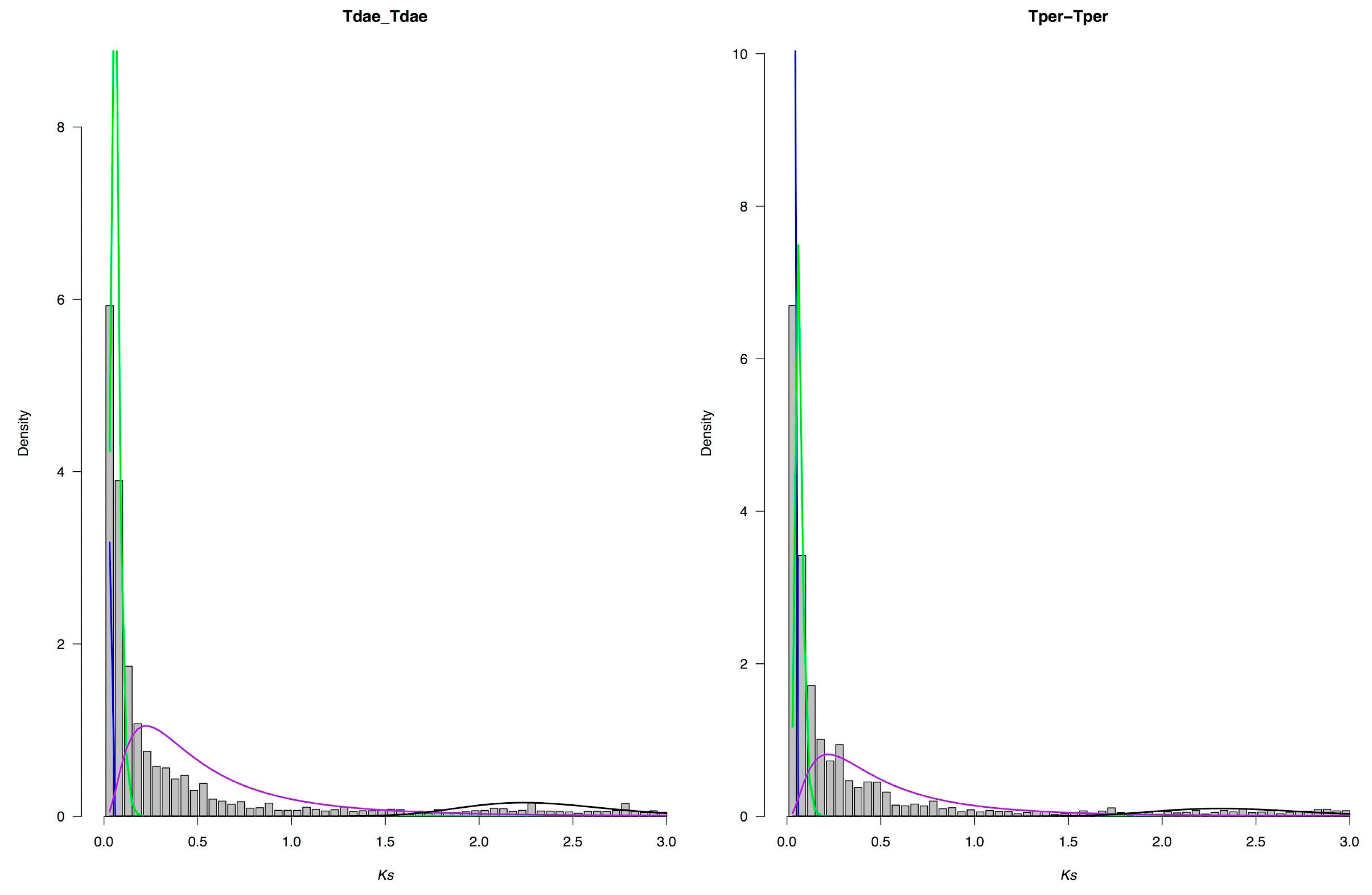
2.8. Ks Analysis for Whole Genome Duplication (WGD)
3. Results and Discussion
3.1. Genome Size Variation across the Genus Thymus
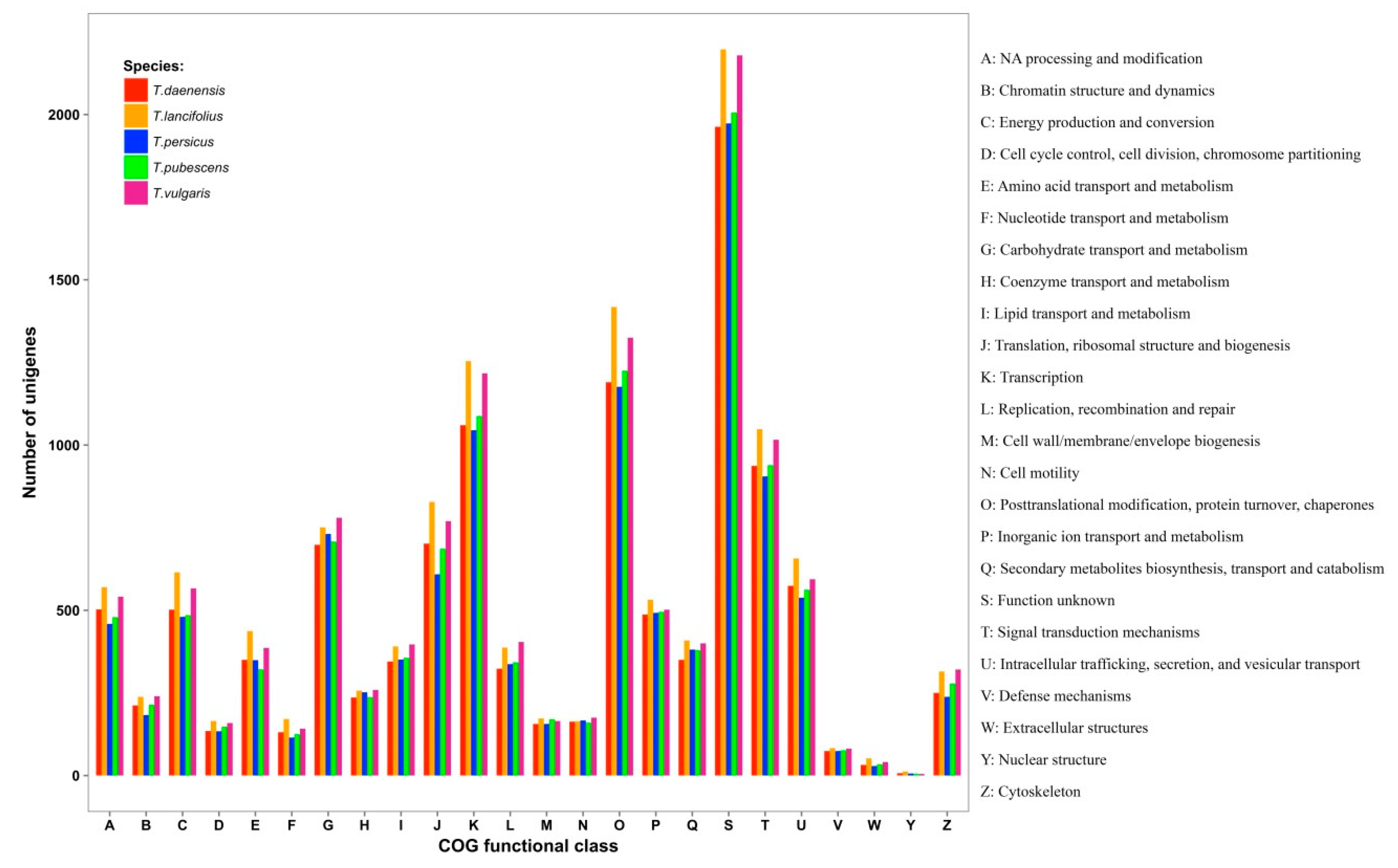
3.2. RNA Sequencing and De Novo Transcriptome Assembly
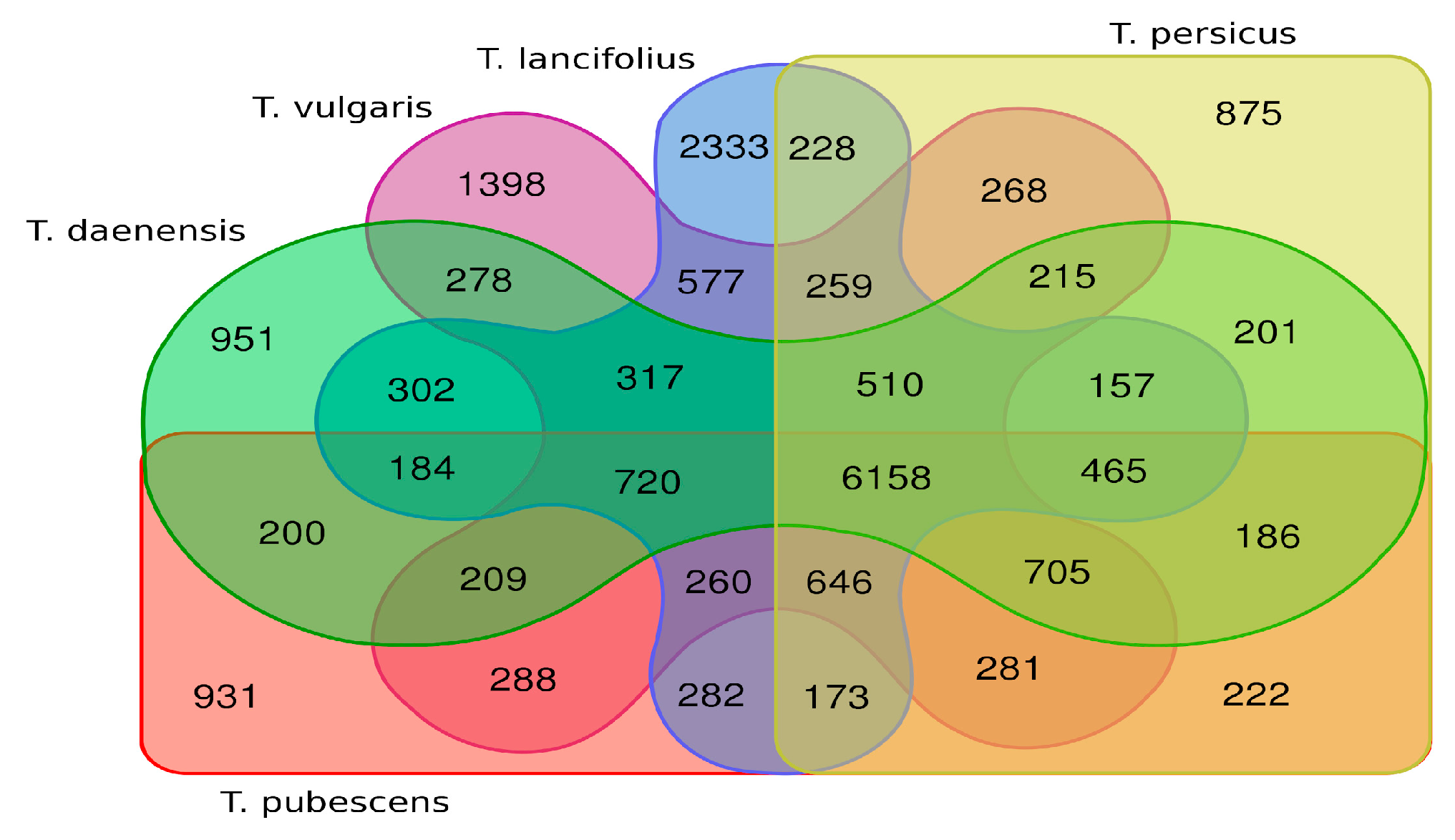
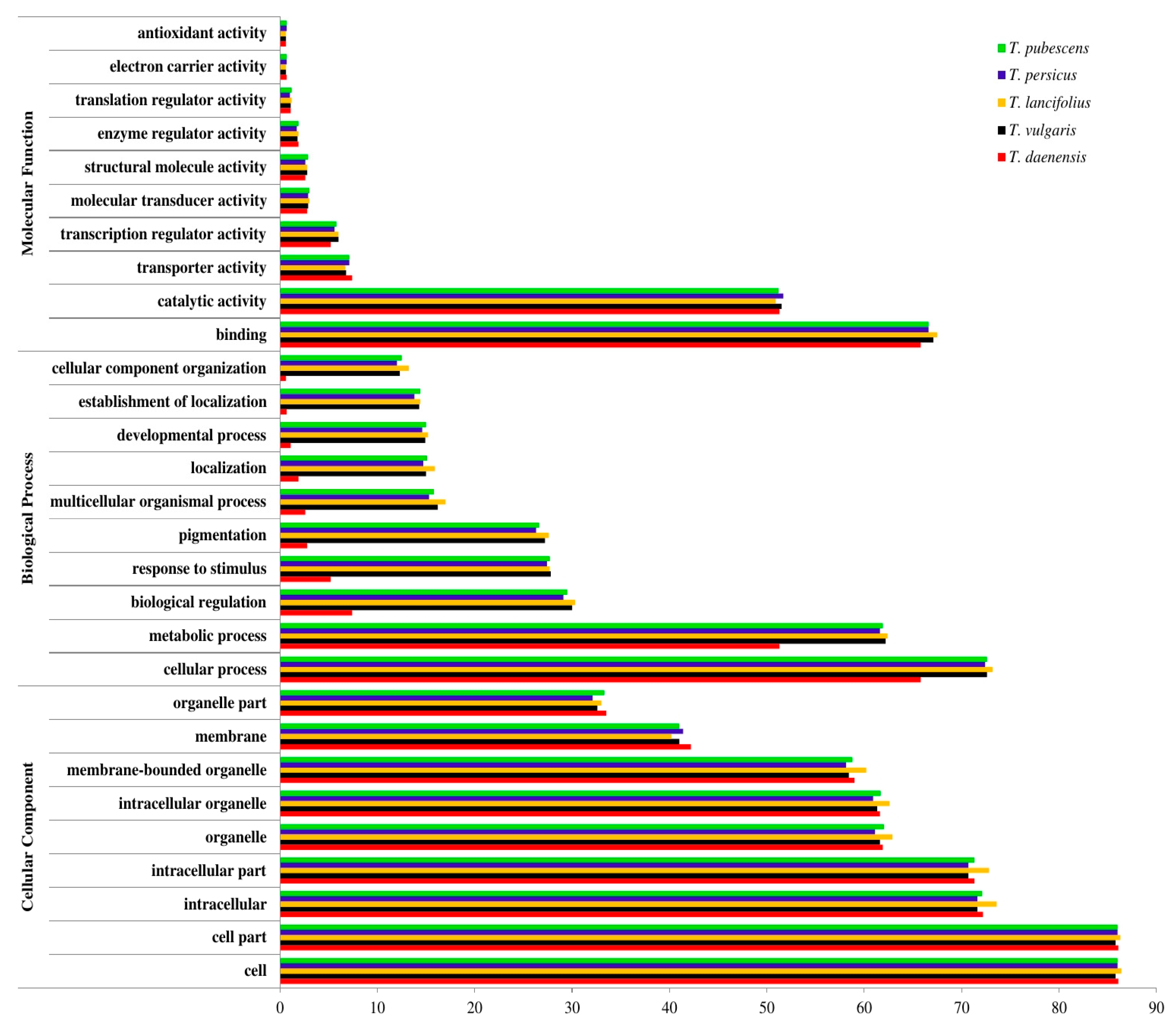
3.3. Database Searches and Gene Ontology
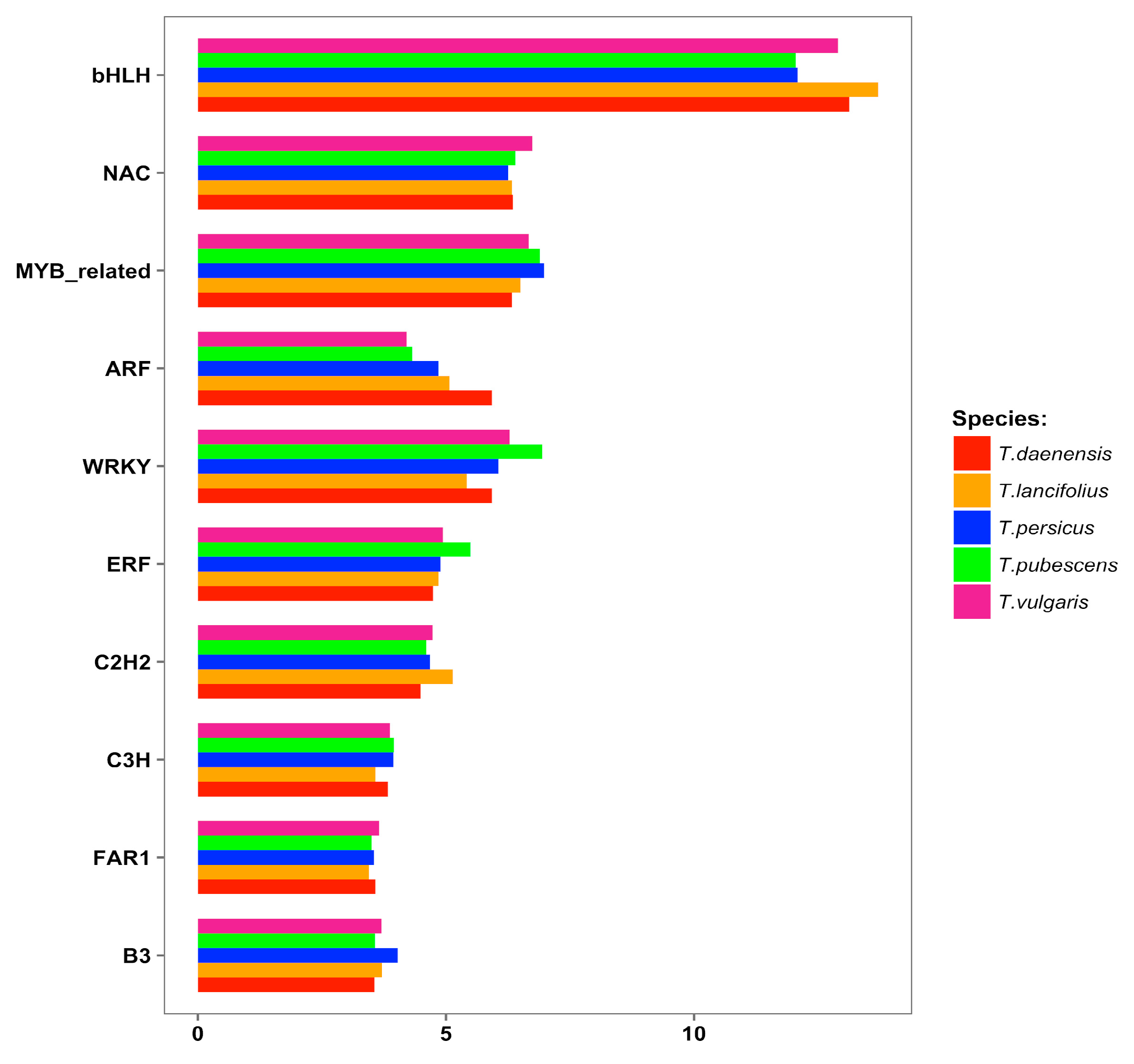
3.4. Identification of Transcription Factors
3.5. Thymol Biosynthetic Pathway
3.6. Orthogroups Identified, Phylogenetic Analysis and Finding Genes under Positive Selection
3.7. Whole Genome Duplication Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- World Checklist of Selected Plant Families. Available online: http://apps.kew.org/wcsp/incfamilies.do (accessed on 5 August 2018).
- Harley, M.R.; Atkins, S.; Budantsev, L.A.; Cantino, P.D.; Conn, B.J.; Grayer, R.; Harley, M.M.; de Kok, R.; Krestovskaja, T.; Morales, R.; et al. The Families and Genera of Vascular Plants; Kubitzki, K., Kadereit, J.W., Eds.; Springer: Berlin, Germany, 2004; Volume 6, pp. 167–275. [Google Scholar]
- Morales, R. The history, botany and taxonomy of the genus Thymus. In Stahl-Biskup; Saez, F., Ed.; Taylor & Francis: London, UK, 2002; pp. 1–43. [Google Scholar]
- Lajayer, H.M.; Zakizadeh, H.; Hamidoghli, Y.; Bigluei, M.H.; Chamani, E. Ornamental potential and freezing tolerance of six Thymus spp. species as ground-covering plants in the landscape. Zemdirb. Agric. 2018, 105, 79–88. [Google Scholar] [CrossRef]
- Leal, F.; Taghouti, M.; Nunes, F.; Silva, A.; Coelho, A.C.; Matos, M. Thymus Plants: A Review—Micropropagation, Molecular and Antifungal Activity. In Active Ingredients from Aromatic and Medicinal Plants; IntechOpen: Rijeka, Croatia, 2017. [Google Scholar]
- Hazzit, M.; Baaliouamer, A.; Verissimo, A.R.; Faleiro, M.L.; Miguel, M.G. Chemical composition and biological activities of Algerian Thymus oils. Food Chem. 2009, 116, 714–721. [Google Scholar] [CrossRef]
- Basch, E.; Ulbricht, C.; Hammerness, P.; Bevins, A.; Sollars, D. Thyme (Thymus vulgaris L.), thymol. J. Herbal Pharma 2004, 4, 49–67. [Google Scholar] [CrossRef]
- Thompson, J.D.; Chalchat, J.-C.; Michet, A.; Linhart, Y.B.; Ehlers, B. Qualitative and Quantitative Variation in Monoterpene Co-Occurrence and Composition in the Essential Oil of Thymus vulgaris Chemotypes. J. Chem. Ecol. 2003, 29, 859–880. [Google Scholar] [CrossRef] [PubMed]
- Karami-Osboo, R.; Miri, R.; Asadollahi, M.; Jassbi, A.R. Comparison Between Head-Space SPME and Hydrodistillation-GC-MS of the Volatiles of Thymus daenensis. J. Essent. Oil Bear. Plants 2015, 18, 925–930. [Google Scholar] [CrossRef]
- Sajjadi, S.E.; Khatamsaz, M. Composition of the Essential Oil of Thymus daenensis Celak. ssp. lancifolius (Celak.) Jalas. J. Essent. Oil Res. 2003, 15, 34–35. [Google Scholar] [CrossRef]
- Rasooli, I.; Mirmostafa, S.A. Antibacterial properties of Thymus pubescens and Thymus serpyllum essential oils. Fitoterapia 2002, 73, 244–250. [Google Scholar] [CrossRef]
- Hashemi-Moghaddam, H.; Mohammadhosseini, M.; Basiri, M. Optimization of Microwave Assisted Hydrodistillationon Chemical Compositions of the Essential Oils from the Aerial Parts of Thymus pubescens and Comparison with Conventional Hydrodistllation. TEoP 2015, 18, 884–893. [Google Scholar] [CrossRef]
- Rasooli, I.; Mirmostafa, S.A. Bacterial susceptibility to and chemical composition of essential oils from Thymus kotschyanus and Thymus persicus. J. Agric. Food Chem. 2003, 51, 2200–2205. [Google Scholar] [CrossRef]
- Thottathil, G.P.; Jayasekaran, K.; Othman, A.S. Sequencing Crop Genomes: A Gateway to Improve Tropical Agriculture. Trop Life Sci. Res. 2016, 27, 93–114. [Google Scholar]
- Pérez-de-Castro, A.M.; Vilanova, S.; Cañizares, J.; Pascual, L.; Blanca, J.M.; Díez, M.J.; Prohens, J.; Picó, B. Application of genomic tools in plant breeding. Curr. Genom. 2012, 13, 179–195. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Jain, M. RNA-Seq for Transcriptome Analysis in Non-model Plants. Adv. Struct. Saf. Stud. 2013, 1069, 43–58. [Google Scholar] [CrossRef]
- He, B.; Zhao, S.; Chen, Y.; Cao, Q.; Wei, C.; Cheng, X.; Zhang, Y. Optimal assembly strategies of transcriptome related to ploidies of eukaryotic organisms. BMC Genom. 2015, 16, 65. [Google Scholar] [CrossRef] [PubMed]
- De Laat, A.M.M.; Blaas, J. Flow-cytometric characterization and sorting of plant chromosomes. Theor. Appl. Genet. 1984, 67, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Dolezel, L.; Dolezel, J.; Bartos, J.; Voglmayr, H.; Greilhuber, J. Nuclear DNA content and genome size of trout and human. Cytom. Part A 2003, 128, 127–128. [Google Scholar] [CrossRef]
- Hunt, A.G. A rapid, simple and inexpensive method for the preparation of strand-specific RNA-Seq libraries. Methods Mol. Biol. 2015, 195–207. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Grabherr, M.G. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef]
- Schulz, M.H.; Zerbino, D.R.; Vingron, M.; Birney, E. Oases: Robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 2012, 28, 1086–1092. [Google Scholar] [CrossRef]
- Liu, J.; Li, G.; Chang, Z.; Yu, T.; Liu, B.; McMullen, R.; Chen, P.; Huang, X. BinPacker: Packing-Based De Novo Transcriptome Assembly from RNA-seq Data. PLoS Comput. Biol. 2016, 12, 1–15. [Google Scholar] [CrossRef]
- Chang, Z.; Li, G.; Liu, J.; Zhang, Y.; Ashby, C.; Liu, D.; Cramer, C.L.; Huang, X. Bridger: A new framework for de novo transcriptome assembly using RNA-seq data. Genome Biol 2015, 16, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Fillmore, N.; Bai, Y.; Collins, M.; Thomson, J.; Stewart, R.; Dewey, C.N. Evaluation of de novo transcriptome assemblies from RNA-Seq data. Genome Boil. 2014, 15, 663. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Punta, M.; Coggill, P.C.; Eberhardt, R.Y.; Mistry, J.; Tate, J.; Boursnell, C.; Pang, N.; Forslund, K.; Ceric, G.; Clements, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2011, 40, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Petersen, T.N.; Brunak, S.; Von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, È.; Von Heijne, G.; Sonnhammer, E.L.L. Predicting Transmembrane Protein Topology with a Hidden Markov Model: Application to Complete Genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; et al. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. eggnog 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016, 44, D286–D293. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015, 16, 1–14. [Google Scholar] [CrossRef]
- Ge, X.X.; Chen, H.W.; Wang, H.L.; Shi, A.P.; Liu, K.F. De novo assembly and annotation of Salvia splendens transcriptome using the illumina platform. PLoS ONE 2014, 9, e87693. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A.; Ludwig, T.; Meier, H. RAxML: A fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics 2005, 21, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Mirarab, S.; Warnow, T. ASTRAL-II: Coalescent-based species tree estimation with many hundreds of taxa and thousands of genes. Bioinformatics 2015, 31. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A.J. BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar]
- Rambaut, A.; Drummond, A.J. FigTree v1.4.0 in Computer Program and Documentation Distributed by the Author. 2012. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 5 August 2018).
- Suyama, M.; Torrents, D.; Bork, P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006, 34, 609–612. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Du, Z.; Zhou, X.; Ling, Y.; Zhang, Z.; Su, Z. agriGO: A GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010, 38, 64–70. [Google Scholar] [CrossRef]
- McKain, M.R.; Tang, H.; McNeal, J.R.; Ayyampalayam, S.; Davis, J.I.; dePamphilis, C.W.; Givnish, T.J.; Pires, J.C.; Stevenson, D.W.; Leebens-Mack, J.H. A Phylogenomic Assessment of Ancient Polyploidy and Genome Evolution across the Poales. Genome Biol. Evol. 2016, 8, 1150–1164. [Google Scholar] [CrossRef]
- Yang, Z.; Niele, R. Estimating Synonymous and Nonsynonymous Substitution Rates Under Realistic Evolutionary Models. Mol. Biol. Evol. 2000, 17, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Goldman, N.; Yang, Z. A Codon-based Model of Nucleotide Substitution for Protein-coding DNA Sequences. Mol. Bid. Evol. 1980, 11, 725–736. [Google Scholar] [CrossRef]
- Fraley, C.; Raftery, A.E.; Murphy, T.B.; Scrucca, L. mclust Version 4 for R: Normal Mixture Modeling for Model-Based Clustering, Classification, and Density Estimation. Tech. Rep. 2012, 597, 1–50. [Google Scholar]
- Marie, D.; Brown, S.C. A cytometric exercise in plant DNA histograms, with 2C values for 70 species. Biol. Cell 1993, 78, 41–51. [Google Scholar] [CrossRef]
- Mahdavi, S.; Karimzadeh, G. Karyological and Nuclear DNA Content Variation in Some Iranian Endemic Thymus Species (Lamiaceae). J. Agric. Sci. Technol 2014, 12, 447–458. [Google Scholar]
- Šmarda, P.; Bureš, P.; Horová, L.; Rotreklová, O. Intrapopulation Genome Size Dynamics in Festuca pallens. Ann. Bot. 2008, 102, 599–607. [Google Scholar] [CrossRef]
- Jamzad, Z. New species and new plant records of lamiaceae from Iran. Iran J. Bot. 2009, 15, 51–56. [Google Scholar]
- Ohri, D. Genome Size Variation and Plant Systematics. Ann. Bot. 1998, 82, 75–83. [Google Scholar] [CrossRef]
- Moreton, J.; Dunham, S.P.; Emes, R.D. A consensus approach to vertebrate de novo transcriptome assembly from RNA-seq data: Assembly of the duck (Anas platyrhynchos) transcriptome. Front. Genet. 2014, 5, 1–6. [Google Scholar] [CrossRef]
- Wang, S.; Gribskov, M. Comprehensive evaluation of de novo transcriptome assembly programs and their effects on differential gene expression analysis. Bioinformatics 2016, 33, 327–333. [Google Scholar] [CrossRef]
- Rana, S.B.; Zadlock, F.J.; Zhang, Z.; Murphy, W.R.; Bentivegna, C.S. Comparison of de Novo transcriptome assemblers and k-mer strategies using the killifish, Fundulus heteroclitus. PLoS ONE 2016, 11, e0153104. [Google Scholar] [CrossRef] [PubMed]
- Smith-Unna, R.D.; Boursnell, C.; Patro, R.; Hibberd, J.M.; Kelly, S.; Street, D.; Brook, S.; Road, S.P. TransRate: Reference free quality assessment of de novo transcriptome assemblies. Genome Res. 2016, 26, 021626. [Google Scholar] [CrossRef] [PubMed]
- Zeng, V.; Villanueva, K.E.; Ewen-Campen, B.S.; Alwes, F.; Browne, W.E.; Extavour, C.G. De novo assembly and characterization of a maternal and developmental transcriptome for the emerging model crustacean Parhyale hawaiensis. BMC Genom. 2011, 12, 581. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, S.T.; Emrich, S.J. Assessing De Novo transcriptome assembly metrics for consistency and utility. BMC Genom. 2013, 14, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chopra, R.; Burow, G.; Farmer, A.; Mudge, J.; Simpson, C.E.; Burow, M.D. Comparisons of de novo transcriptome assemblers in diploid and polyploid species using peanut (Arachis spp.) RNA-Seq data. PLoS ONE 2014, 9, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Huang, Q.; Wu, X.; Zhou, Z.; Ding, M.; Shi, M.; Huang, F.; Li, S.; Wang, Y.; Kai, G. Comprehensive transcriptome profiling of Salvia miltiorrhiza for discovery of genes associated with the biosynthesis of tanshinones and phenolic acids. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bustos-Segura, C.; Padovan, A.; Kainer, D.; Foley, W.J.; Külheim, C.; Bustos-Segura, C. Transcriptome analysis of terpene chemotypes of Melaleuca alternifolia across different tissues. Plant Cell Environ. 2017, 40, 2406–2425. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zheng, Y.; Zhong, Y.; Wu, Y.; Li, Z.; Xu, L.A.; Xu, M. Transcriptome analysis and identification of genes related to terpenoid biosynthesis in Cinnamomum camphora. BMC Genom. 2018, 19, 1–15. [Google Scholar] [CrossRef]
- Meena, S.; Rajeev Kumar, S.; Dwivedi, V.; Kumar Singh, A.; Chanotiya, C.S.; Akhtar, M.Q.; Kumar, K.; Kumar Shasany, A.; Nagegowda, D.A. Transcriptomic insight into terpenoid and carbazole alkaloid biosynthesis, and functional characterization of two terpene synthases in curry tree (Murraya koenigii). Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Ashour, M.; Wink, M.; Gershenzon, J. Biochemistry of Terpenoids: Monoterpenes, Sesquiterpenes and Diterpenes. Annu. Plant Rev. 2010, 40, 258–303. [Google Scholar] [CrossRef]
- Singh, B.; Sharma, R.A. Plant terpenes: Defense responses, phylogenetic analysis, regulation and clinical applications. Biotech 2015, 5, 129–151. [Google Scholar] [CrossRef] [PubMed]
- Abousaber, M.; Khanavi, M.; Khoshchehreh, M.; Hadjiakhoondi, A.; Shams Ardekani, M.R. Composition of the Essential Oils of Thymus deanensis Celak Var. deanensis from Different Regions of Iran. J. Med. Plants 2012, 11, 34–39. [Google Scholar]
- Tohidi, B.; Rahimmalek, M.; Arzani, A. Essential oil composition, total phenolic, flavonoid contents, and antioxidant activity of Thymus species collected from different regions of Iran. Food Chem. 2017, 220, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Hong, G.J.; Xue, X.Y.; Mao, Y.B.; Wang, L.J.; Chen, X.Y. Arabidopsis MYC2 Interacts with DELLA Proteins in Regulating Sesquiterpene Synthase Gene Expression. Plant Cell 2012, 24, 2635–2648. [Google Scholar] [CrossRef] [PubMed]
- Zvi, M.M. B.; Shklarman, E.; Masci, T.; Kalev, H.; Debener, T.; Shafir, S.; Ovadis, M.; Vainstein, A. PAP1 transcription factor enhances production of phenylpropanoid and terpenoid scent compounds in rose flowers. New Phytol. 2012, 195, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Xiao, J.; Shen, Y.; Ma, D.; Li, Z.; Pu, G.; Li, X.; Huang, L.; Liu, B.; Ye, H.; et al. Cloning and characterization of AabHLH1, a bHLH transcription factor that positively regulates artemisinin biosynthesis in Artemisia annua. Plant Cell Physiol. 2014, 55, 1592–1604. [Google Scholar] [CrossRef] [PubMed]
- Spyropoulou, E.A.; Haring, M.A.; Schuurink, R.C. RNA sequencing on Solanum lycopersicum trichomes identifies transcription factors that activate terpene synthase promoters. BMC Genom. 2014, 15, 1–16. [Google Scholar] [CrossRef]
- Mertens, J.; Pollier, J.; Vanden Bossche, R.; Lopez-Vidriero, I.; Franco-Zorrilla, J.M.; Goossens, A. The bHLH Transcription Factors TSAR1 and TSAR2 Regulate Triterpene Saponin Biosynthesis in Medicago truncatula. Plant Physiol. 2016, 170, 194–210. [Google Scholar] [CrossRef]
- Mertens, J.; Van Moerkercke, A.; Bossche, R.V.; Pollier, J.; Goossens, A. Clade IVa basic helix-loop-helix transcription factors form part of a conserved jasmonate signaling circuit for the regulation of bioactive plant terpenoid biosynthesis. Plant Cell Physiol. 2016, 57, 2564–2575. [Google Scholar] [CrossRef]
- Xu, Y.H. Characterization of GaWRKY1, a Cotton Transcription Factor That Regulates the Sesquiterpene Synthase Gene (+)–Cadinene Synthase-A. Plant Physiol. 2004, 135, 507–515. [Google Scholar] [CrossRef]
- Yokotani, N.; Sato, Y.; Tanabe, S.; Chujo, T.; Shimizu, T.; Okada, K.; Yamane, H.; Shimono, M.; Sugano, S.; Takatsuji, H.; et al. WRKY76 is a rice transcriptional repressor playing opposite roles in blast disease resistance and cold stress tolerance. J. Exp. Bot. 2013, 64, 5085–5097. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Allan, A.C.; Li, C.; Wang, Y.; Yao, Q. De Novo assembly and characterization of the transcriptome of the Chinese medicinal herb, Gentiana rigescens. Int. J. Mol. Sci. 2015, 16, 11550–11573. [Google Scholar] [CrossRef] [PubMed]
- Nieuwenhuizen, N.J.; Chen, X.; Wang, M.Y.; Matich, A.J.; Perez, R.L.; Allan, A.C.; Green, S.A.; Atkinson, R.G. Natural Variation in Monoterpene Synthesis in Kiwifruit: Transcriptional Regulation of Terpene Synthases by NAC and ETHYLENE-INSENSITIVE3-Like Transcription Factors. Plant Physiol. 2015, 167, 1243–1258. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.X.; Li, J.X.; Yang, C.Q.; Hu, W.L.; Wang, L.J.; Chen, X.Y. The jasmonate-responsive AP2/ERF transcription factors AaERF1 and AaERF2 positively regulate artemisinin biosynthesis in Artemisia annua L. Mol. Plant 2012, 5, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, K.; Parthier, C.; Egerer-Sieber, C.; Geiger, D.; Muller, Y.A.; Kreis, W.; Müller-Uri, F. Expression, crystallization and structure elucidation of γ-terpinene synthase from Thymus vulgaris. Acta Crystallogr. Sect. F Struct. Boil. Commun. 2016, 72, 16–23. [Google Scholar] [CrossRef]
- Trindade, H.; Pedro, L.G.; Figueiredo, A.C.; Barroso, J.G. Chemotypes and terpene synthase genes in Thymus genus: State of the art. Ind. Crop Prod. 2018, 124, 530–547. [Google Scholar] [CrossRef]
- Chen, F.; Tholl, D.; Bohlmann, J.; Pichersky, E. The family of terpene synthases in plants: A mid-size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Plant J. 2011, 66, 212–229. [Google Scholar] [CrossRef] [PubMed]
- Mendes, M.D.D.S.; Barroso, J.G.; Oliveira, M.M.; Trindade, H. Identification and characterization of a second isogene encoding γ-terpinene synthase in Thymus caespititius. J. Plant Physiol. 2014, 171, 1017–1027. [Google Scholar] [CrossRef]
- Lima, A.S.; Schimmel, J.; Lukas, B.; Novak, J.; Barroso, J.; Figueiredo, A.C.; Pedro, L.; Degenhardt, J.; Trindade, H. Genomic characterization, molecular cloning and expression analysis of two terpene synthases from Thymus caespititius (Lamiaceae). Planta 2013, 238, 191–204. [Google Scholar] [CrossRef]
- Julia, A.; Christina, N. The Molecular Regulation of Terpene Polymorphism in Thymus Vulgaris. Ph.D. Thesis, Friedrich-Schiller-Universität, Jena, Germany, 2014. [Google Scholar]
- Filipe, A.; Cardoso, J.C.; Miguel, G.; Anjos, L.; Trindade, H.; Figueiredo, A.C.; Barroso, J.; Power, D.M.; Marques, N.T. Molecular cloning and functional characterization of a monoterpene synthase isolated from the aromatic wild shrub Thymus albicans. J. Plant Physiol. 2017, 218, 35–44. [Google Scholar] [CrossRef]
- He, S.; Wang, X.; Yang, S.; Dong, Y.; Zhao, Q.; Yang, J.; Cong, K.; Zhang, J.; Zhang, G.; Wang, Y.; et al. De novo Transcriptome Characterization of Rhodomyrtus tomentosa Leaves and Identification of Genes Involved in α/β-Pinene and β-Caryophyllene Biosynthesis. Front Plant Sci. 2018, 9, 1231. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Yu, R.; Fan, Y. Transcriptome profiling provides new insights into the formation of floral scent in Hedychium coronarium. BMC Genom. 2015, 16, 470. [Google Scholar] [CrossRef] [PubMed]
- Zapata, F.; Fine, P.V.A. Diversification of the monoterpene synthase gene family (TPSb) in Protium, a highly diverse genus of tropical trees. Mol. Phylo Genet. Evol. 2013, 68, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Priya, P.; Yadav, A.; Chand, J.; Yadav, G. Terzyme: A tool for identification and analysis of the plant terpenome. Plant Methods 2018, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, J.; Kollner, T.G.; Gershenzon, J. Monoterpene and sesquiterpene synthases and the origin of terpene skeletal diversity in plants. Phytochemistry 2009, 70, 1621–1637. [Google Scholar] [CrossRef] [PubMed]
- Boutanaev, A.M.; Moses, T.; Zi, J.; Nelson, D.R.; Mugford, S.T.; Peters, R.J.; Osbourn, A. Investigation of terpene diversification across multiple sequenced plant genomes. Proc. Natl. Acad. Sci. USA 2015, 112, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Krause, S. Biosynthesis of Oxygenated Monoterpenes in Thyme, Melaleuca, and Eucalyptus Species. Ph.D. Dissertation, Martin-Luther-Universität Halle-Wittenberg, Halle (Saale), Germany, 2016. [Google Scholar]
- Schäferhoff, B.; Fleischmann, A.; Fischer, E.; Albach, D.C.; Borsch, T.; Heubl, G.; Müller, K.F. Towards resolving Lamiales relationships: Insights from rapidly evolving chloroplast sequences. BMC Evol. Biol. 2010, 10, 352. [Google Scholar] [CrossRef] [PubMed]
- Federici, S.; Galimberti, A.; Bartolucci, F.; Bruni, I.; De mattia, F.; Cortis, P. DNA barcoding to analyse taxonomically complex groups in plants: The case of Thymus (Lamiaceae). Bot. J. Linn. Soc. 2013, 171, 687–699. [Google Scholar] [CrossRef]
- Trachana, K.; Larsson, T.A.; Powell, S.; Chen, W.H.; Doerks, T.; Muller, J.; Bork, P. Orthology prediction methods: A quality assessment using curated protein families. Bioessays 2011, 33, 769–780. [Google Scholar] [CrossRef]
- Rustaiee, AR.; Yavari, A.; Nazeri, V.; Shokrpour, M.; Sefidkon, F.; Rasouli, M. Genetic diversity and chemical polymorphism of some Thymus species. Chem. Biodivers. 2013, 10, 1088–1098. [Google Scholar] [CrossRef]
- Drew, B.T.; Sytsma, K.J. Phylogenetics, biogeography, and staminal evolution in the tribe Mentheae (Lamiaceae). Am. J. Bot. 2012, 99, 933–953. [Google Scholar] [CrossRef]
- Parham, J.F.; Donoghue, P.C.J.; Bell, C.J.; Calway, T.D.; Head, J.J.; Holroyd, P.A. Best practices for justifying fossil calibrations. Syst. Biol. 2012, 61, 346–359. [Google Scholar] [CrossRef] [PubMed]
- Buckley, T.R.; Simon, C.; Chambers, K. Exploring among-site rate variation models in a maximum likelihood framework sing empirical data: Effects of model assumptions on estimates of topology, branch lengths, and bootstrap support. Syst. Biol. 2001, 50, 67–86. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.Y.W.; Duchene, S. Molecular clock methods for estimating evolutionary rates and timescales. Mol. Ecol. 2014, 23, 5947–5965. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.H. Adaptive evolution in two large families of ubiquitin-ligase adapters in nematodes and plants. Genome Res. 2006, 16, 1017–1030. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Quezada, A.; Schumann, N.; Quint, M. Plant F-Box protein evolution is determined by lineage-specific timing of major gene family expansion waves. PLoS ONE 2013, 8, e68672. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Kalluri, U.C.; Jawdy, S.; Gunter, L.E.; Yin, T.; Tschaplinski, T.J.; Weston, D.J.; Ranjan, P.; Tuskan, G.A. The F-Box Gene Family Is Expanded in Herbaceous Annual Plants Relative to Woody Perennial Plants1[W][OA]. Plant Physiol. 2008, 148, 1189–1200. [Google Scholar] [CrossRef]
- Cushing, D.A.; Forsthoefel, N.R.; Gestaut, D.R.; Vernon, D.M. Arabidopsis emb175 and other ppr knockout mutants reveal essential roles for pentatricopeptide repeat (PPR) proteins in plant embryogenesis. Planta 2005, 221, 424–436. [Google Scholar] [CrossRef]
- Manicacci, D.; Atlan, A.; Rossello, J.A.E.; Couvet, D. Gynodioecy and reproductive trait variation in three Thymus species (Lamiaceae). Int. J. Plant Sci. 1998, 159, 948–957. [Google Scholar] [CrossRef]
- Geddy, R.; Brown, G.G. Genes encoding pentatricopeptide repeat (PPR) proteins are not conserved in location in plant genomes and may be subject to diversifying selection. BMC Genom. 2007, 8, 130. [Google Scholar] [CrossRef]
- Fujii, S.; Bond, C.S.; Small, I.D. Selection patterns on restorer-like genes reveal a conflict between nuclear and mitochondrial genomes throughout angiosperm evolution. Proc. Natl. Acad. Sci. USA 2011, 108, 1723–1728. [Google Scholar] [CrossRef] [PubMed]
- Blanc, G.; Wolfe, K.H. Widespread Paleopolyploidy in Model Plant Species Inferred from Age Distributions of Duplicate Genes. Plant Cell 2004, 16, 1667–1678. [Google Scholar] [CrossRef] [PubMed]
- Mint Evolutionary Genomics Consortium. Phylogenomic mining of the mints reveals multiple mechanisms contributing to the evolution of chemical diversity in Lamiaceae. Mol. Plant 2018, 11, 1084–1096. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Location (Province) | 1C-Value ± SD (pg) | Haploid Size (Mbp) | Estimated Polyploidy |
|---|---|---|---|---|
| Thymus pubescens | East Azerbaijan | 0.63 ± 0.02 | 616.14 | 2n = 2x |
| T. pubescens | West Azerbaijan | 1.11 ± 0.01 | 1085.84 | 2n = 4x |
| Thymus migricus | West Azerbaijan | 0.61 ± 0.01 | 596.72 | 2n = 2x |
| Thymus daenensis | Lorestan | 0.56 ± 0.01 | 547.81 | 2n = 2x |
| T. daenensis | Fereydunshahr | 0.55 ± 0.27 | 538.03 | 2n = 2x |
| T. daenensis | Arak | 0.54 ± 0.28 | 528.25 | 2n = 2x |
| Thymus persicus | West Azerbaijan | 1.08 ± 0.01 | 1056.49 | 2n = 2x |
| Thymus kotschyanus | Qazvin | 1.09 ± 0.05 | 1066.28 | 2n = 2x |
| T. kotschyanus | Qazvin | 1.09 ± 0.04 | 1066.28 | 2n = 2x |
| Thymus lancifolius | Isfahan | 0.56 ± 0.01 | 547.81 | 2n = 2x |
| T. lancifolius | Kordestan | 1.11 ± 0.04 | 1085.84 | 2n = 4x |
| Thymus vulgaris | Cultivated | 0.82 ± 0.02 | 802.15 | 2n = 2n |
| Thymus fedtschenkoi | Semnan | 0.61 ± 0.01 | 596.72 | 2n = 2x |
| T. fedtschenkoi | Yazd | 1.17 ± 0.02 | 1144.54 | 2n = 4x |
| Thymus transcaspicus | Khorasan | 1.09 ± 0.02 | 1066.28 | 2n = 2x |
| T. transcaspicus | Yazd | 1.07 ± 0.02 | 1046.71 | 2n = 2x |
| Thymus fallax | Yazd | 1.14 ± 0.02 | 1115.19 | 2n = 2x |
| Thymus carmanicus | Kerman | 1.08 ± 0.02 | 1056.49 | 2n = 2x |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soorni, A.; Borna, T.; Alemardan, A.; Chakrabarti, M.; Hunt, A.G.; Bombarely, A. Transcriptome Landscape Variation in the Genus Thymus. Genes 2019, 10, 620. https://doi.org/10.3390/genes10080620
Soorni A, Borna T, Alemardan A, Chakrabarti M, Hunt AG, Bombarely A. Transcriptome Landscape Variation in the Genus Thymus. Genes. 2019; 10(8):620. https://doi.org/10.3390/genes10080620
Chicago/Turabian StyleSoorni, Aboozar, Tahereh Borna, Ali Alemardan, Manohar Chakrabarti, Arthur G. Hunt, and Aureliano Bombarely. 2019. "Transcriptome Landscape Variation in the Genus Thymus" Genes 10, no. 8: 620. https://doi.org/10.3390/genes10080620
APA StyleSoorni, A., Borna, T., Alemardan, A., Chakrabarti, M., Hunt, A. G., & Bombarely, A. (2019). Transcriptome Landscape Variation in the Genus Thymus. Genes, 10(8), 620. https://doi.org/10.3390/genes10080620