1. Introduction
Inherited forms of recurrent pneumonia or inflammatory airway disease are often due to primary ciliary dyskinesia (PCD) in humans and domestic animals. PCD is characterized by a loss of function of the motile cilia, not only in the lung, but also in the paranasal sinuses, the middle ear, sperm, the female reproductive tract and the ependyma of the brain. A defect in the structure or the function of the cilia typically leads to recurring infections of the upper and lower respiratory tract. The underlying pathogenesis is a decreased mucociliary clearance of dust and infectious agents [
1]. Variants in over 40 genes are known to cause PCD in humans [
2,
3,
4]. PCD causative genetic variants were also reported in purebred dogs. Variants in
CCDC39 [
5] and
NME5 [
6] were identified in PCD affected Old English Sheepdogs and Alaskan Malamutes, respectively. Despite the extensive knowledge about PCD, there are also clinically described forms of inherited recurrent inflammatory airway disease in humans and dogs, whose underlying genetic cause is still unknown [
7,
8]. Changes to the cilia may also result from respiratory disease and are then termed secondary ciliary dyskinesia (SCD). Discrimination of PCD and SCD based on clinical signs and/or pathological findings can be challenging and is not routinely available in veterinary medicine [
9].
Dog owners recently noticed several closely related Rough Collies with recurrent pulmonary disease, which clinically resembled canine forms of PCD. The goal of this study was to identify the underlying causative genetic defect.
2. Materials and Methods
2.1. Ethics Statement
All animal experiments were performed according to local regulations. All dogs in this study were privately owned and examined with the consent of their owners. The “Cantonal Committee for Animal Experiments” approved the collection of blood samples (Canton of Bern; permit 75/16).
2.2. Animal Selection
This study included 88 Rough Collies. Three related Rough Collies, two of them being half siblings, had recurrent pulmonary disease as reported by the owners. The other 85 dogs were reported as unaffected by their owners. EDTA blood samples were collected for DNA isolation. We also used 539 dogs of various other breeds, which had been donated to the Vetsuisse Biobank (
Table S1). They represented population controls without reports of recurrent pulmonary diseases.
2.3. DNA Extraction and Genotyping
Genomic DNA was isolated from EDTA blood with the Maxwell RSC Whole Blood Kit using a Maxwell RSC instrument (Promega, Dübendorf, Switzerland). Ten Rough Collies consisting of 3 cases and 7 control dogs from an extended family were genotyped on the Illumina canine_HD BeadChips containing 220,853 markers (Neogen, Lincoln, NE, USA).
2.4. Linkage and Homozygosity Mapping
The genotype data of 3 non-affected parents, 2 affected and 4 non-affected offspring in two litters were used for a parametric linkage analysis. Using PLINK v 1.09 [
10], markers that were non-informative, located on the sex chromosomes, or missing in any of the nine dogs, containing Mendel errors, or having a minor allele frequency <0.05 were excluded. The final dataset contained 65,446 markers. Parametric linkage analysis using an autosomal recessive inheritance model with full penetrance and a disease allele frequency of 0.4 was performed with the Merlin software [
11].
For homozygosity mapping, the genotype data of the three affected dogs were used. The --homozyg and --homozyg -group options in PLINK were used to search for extended regions of homozygosity >1 Mb. The output intervals were matched against the intervals from linkage analysis in Excel spreadsheets to find overlapping regions. The raw single nucleotide variant (SNV) genotypes are available in S1. The output of the linkage and homozygosity mapping is given in
Table S2.
2.5. Whole Genome Sequencing of an Affected Rough Collie
An Illumina TruSeq PCR-free DNA library with 500 bp insert size of an affected Rough Collie (CL013) was prepared. We collected 227 million 2 × 150 bp paired-end reads on a NovaSeq 6000 instrument (26.9× coverage). Mapping and alignment were performed as described [
12]. The sequence data were deposited under study accession PRJEB16012 and sample accession SAMEA4867926 at the European Nucleotide Archive.
2.6. Variant Calling
Variant calling was performed using GATK HaplotypeCaller [
13] in gVCF mode as described [
9]. To predict the functional effects of the called variants, SnpEff [
14] software together with NCBI annotation release 105 for the CanFam 3.1 genome reference assembly was used. For variant filtering we used 601 control genomes, which were either publicly available [
15] or produced during other projects of our group [
16] (
Table S3).
2.7. Gene Analysis
We used the dog CanFam 3.1 reference genome assembly for all analyses. Numbering within the canine AKNA gene corresponds to the NCBI RefSeq accessions XM_014117950.2 (mRNA) and XP_013973425.1 (protein).
2.8. Sanger Sequencing
The AKNA:c.2717_2720delACAG variant was genotyped by direct Sanger sequencing of PCR amplicons. A 305 bp (or 301 bp for the deletion allele) PCR product was amplified from genomic DNA using AmpliTaqGold360Mastermix (Thermo Fisher Scientific, Waltham, MA, USA) together with primers 5′-CCT GTG AGC CTC TGG AAT GT-3′ (Primer F) and 5′-CTG AGA ATG CCC AGA CCA TC-3′ (Primer R). After treatment with exonuclease I and alkaline phosphatase, amplicons were sequenced on an ABI 3730 DNA Analyzer (Thermo Fisher Scientific). Sanger sequences were analyzed using the Sequencher 5.1 software (GeneCodes, Ann Arbor, MI, USA).
3. Results
3.1. Phenotype Description
The owners reported recurrent foamy vomiting, shallow breathing, coughing, increased breathing sounds, nasal discharge and fever in three related Rough Collies. Bronchopneumonia was diagnosed by a private veterinarian. The clinical signs started at a few days of age in two affected dogs. The dogs responded to therapy with antibiotics and secretolytics. However, they tended to relapse quickly. Two dogs were reported to be alive at three years with frequent yellowish nasal discharge. No information on the course of the disease is available for the third affected dog.
3.2. Genetic Analysis
Three related cases were reported. The pedigree of these dogs is shown in
Figure 1 and suggested autosomal recessive inheritance. Linkage analysis using two litters with six offspring (two affected, four non-affected) and their three parents showed 18 linked genome segments totaling 246 Mb. We additionally performed homozygosity mapping in the three affected dogs. They shared 87 homozygous segments with identical alleles totaling 354 Mb. Intersecting the linked and homozygous regions delineated critical intervals comprising 19 segments on 10 chromosomes spanning 99 Mb or roughly 4.1% of the 2.4 Gb dog genome. The two known canine PCD causative variants,
CCDC39:c.286C>T [
5] and
NME5:c.43delA [
6] were not located in the critical intervals and could thus be excluded.
We sequenced the genome of an affected dog at 26.9× coverage and called SNVs and short indels with respect to the CanFam3.1 genome assembly. We then searched for private homozygous variants in the genome sequence of the affected dog that were not present in 601 control dogs of different breeds (
Table 1). We identified only one private homozygous protein-changing variant in the critical intervals (
Table S4).
This variant represented a 4 bp deletion that can be designated as Chr11:68,576,241_68,576,244delCTGT (CanFam 3.1 assembly). It was located in the
AKNA gene encoding AT-hook transcription factor and is predicted to result in a frameshift of the coding sequence, XM_014117950.2:c.2717_2720delACAG. The variant introduces a premature stop codon. Due to a lack of suitable RNA samples we did not experimentally investigate whether the mutant transcripts are subject to nonsense-mediated decay and/or nonsense-mediated alternative splicing. If expressed, the predicted translation product of the mutant transcript would lack 529 amino acids of the 1434 amino acid wildtype AKNA protein and contain 172 new amino acids at its C-terminus. The formal variant designation is XP_013973425.1:p.(Asp906Alafs*173). We confirmed the presence of the
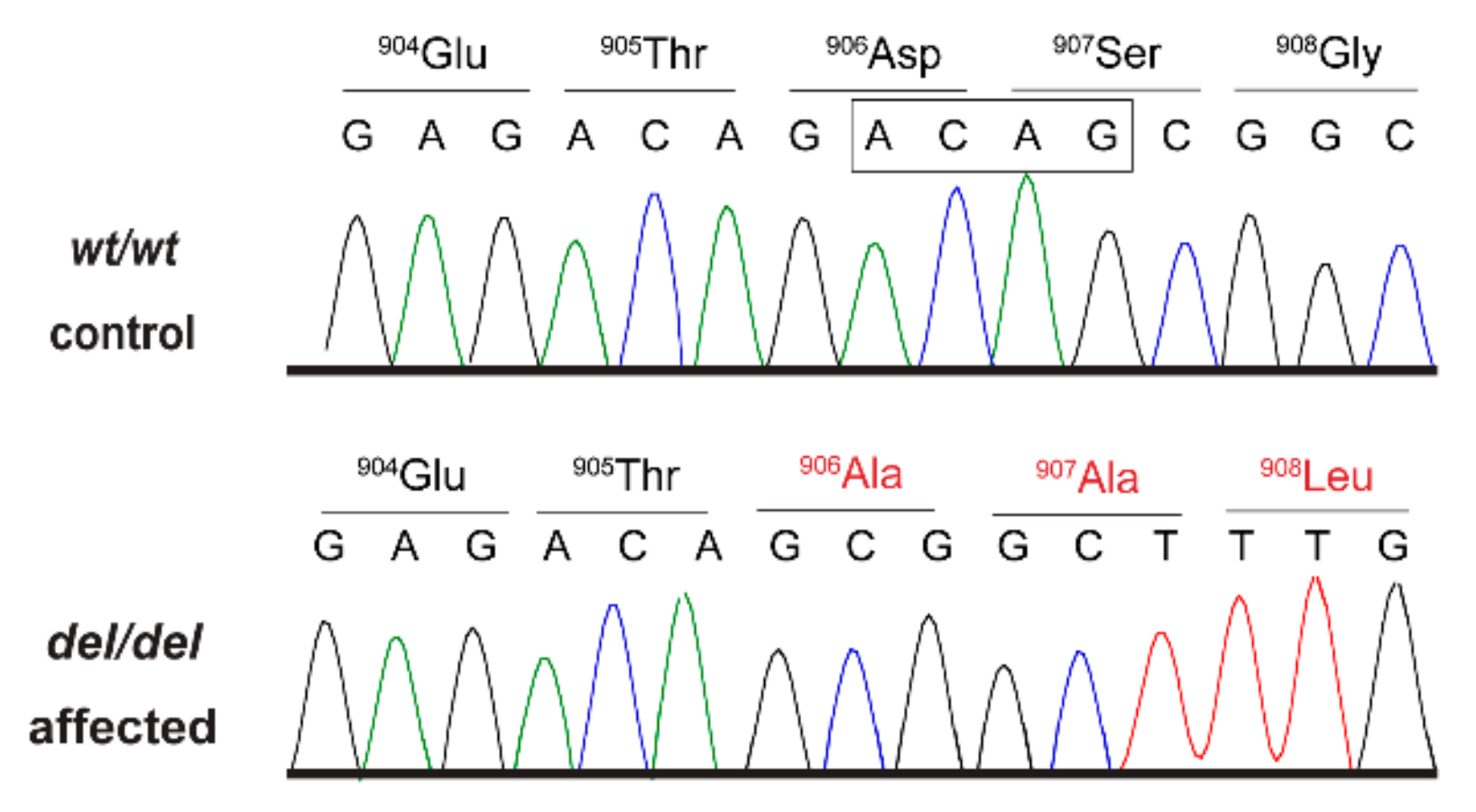
AKNA:c.2717_2720delACAG variant in genomic DNA samples by Sanger sequencing (
Figure 2).
We genotyped a total of 627 dogs for the variant and observed perfect genotype-phenotype concordance (
Table 2). All three affected Rough Collies carried the deletion in the homozygous state. Eighteen non-affected dogs were heterozygous and presumed carriers. The remaining 67 non-affected Rough Collies were homozygous wildtype. As we tested only a relatively small number of Rough Collies including many close relatives of the affected dogs, we cannot reliably estimate the allele frequency in the breed. The mutant allele at the
AKNA:c.2717_2720delACAG variant was absent from 539 additionally genotyped dogs of various breeds (
Table S1).
4. Discussion
In this study, we identified AKNA:c.2717_2720delACAG as a candidate causative variant for an inherited form of recurrent inflammatory pulmonary disease in dogs. The affected dogs were initially submitted with a suspected diagnosis of PCD. We therefore were surprised that the candidate causative variant turned out to be unrelated to cilial function. The clinical phenotype has not yet been characterized in detail. We putatively propose the term recurrent inflammatory pulmonary disease to emphasize the phenotypic similarities of the affected dogs with Akna−/− knockout mice (see below).
AKNA encodes the AT-hook transcription factor, which has an important regulatory function in the immune system. It modulates the transcription of CD40 and its ligand CD154 (also called CD40L) in immune cells [
17]. The interaction of CD40 and CD154 has been shown to be involved in the negative regulation of T cell autoreactivity and abnormalities in their interaction may lead to autoimmunity. In human systemic autoimmune diseases such as Vogt-Koyanagi-Harada syndrome, AKNA was found to be downregulated in CD4
+ T-lymphocytes [
18]. Furthermore, associations between
AKNA gene variants with knee osteoarthritis [
19] and an elevated risk for cervical cancer due to a deregulation in the inflammasome network were reported [
20,
21]. To the best of our knowledge, no human patients affected by recurrent inflammatory pulmonary disease or any other pathology with germline coding variants in
AKNA have been described in the scientific literature. Data from more than 120,000 humans in the gnomAD browser [
22] indicate that
AKNA loss of function (LoF) variants are less frequent than expected (19 observed vs 61.4 theoretically expected LoF variants). No individuals carrying an
AKNA LoF variant in homozygous state have been identified so far.
Akna−/− knockout mice are smaller than their wildtype littermates and they die within 10 days after birth due to systemic inflammation, predominantly seen in the lungs [
23]. Enhanced leukocyte infiltration and a massive overproduction of pro-inflammatory cytokines and matrix metallproteinases lead to alveolar destruction in the
Akna−/− knockout mice. The murine
Akna gene contains 22 exons and two different knockout strains were investigated. The initial strain was constructed by deleting exons 19–21. It was not assessed whether a C-terminally truncated protein might be expressed in this particular strain [
23]. This knockout strain closely resembles the situation in homozygous mutant Rough Collies, which have a shift of the reading frame starting in exon 13. Later, a second mouse knockout strain with a deletion of exon 3 was constructed. Exon 3 is the largest coding exon and encodes the first AT-hook motif of AKNA. Both
Akna−/− mouse knockout strains exhibited comparable phenotypes [
23].
The genetic investigations in dogs showed a perfect genotype-phenotype correlation of the AKNA variant with recurrent inflammatory pulmonary disease. This association supports a causative role of AKNA:c.2717_2720delACAG, however cannot provide definitive proof. Nonetheless, the known role of AKNA as an important anti-inflammatory regulator of the immune response and the striking phenotypic similarities of the affected dogs with Akna−/− knockout mice in combination with the genetic association strongly suggests that AKNA:c.2717_2720delACAG is the causative variant.
5. Conclusions
We identified AKNA:c.2717_2720delACAG as a candidate causative variant for an inherited, autosomal recessive, recurrent inflammatory pulmonary disease in Rough Collies. The findings from knockout mice and these dogs suggest that AKNA should be considered as a candidate gene for human patients with unexplained recurrent inflammatory pulmonary disease. Our findings enabled genetic testing for Rough Collies, which can be used to avoid the unintentional breeding of affected puppies.
Supplementary Materials
The following are available online at
https://www.mdpi.com/2073-4425/10/8/567/s1, S1: Illumina canine_HD SNV genotypes of 10 Rough Collies, Table S1: AKNA:c.2717_2720delACAG genotypes of 539 control dogs. Table S2: Linkage and homozygosity mapping, Table S3: Accession numbers of 594 dog and 8 wolf genome sequences, Table S4: Private protein changing variants in the sequenced Rough Collie with recurrent inflammatory pulmonary disease.
Author Contributions
Conceptualization, T.L.; data curation, V.J.; investigation, P.H., L.A., A.K., V.J., and T.L.; methodology, V.J.; resources, A.K.; supervision, T.L.; visualization, P.H.; writing—original draft, P.H. and T.L.; writing—review and editing, P.H., L.A., A.K., V.J. and T.L.
Funding
This research received no external funding.
Acknowledgments
The authors are grateful to the dog owners who donated samples and shared health and pedigree data of their dogs. We thank Eva Andrist, Nathalie Besuchet Schmutz, Muriel Fragnière, and Sabrina Schenk for expert technical assistance, the Next Generation Sequencing Platform of the University of Bern for performing the high-throughput sequencing experiments, and the Interfaculty Bioinformatics Unit of the University of Bern for providing high performance computing infrastructure. We thank the Dog Biomedical Variant Database Consortium (Gus Aguirre, Catherine André, Danika Bannasch, Doreen Becker, Brian Davis, Cord Drögemüller, Kari Ekenstedt, Kiterie Faller, Oliver Forman, Steve Friedenberg, Eva Furrow, Urs Giger, Christophe Hitte, Marjo Hytönen, Vidhya Jagannathan, Tosso Leeb, Hannes Lohi, Cathryn Mellersh, Jim Mickelson, Leonardo Murgiano, Anita Oberbauer, Sheila Schmutz, Jeffrey Schoenebeck, Kim Summers, Frank van Steenbeek, Claire Wade) for sharing whole genome sequencing data from control dogs. We also acknowledge all canine researchers who deposited dog whole genome sequencing data into public databases.
Conflicts of Interest
A.K. is an employee of Laboklin, a company that offers genetic testing for dogs.
References
- Knowles, M.; Zariwala, M.; Leigh, M. Primary ciliary dyskinesia. Clin. Chest Med. 2016, 37, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Horani, A.; Ferkol, T.W. Advances in the genetics of primary ciliary dyskinesia: Clinical implications. Chest 2018, 154, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Bonnefoy, S.; Watson, C.M.; Kernohan, K.D.; Lemos, M.; Hutchinson, S.; Poulter, J.A. Biallelic mutations in LRRC56, encoding a protein associated with intraflagellar transport, cause mucociliary clearance and laterality defects. Am. J. Hum. Genet. 2018, 103, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Fassad, M.R.; Shoemark, A.; Legendre, M.; Hirst, R.A.; Koll, F.; le Borgne, P. Mutations in outer dynein arm heavy chain DNAH9 cause motile cilia defects and situs inversus. Am. J. Hum. Genet. 2018, 103, 984–994. [Google Scholar] [CrossRef] [PubMed]
- Merveille, A.C.; Davis, E.E.; Becker-Heck, A.; Legendre, M.; Amirav, I.; Bataille, G.; Belmont, J.; Beydon, N.; Billen, F.; Clément, A.; et al. CCDC39 is required for assembly of inner dynein arms and the dynein regulatory complex and for normal ciliary motility in humans and dogs. Nat. Genet. 2011, 43, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Anderegg, L.; Im Hof Gut, M.; Hetzel, U.; Howerth, E.W.; Leuthard, F.; Jagannathan, V.; Leeb, T. NME5 frameshift variant in Alaskan Malamutes with primary ciliary dyskinesia. PLoS Genet. 2019. in revision. [Google Scholar]
- Andjelkovic, M.; Minic, P.; Vreca, M.; Stojiljkovic, M.; Skakic, A.; Sovtic, A.; Rodic, M.; Skodric-Trifunovic, V.; Maric, N.; Visekruna, J.; et al. Genomic profiling supports the diagnosis of primary ciliary dyskinesia and reveals novel candidate genes and genetic variants. PLoS ONE 2018, 13, e0205422. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.T.; Griffin, P.; Martinello, P.; Robinson, P. Primary ciliary dyskinesia in two English Cocker Spaniels. Aust. Vet. J. 2016, 94, 149–153. [Google Scholar] [CrossRef]
- Miranda, I.C.; Granick, J.L.; Armién, A.G. Histologic and ultrastructural findings in dogs with chronic respiratory disease suspected of ciliary dyskinesia. Vet. Pathol. 2017, 54, 802–812. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Abecasis, G.R.; Cherny, S.S.; Cookson, W.O.; Cardon, L.R. Merlin--rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002, 30, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Bauer, A.; Jagannathan, V.; Högler, S.; Richter, B.; McEwan, N.A.; Thomas, A.; Cadieu, E.; André, C.; Hytönen, M.K.; Lohi, H.; et al. MKLN1 splicing defect in dogs with lethal acrodermatitis. PLoS Genet. 2018, 14, e1007264. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics 2013, 43, 11. [Google Scholar] [PubMed]
- Cingolani, P.; Platts, A.; Wang le, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Bai, B.; Zhao, W.M.; Tang, B.X.; Wang, Y.Q.; Wang, L.; Zhang, Z.; Yang, H.C.; Liu, Y.H.; Zhu, J.W.; Irwin, D.M.; et al. DoGSD: The dog and wolf genome SNP database. Nucleic Acids Res. 2015, 43, 777–783. [Google Scholar] [CrossRef]
- Jagannathan, V.; Drögemüller, C.; Leeb, T. Dog Biomedical Variant Database Consortium (DBVDC). A comprehensive biomedical variant catalogue based on whole genome sequences of 582 dogs and 8 wolves. Anim Genet. 2019. [Google Scholar] [CrossRef]
- Siddiqa, A.; Sims-Mourtada, J.C.; Guzman-Rojas, L. Regulation of CD40 and CD40 ligand by the AT-hook transcription factor AKNA. Nature 2001, 410, 383–387. [Google Scholar] [CrossRef]
- Mao, L.; Yang, P.; Hou, S.; Li, F.; Kijlstra, A. Label-free proteomics reveals decreased expression of CD18 and AKNA in peripheral CD4+ T cells from patients with Vogt-Koyanagi-Harada Syndrome. PLoS ONE 2011, 6, e14616. [Google Scholar] [CrossRef]
- Martínez-Nava, G.A.; Fernández-Torres, J.; Martínez-Flores, K.; Zamudio-Cuevas, Y.; Clavijo-Cornejo, D.; Espinosa-Morales, R.; Lozada, C.A.; Gutierrez, M.; Granados, J.; Pineda, C.; et al. The association of AKNA gene polymorphisms with knee osteoarthritis suggests the relevance of this immune response regulator in the disease genetic susceptibility. Mol. Biol. Rep. 2018, 45, 151–161. [Google Scholar] [CrossRef]
- Manzo-Merino, J.; Lagunas-Martínez, A.; Contreras-Ochoa, C.O.; Lizano, M.; Castro-Muñoz, L.J.; Calderón-Corona, C.; Torres-Poveda, K.; Román-Gonzalez, A.; Hernández-Pando, R.; Bahena-Román, M.; et al. The human papillomavirus (HPV) E6 oncoprotein regulates CD40 expression via the AT-hook transcription factor AKNA. Cancers 2018, 10, 521. [Google Scholar] [CrossRef]
- Perales, G.; Burguete-García, A.I.; Dimas, J.; Bahena-Román, M.; Bermúdez-Morales, V.H.; Moreno, J.; Madrid-Marina, V. A polymorphism in the AT-hook motif of the transcriptional regulator AKNA is a risk factor for cervical cancer. Biomarkers 2010, 15, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Ortiz-Quintero, B.; Rangel, R.; McKeller, M.R.; Herrera-Rodriguez, S.; Castillo, E.F.; Schluns, K.; Hall, M.; Zhang, H.; Suh, W.K.; et al. Coordinate activation of inflammatory gene networks, alveolar destruction and neonatal death in AKNA deficient mice. Cell Res. 2011, 21, 1564–1577. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).






{kind=link}
{kind=link}