Genetic Analyses of Tanzanian Local Chicken Ecotypes Challenged with Newcastle Disease Virus

 ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Design
2.2. Genotyping and Quality Control
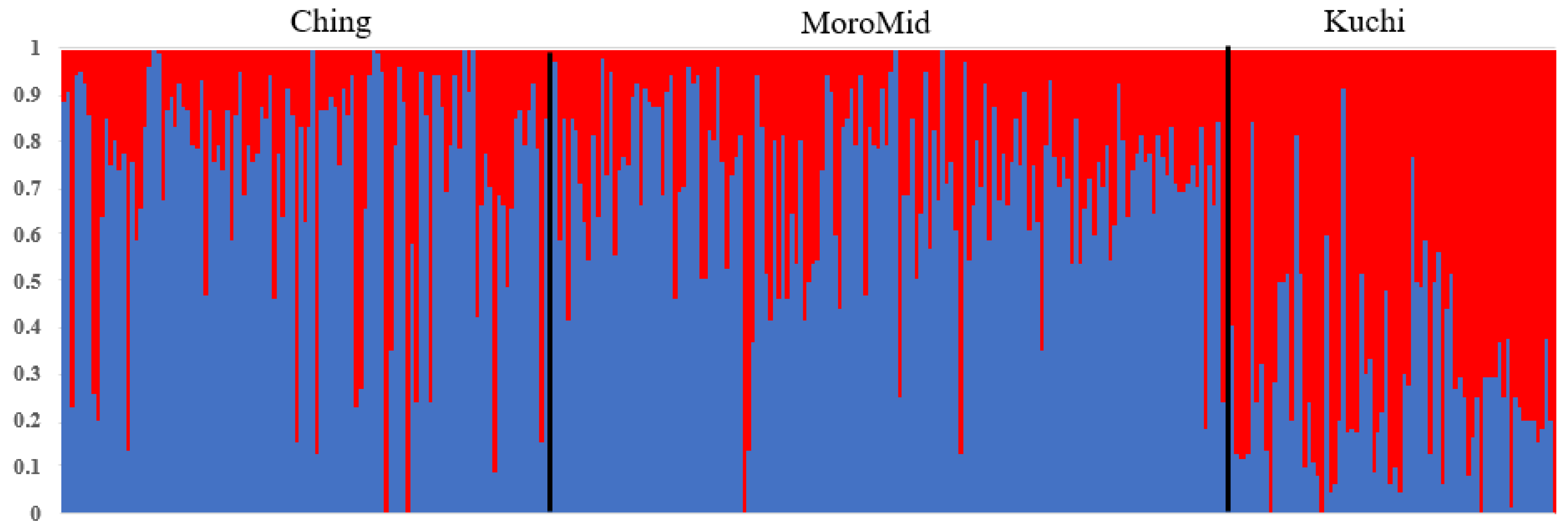
2.3. Population Stratification
2.4. Genetic Parameters and Correlations
2.5. Genome-Wide Association Analyses
2.6. Multiple Test Correction
2.7. Bioinformatics Analyses
3. Results
3.1. Population Stratification and Phenotypic Data
3.2. Genetic Parameter Estimates
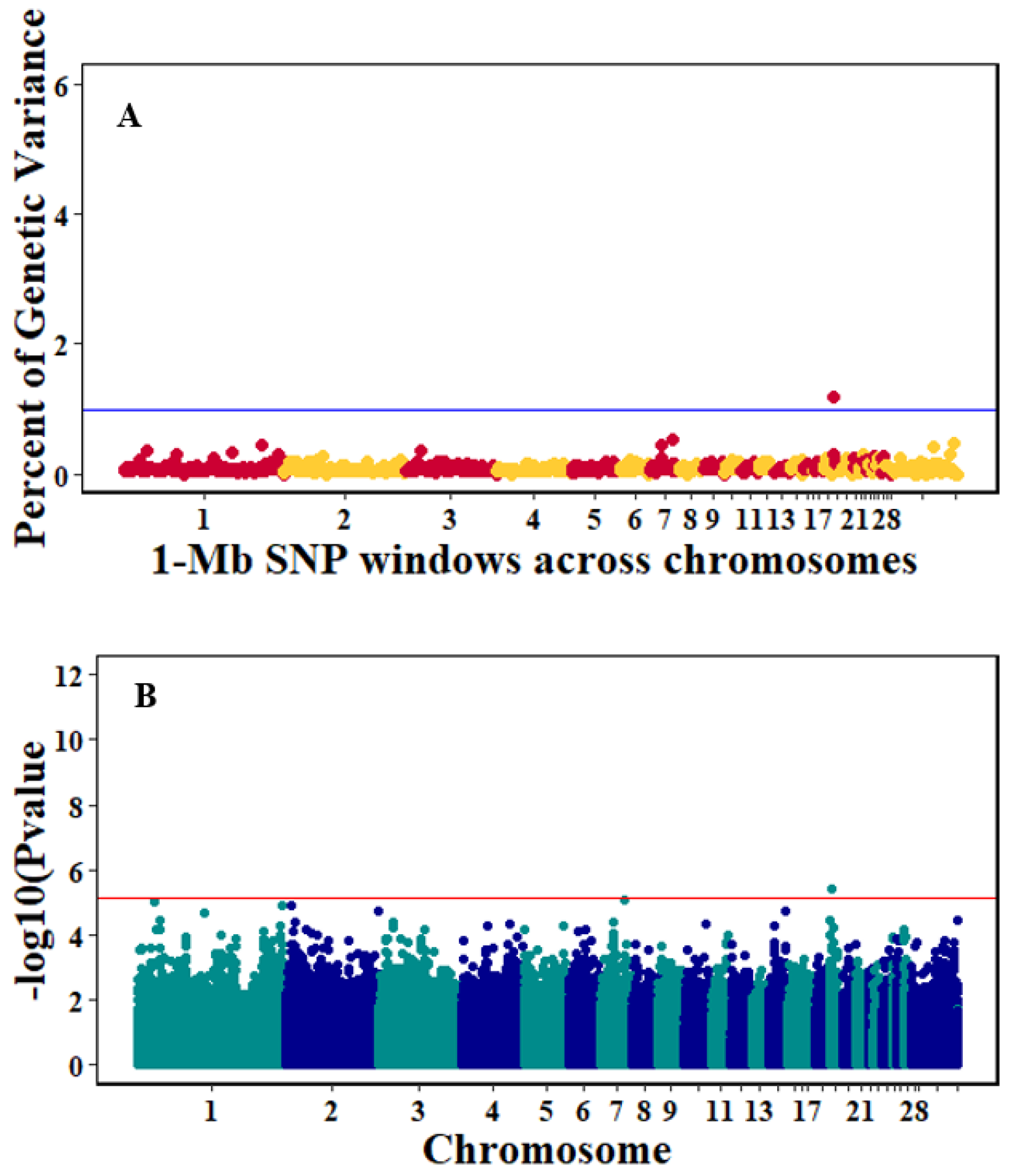
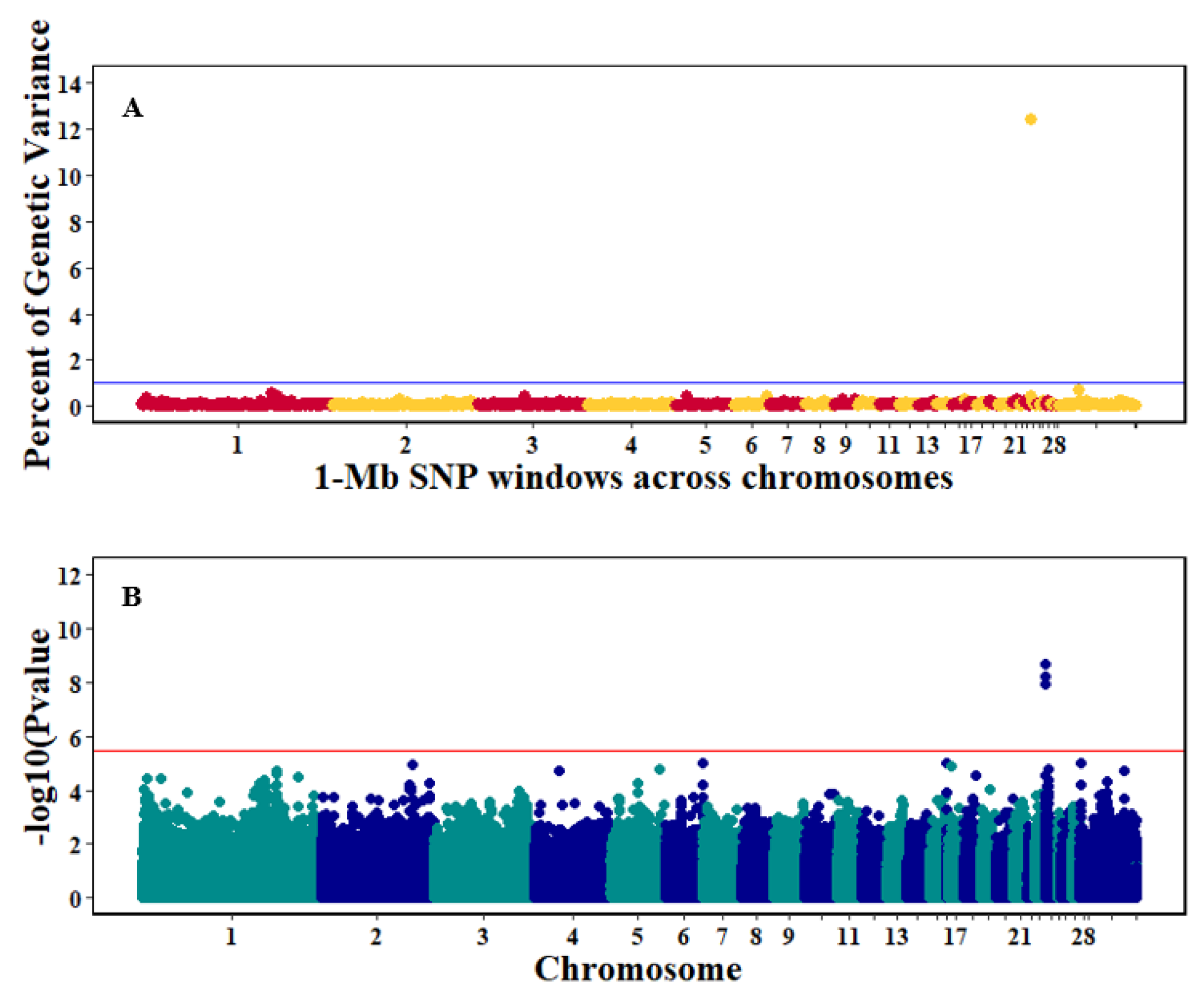
3.3. Genome-Wide Association Studies
4. Discussion
4.1. Population Stratification
4.2. Genetic Parameters
4.3. Genome-Wide Association Analyses
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- GuÈye, E.H.F. Village egg and fowl meat production in Africa. World’s Poult. Sci. J. 1998, 54, 73–86. [Google Scholar] [CrossRef]
- Goromela, E.; Kwakkel, R.; Verstegen, M.W.A.; Katule, A.M. Strategies to optimize the use of scavengeable feed resource base by smallholders in traditional poultry production systems in Africa: A review. Afr. J. 2006, 1, 91–100. [Google Scholar]
- Alabi, R.A.; Esobhawan, A.O.; Aruna, M.B. Econometric determination of contribution of family poultry to women’s income in Niger-delta, Nigeria. J. Cent. Eur. Agric. 2006, 7, 753–760. [Google Scholar]
- Sambo, E.; Bettridge, J.; Dessie, T.; Amare, A.; Habte, T.; Wigley, P.; Christley, R.M. Participatory evaluation of chicken health and production constraints in Ethiopia. Prev. Vet. Med. 2015, 118, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Guèye, E.H.F. Family poultry research and development in low-income food-deficit countries: Approaches and prospects. Outlook Agric. 2002, 31, 13–21. [Google Scholar] [CrossRef]
- Copland, J.W.; Alders, R.G. The Australian village poultry development programme in Asia and Africa. World’s Poult. Sci. J. 2005, 61, 31–38. [Google Scholar] [CrossRef][Green Version]
- Alders, R.G. Making Newcastle disease vaccines available at village level. Vet. Rec. 2014, 174, 502–503. [Google Scholar] [CrossRef]
- Mayo, M.A. Virus Taxonomy Houston 2002. Arch. Virol. 2002, 147, 1071–1076. [Google Scholar]
- Fuller, C.M.; Collins, M.S.; Alexander, D.J. Development of a real-time reverse-transcription PCR for the detection and simultaneous pathotyping of Newcastle disease virus isolates using a novel probe. Arch. Virol. 2009, 154, 929–937. [Google Scholar] [CrossRef]
- Songhua, S.; Chaogang, S.; Caozhe, X.; Jian, Z.; Yongqiang, H.; Jianhua, W.; Zuxun, G. Differentiation of velogenic, mesogenic and lentogenic strains of Newcastle disease virus by multiplex RT-PCR. Ann. Appl. Biol. 2003, 142, 49–54. [Google Scholar] [CrossRef]
- Brown, V.R.; Bevins, S.N. A review of virulent Newcastle disease viruses in the United States and the role of wild birds in viral persistence and spread. Vet. Res. 2017, 48, 1–15. [Google Scholar] [CrossRef]
- Cornax, I.; Miller, P.J.; Afonso, C.L. Characterization of Live LaSota Vaccine Strain–Induced Protection in Chickens upon Early Challenge with a Virulent Newcastle Disease Virus of Heterologous Genotype. Avian Dis. Dig. 2012, 7, e7–e8. [Google Scholar] [CrossRef]
- Miller, P.J.; Decanini, E.L.; Afonso, C.L. Newcastle disease: Evolution of genotypes and the related diagnostic challenges. Infect. Genet. Evol. 2010, 10, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Fleming, D.S.; Koltes, J.E.; Markey, A.D.; Schmidt, C.J.; Ashwell, C.M.; Rothschild, M.F.; Persia, M.E.; Reecy, J.M.; Lamont, S.J. Genomic analysis of Ugandan and Rwandan chicken ecotypes using a 600 k genotyping array. BMC Genom. 2016, 17, 407. [Google Scholar] [CrossRef] [PubMed]
- Walugembe, M.; Bertolini, F.; Dematawewa, C.M.B.; Reis, M.P.; Elbeltagy, A.R.; Schmidt, C.J.; Lamont, S.J.; Rothschild, M.F. Detection of Selection Signatures Among Brazilian, Sri Lankan, and Egyptian Chicken Populations Under Different Environmental Conditions. Front. Genet. 2019, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Qu, H.; Ma, J.; Wang, J.; Li, C.; Yang, C.; Hu, X.; Li, N.; Shu, D. Genome-wide association study of antibody response to Newcastle disease virus in chicken. BMC Genet. 2013, 14, 42. [Google Scholar] [CrossRef] [PubMed]
- Yonash, N.; Cheng, H.H.; Hillel, J.; Heller, D.E.; Cahaner, A. DNA microsatellites linked to quantitative trait loci affecting antibody response and survival rate in meat-type chickens. Poult. Sci. 2001, 80, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Rowland, K.; Wolc, A.; Gallardo, R.A.; Kelly, T.; Zhou, H.; Dekkers, J.C.M.; Lamont, S.J. Genetic Analysis of a Commercial Egg Laying Line Challenged with Newcastle Disease Virus. Front. Genet. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Saelao, P.; Wang, Y.; Chanthavixay, G.; Gallardo, R.; Wolc, A.; Dekkers, J.; Lamont, S.; Kelly, T.; Zhou, H. Genetics and Genomic Regions Affecting Response to Newcastle Disease Virus Infection under Heat Stress in Layer Chickens. Genes 2019, 10, 61. [Google Scholar] [CrossRef]
- Lwelamira, J.; Kifaro, G.C.; Gwakisa, P.S. Genetic parameters for body weights egg traits and antibody response against Newcastle Disease Virus (NDV) vaccine among two Tanzania chicken ecotypes. Trop. Anim. Health Prod. 2009, 41, 51–59. [Google Scholar] [CrossRef]
- Lwelamira, J. Phenotypic and genetic parameters for body weights and antibody response against Newcastle disease virus (NDV) vaccine for Kuchi chicken ecotype of Tanzania under extensive management. Trop. Anim. Health Prod. 2012, 44, 1529–1534. [Google Scholar] [CrossRef] [PubMed]
- Mushi, J.R.; Chiwanga, G.C.; Mauch, E.; Gallardo, R.; Lamont, S.J.; Kelly, T.R.; Zhou, H.; Muhairwa, A.P. Phenotypic and genotypic diversity of Free Range Local Chickens in Tanzania. In Proceedings of the World Congress on Genetics Applied to Livestock Production, Auckland, New Zealand, 11–16 February 2018. [Google Scholar]
- Zhang, J.; Kaiser, M.G.; Deist, M.S.; Gallardo, R.A.; Bunn, D.A.; Kelly, T.R.; Dekkers, J.C.M.; Zhou, H.; Lamont, S.J. Transcriptome analysis in spleen reveals differential regulation of response to newcastle disease virus in two chicken lines. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sargolzaei, M.; Chesnais, J.P.; Schenkel, F.S. A new approach for efficient genotype imputation using information from relatives. BMC Genom. 2014, 15, 478. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Gilmour, A.R.; Gogel, B.J.; Cullis, B.R.; Welham, S.J.; Thompson, R. ASReml User Guide Release 4.1 Structural Specification. VSN Int. Ltd. Hemel Hempstead 2015, 1–30. [Google Scholar] [CrossRef]
- VanRaden, P.M. Efficient Methods to Compute Genomic Predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [PubMed]
- Habier, D.; Pioneer, D.; Fernando, R.L.; Kizilkaya, K.; Garrick, D.J. Extension of the Bayesian alphabet for genomic selection. BMC 2011, 12, 186. [Google Scholar] [CrossRef] [PubMed]
- Fernando, R.L.; Garrick, D. GenSel–User manual for a portfolio of genomic selection related analyses. Iowa State Univ. In Bayesian Methods Applied to GWAS; Humana Press: Totowa, NJ, USA, 2013. [Google Scholar]
- Aulchenko, Y. Package GenABEL R Package Reference Manual. Available online: https://cran.r-project.org/src/contrib/Archive/GenABEL/ (accessed on 20 July 2018).
- Rönnegård, L.; Shen, X.; Alam, M. Hglm: A Package for Fitting Hierarchical Generalized Linear Models. R J. 2010, 2, 20–28. [Google Scholar] [CrossRef]
- Waide, E.H.; Tuggle, C.K.; Serão, N.V.L.; Schroyen, M.; Hess, A.; Rowland, R.R.R.; Lunney, J.K.; Plastow, G.; Dekkers, J.C.M. Genomewide association of piglet responses to infection with one of two porcine reproductive and respiratory syndrome virus isolates. J. Anim. Sci. 2017, 95, 16–38. [Google Scholar] [CrossRef]
- Oksenberg, N.; Stevison, L.; Wall, J.D.; Ahituv, N. Function and Regulation of AUTS2, a Gene Implicated in Autism and Human Evolution. PLoS Genet. 2013, 9, e1003221. [Google Scholar] [CrossRef] [PubMed]
- Beunders, G.; Voorhoeve, E.; Golzio, C.; Pardo, L.M.; Rosenfeld, J.A.; Talkowski, M.E.; Simonic, I.; Lionel, A.C.; Vergult, S.; Pyatt, R.E.; et al. Exonic deletions in AUTS2 cause a syndromic form of intellectual disability and suggest a critical role for the C terminus. Am. J. Hum. Genet. 2013, 92, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Leung, R.; Cuddy, K.; Wang, Y.; Rommens, J.; Glogauer, M. Sbds is required for Rac2-mediated monocyte migration and signaling downstream of RANK during osteoclastogenesis. Blood 2011, 117, 2044–2053. [Google Scholar] [CrossRef] [PubMed]
- Austin, K.M.; Leary, R.J.; Shimamura, A. The Shwachman-Diamond SBDS protein localizes to the nucleolus. Blood 2005, 106, 1253–1258. [Google Scholar] [CrossRef] [PubMed]
- Wendorff, A.A.; Koch, U.; Wunderlich, F.T.; Wirth, S.; Dubey, C.; Brüning, J.C.; MacDonald, H.R.; Radtke, F. Hes1 is a critical but context-dependent mediator of canonical notch signaling in lymphocyte development and transformation. Immunity 2010, 33, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Chiba, S.; Ichikawa, M.; Kunisato, A.; Asai, T.; Shimizu, K.; Yamaguchi, T.; Yamamoto, G.; Seo, S.; Kumano, K.; et al. Notch2 is preferentially expressed in mature B cells and indispensable for marginal zone B line development. Immunity 2003, 18, 675–685. [Google Scholar] [CrossRef]
- Yu, X.; Alder, J.K.; Chun, J.H.; Friedman, A.D.; Heimfeld, S.; Cheng, L.; Civin, C.I. HES1 Inhibits Cycling of Hematopoietic Progenitor Cells via DNA Binding. Stem Cells 2006, 24, 876–888. [Google Scholar] [CrossRef]
- Brown, K.R.; Jurisica, I. Unequal evolutionary conservation of human protein interactions in interologous networks. Genome Biol. 2007, 8, 1–11. [Google Scholar] [CrossRef]
- Wang, K.T.; Wang, H.H.; Wu, Y.Y.; Su, Y.L.; Chiang, P.Y.; Lin, N.Y.; Wang, S.C.; Chang, G.D.; Chang, C.J. Functional regulation of Zfp36l1 and Zfp36l2 in response to lipopolysaccharide in mouse RAW264.7 macrophages. J. Inflamm. 2015, 12, 1–13. [Google Scholar] [CrossRef]
- Taniguchi, M.; Fukunaka, A.; Hagihara, M.; Watanabe, K.; Kamino, S.; Kambe, T.; Enomoto, S.; Hiromura, M. Essential Role of the Zinc Transporter ZIP9/SLC39A9 in Regulating the Activations of Akt and Erk in B-Cell Receptor Signaling Pathway in DT40 Cells. PLoS ONE 2013, 8, 1–10. [Google Scholar] [CrossRef]
- Talebi, R.; Ahmadi, A.; Afraz, F. Analysis of protein-protein interaction network based on transcriptome profiling of ovine granulosa cells identifies candidate genes in cyclic recruitment of ovarian follicles. J. Anim. Sci. Technol. 2018, 60, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Horng, T.; Barton, G.M.; Medzhitov, R. TIRAP: An adapter molecule in the Toll signaling pathway. Nat. Immunol. 2001, 2, 835–841. [Google Scholar] [CrossRef] [PubMed]
- Burroughs, A.M.; Zhang, D.; Aravind, L. The eukaryotic translation initiation regulator CDC123 defines a divergent clade of ATP-grasp enzymes with a predicted role in novel protein modifications. Biol. Direct 2015, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Haynes, L.M.; Freeman, M.W.; Walsh, E.E.; Tripp, R.A.; Kwinn, L.; Finberg, R.W.; Jones, L.P.; Golenbock, D.T.; Popova, L.; Anderson, L.J.; et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat. Immunol. 2002, 1, 398–401. [Google Scholar] [CrossRef]
- Wasylyk, C.; Schlumberger, S.E.; Criqui-Filipe, P.; Wasylyk, B. Sp100 Interacts with ETS-1 and Stimulates Its Transcriptional Activity. Mol. Cell. Biol. 2002, 22, 2687–2702. [Google Scholar] [CrossRef] [PubMed]
- Walunas, T.L.; Wang, B.; Wang, C.; Leiden, J.M.; Walunas, T.L.; Wang, B.; Wang, C.; Leiden, J.M. Cutting Edge: The Ets1 Transcription Factor Is Required for the Development of NK T Cells in Mice. J. Immunol. 2019, 164, 2857–2860. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Affymetrix Genotype Metric | Metric Description | Requirement |
|---|---|---|
| Nclus | Number of genotype clusters | ≥2 |
| CR | % of samples with genotype call other than "No call" for SNP | ≥99% |
| MinorAlleleFrequency | Min (PA, PB), where PA is frequency of Allele A, and PB = 1−PA | ≥0.05 |
| FLD | Measure of the cluster quality of a probeset | ≥5.12 |
| HomRO | Distance to zero in the Contrast dimension (X position) from the center of the homozygous cluster that is closest to zero | ≥0.47 |
| HomFLD | Version of FLD computed for the homozygous genotype clusters | ≥13.34 |
| HetSO | Measures how far the heterozygous cluster center sits above the homozygous cluster centers in the Size dimension (Y position) | ≥−0.35 |
| ConversionType | Probeset classification | ≠OTV |
| BB.varX | Contrast (X position) variance for BB cluster | ≤0.85 |
| BB.varY | Size (Y position) variance for BB cluster | ≤0.69 |
| AB.varX | Contrast (X position) variance for AB cluster | ≤0.75 |
| AB.varY | Size (Y position) variance for AB cluster | ≤0.78 |
| AA.varX | Contrast (X position) variance for AA cluster | ≤0.79 |
| AA.varY | Size (Y position) variance for AA cluster | ≤0.51 |
| Trait | N3 | Mean4 | SD5 | Heritability + SE | Maternal6 | Residual |
|---|---|---|---|---|---|---|
| Pre-infection GR1 | 1392 | 5.12 | 1.31 | 0.35 ± 0.07 | 0.02 | 0.80 ± 0.06 |
| Post-infection GR1 | 1359 | 6.85 | 2.82 | 0.21 ± 0.06 | - | 3.83 ± 0.24 |
| Antibody titer2 | 1394 | 3.45 | 0.45 | 0.22 ± 0.05 | - | 0.13 ± 0.01 |
| VL2dpi2 | 1375 | 4.72 | 1.03 | 0.18 ± 0.07 | 0.06 | 0.49 ± 0.03 |
| VL6dpi2 | 1365 | 4.25 | 1.18 | 0.29 ± 0.06 | - | 0.66 ± 0.05 |
| Viral clearance | 1342 | 0.06 | 0.68 | 0.04 ± 0.01 | - | 0.41 ± 0.02 |
| Pre-Infection GR3 | Post-Infection GR | Antibody | VL2dpi | VL6dpi | Viral Clearance | |
|---|---|---|---|---|---|---|
| Pre-infection GR1 | 0.74 ± 0.08 | 0.24 ± 0.14 | −0.06 ± 0.14 | −0.23 ± 0.13 | 0.29 ± 0.20 | |
| Post-infection GR1 | 0.54 ± 0.02 | 0.26 ± 0.15 | 0.02 ± 0.17 | −0.13 ± 0.16 | 0.15 ± 0.23 | |
| Antibody2 | 0.16 ± 0.03 | 0.06 ± 0.03 | 0.07 ± 0.17 | −0.04 ± 0.14 | 0.30 ± 0.21 | |
| VL2dpi2 | −0.05 ± 0.03 | −0.07 ± 0.03 | 0.10 ± 0.03 | 0.17 ± 0.15 | 0.06 ± 0.23 | |
| VL6dpi2 | −0.14 ± 0.03 | −0.13 ± 0.03 | 0.03 ± 0.03 | 0.09 ± 0.03 | −0.11 ± 0.21 | |
| Viral Clearance | 0.04 ± 0.03 | 0.01 ± 0.03 | 0.04 ± 0.03 | 0.18 ± 0.03 | −0.29 ± 0.03 |
| Trait | SNP | Position | p-Value | Candidate Genes and Location |
|---|---|---|---|---|
| Pre-infection_GR | AX-76523043 | 3:63366122 | 5.42 × 10−6 | GOPC, downstream, 4697 DCBLD1, downstream, 9312 LOC421740, upstream, 596840 ROS1, upstream, 69755 UNC5D, intron LOC431251, downstream, 400465 ATP6V1B2, upstream, 242578 |
| AX-76262097 | 22:1854894 | 6.75 × 10−6 | ||
| Post-infection_GR | AX-75920682 | 19:1607256 | 3.65 × 10−6 | AUTS2, intron SBDS, upstream, 793675 MIR1567, downstream, 99809 |
| Antibody | AX-76035154 | 2:145809151 | 6.43 × 10−6 | RPLP1, upstream, 3167 MIR6572, upstream, 49226 LPP, intron |
| AX-77135791 | 9: 14444877 | 9.66 × 10−6 | ||
| Log10Viral load, 2dpi | AX-76811433 | 5:28848641 | 5.88 × 10−6 | PLEKHH1, intron TMEM229B, upstream, 82089 PIGH, downstream, 22777 |
| Log10Viral load, 6dpi | AX-76312211 | 24:429611 | 2.25 × 10−9 | TIRAP, downstream, 9853 ETS1, downstream, 448924 TIRAP, downstream, 12699 ETS1, downstream, 446078 TIRAP, downstream, 4299 ETS1, downstream, 454478 |
| AX-76312344 | 24:432457 | 6.16 × 10−9 | ||
| AX-76311970 | 24:424057 | 1.22 × 10−8 |
| Trait | Chr | Position Window (Mb) | #Markers | %TGV1 |
|---|---|---|---|---|
| Pre-infection GR | 22 | 1004589-1997368 | 509 | 1.15 |
| 4 | 71001596-71999395 | 287 | 0.93 | |
| 11 | 18001466-18991342 | 409 | 0.64 | |
| 12 | 11001448-11994345 | 485 | 0.63 | |
| 15 | 4000820-4999664 | 625 | 0.59 | |
| 3 | 63009968-63997299 | 322 | 0.58 | |
| 20 | 76150-998687 | 317 | 0.51 | |
| 3 | 65001841-65999833 | 377 | 0.5 | |
| 2 | 29005343-29996746 | 326 | 0.5 | |
| 1 | 140114736-140998169 | 292 | 0.5 | |
| Post-infection GR | 19 | 1000224-1999134 | 722 | 1.18 |
| 7 | 28002821-28999513 | 472 | 0.55 | |
| Antibody2 | 9 | 13000454-13998539 | 492 | 1.08 |
| 13 | 12000451-12999639 | 472 | 0.67 | |
| 14 | 10000304-10999961 | 635 | 0.65 | |
| 8 | 1000043-1999902 | 446 | 0.63 | |
| 30 | 48000483-48965385 | 175 | 0.56 | |
| 9 | 14001725-14997194 | 537 | 0.54 | |
| 10 | 2000005-2998892 | 581 | 0.54 | |
| Log10Viral load, 2dpi2 | 5 | 28000344-28996407 | 407 | 2 |
| 5 | 41000480-41998371 | 353 | 0.8 | |
| 9 | 5001289-5998634 | 519 | 0.51 | |
| 7 | 8003124-8997158 | 310 | 0.51 | |
| Log10Viral load, 6dpi2 | 24 | 7891-999869 | 740 | 12.4 |
| 30 | 21001186-21998289 | 341 | 0.71 | |
| 1 | 133002233-133996605 | 410 | 0.57 |
| Trait | # SNPs | Chr: Window (Mb) | Genes |
|---|---|---|---|
| Pre-infection_GR | 287 | 4: 71.00–72.0 | PCDH7 |
| 509 | 22: 1.00–2.0 | TNFRSF10B, NEFM, GFRA2, NKX2-6, XPO7, NEFL, TTI2, RHOBTB2, CHMP7, ADAM28, LOXL2, NKX3-1, DOK2, DMTN, LZTS1, SLC18A1, SLC39A14, STC1, MAK16, RNF122, DUSP26, ENTPD4, SLC25A37, DOCK5, ATP6V1B2, EGR3, PBDC1, PHYHIP, C8orf58, SORBS3, NPM2, POLR3D, BIN3, PPP3CC, PEBP4, R3HCC1, LOC107050771 | |
| Post_infection_GR | 722 | 19: 1.00–2.0 | AUTS2, WBSCR17, CALN1, TYW1, MIR1587, MIR1354, MIR1567 |
| Antibody | 492 | 9: 13.0–14.0 | UTS2B, FGF12, ATP13A4, OPA1, CCDC50, GMNC, GP5, LRRC15, OSTN, MB21D2, FCGBP, HRASLS, ATP13A5, CPN2, ATP13A3, HES1 |
| 2 dpi | 407 | 5: 28.0–29.0 | ACTN1, SLC39A9, ZFP36L1, SMOC1, SRSF5, EXD2, TMEM229B, ERH, CCDC177, RAD51B, DCAF5, GALNT16, PLEKHD1, SUSD6, MIR1710, MIR1617, SRSF5A, SLC10A1 |
| 6 dpi | 740 | 24: 0.0–1.0 | ETS1, CHEK1, H2AFX, CDON, PANX3, ST3GAL4, C2CD2L, FAM118B, STT3A, MSANTD2, SRPRA, VSIG10L2, ROBO3, RPUSD4, HYLS1, SIK2, HEPACAM, FEZ1, KIRREL3, DCPS, TIRAP, FOXRED1, PUS3, ESAM, CCDC15, SLC37A2, VPS11, HMBS, DPAGT1, PKNOX2, NRGN, MIR1758, EI24, TMEM218, ROBO4, SPA17, LOC112530272 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walugembe, M.; Mushi, J.R.; Amuzu-Aweh, E.N.; Chiwanga, G.H.; Msoffe, P.L.; Wang, Y.; Saelao, P.; Kelly, T.; Gallardo, R.A.; Zhou, H.; et al. Genetic Analyses of Tanzanian Local Chicken Ecotypes Challenged with Newcastle Disease Virus. Genes 2019, 10, 546. https://doi.org/10.3390/genes10070546
Walugembe M, Mushi JR, Amuzu-Aweh EN, Chiwanga GH, Msoffe PL, Wang Y, Saelao P, Kelly T, Gallardo RA, Zhou H, et al. Genetic Analyses of Tanzanian Local Chicken Ecotypes Challenged with Newcastle Disease Virus. Genes. 2019; 10(7):546. https://doi.org/10.3390/genes10070546
Chicago/Turabian StyleWalugembe, Muhammed, James R. Mushi, Esinam N. Amuzu-Aweh, Gaspar H. Chiwanga, Peter L. Msoffe, Ying Wang, Perot Saelao, Terra Kelly, Rodrigo A. Gallardo, Huaijun Zhou, and et al. 2019. "Genetic Analyses of Tanzanian Local Chicken Ecotypes Challenged with Newcastle Disease Virus" Genes 10, no. 7: 546. https://doi.org/10.3390/genes10070546
APA StyleWalugembe, M., Mushi, J. R., Amuzu-Aweh, E. N., Chiwanga, G. H., Msoffe, P. L., Wang, Y., Saelao, P., Kelly, T., Gallardo, R. A., Zhou, H., Lamont, S. J., Muhairwa, A. P., & Dekkers, J. C. M. (2019). Genetic Analyses of Tanzanian Local Chicken Ecotypes Challenged with Newcastle Disease Virus. Genes, 10(7), 546. https://doi.org/10.3390/genes10070546