Metagenomic Insights into the Bacterial Functions of a Diesel-Degrading Consortium for the Rhizoremediation of Diesel-Polluted Soil

 , , , ,
, , , ,  , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Isolation of the Bacterial Consortium and Growth Conditions
2.2. DNA Extraction, Sequencing, and Assembly
2.3. Diversity Analysis of the 16S rRNA Gene and Coding DNA Sequences (CDSs)
2.4. Identification of CDSs Involved in Alkanes and Aromatic Hydrocarbons Metabolism
2.5. Bioremediation Treatments in Microcosms
2.6. Total Petroleum Hydrocarbon and PAHs Characterization
2.7. Sequence Deposition
3. Results and Discussion
3.1. Diesel Characterization
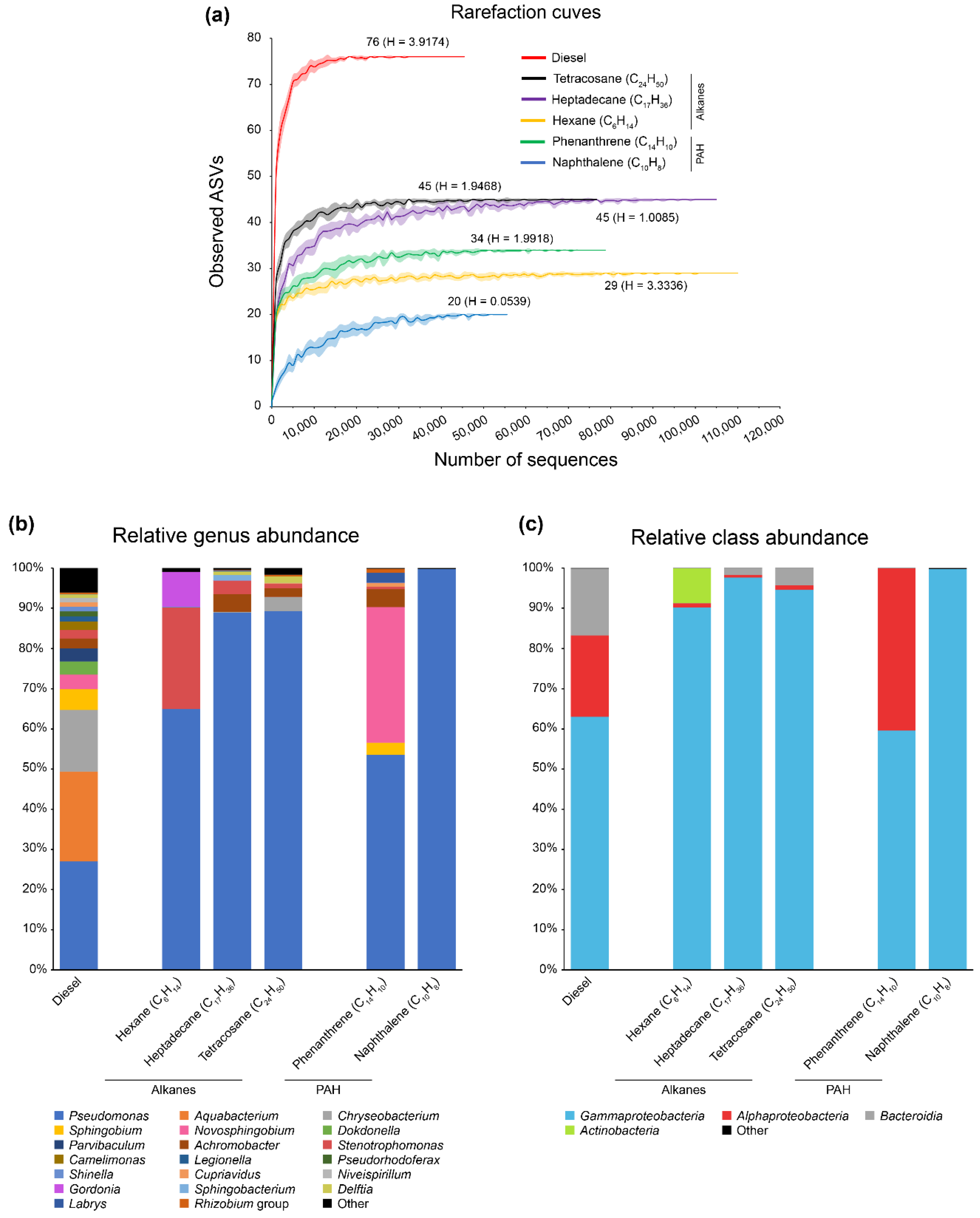
3.2. Bacterial Diversity in the Diesel-Degrading Consortium
3.3. Substrate-Specific Diversity
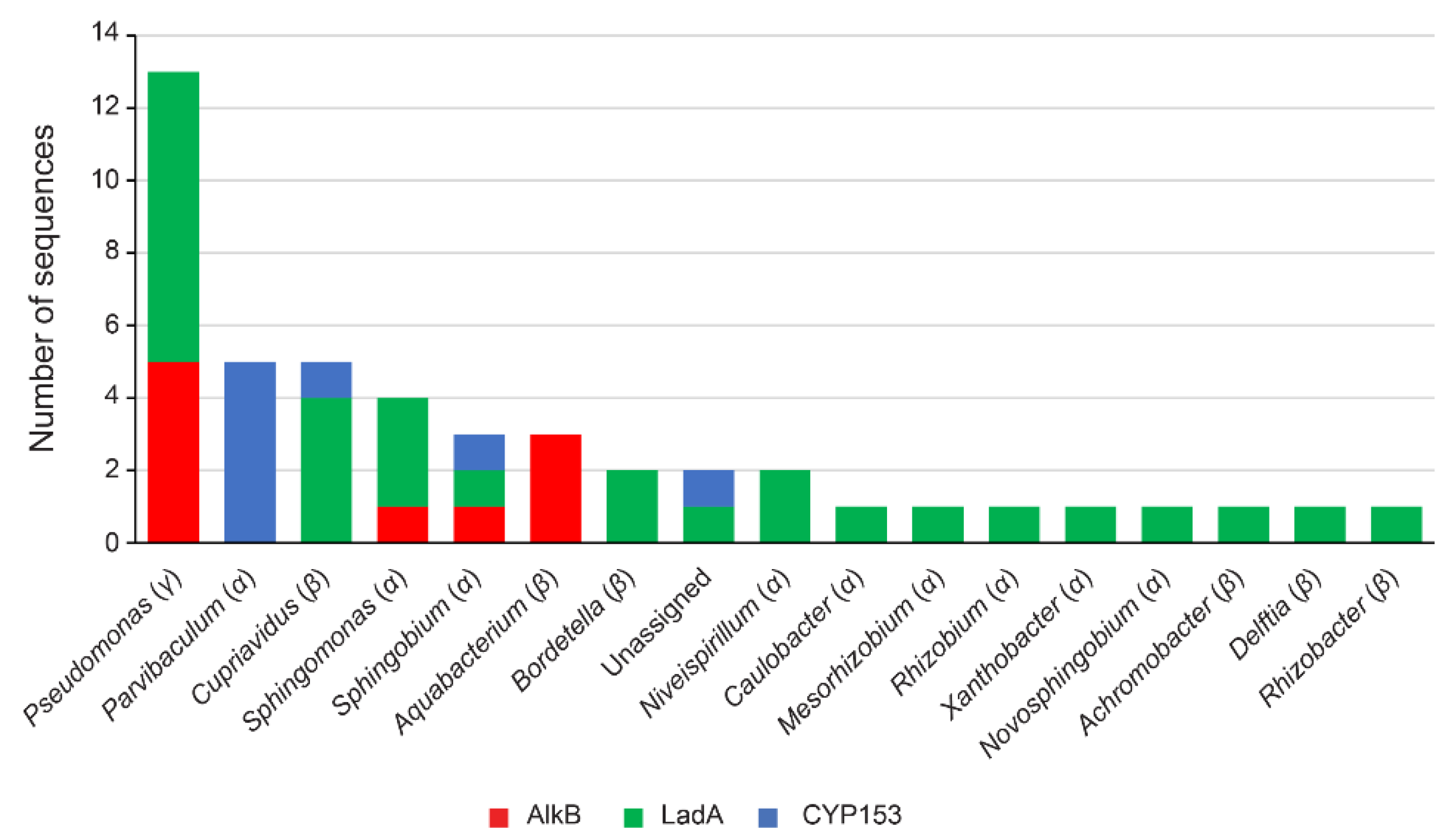
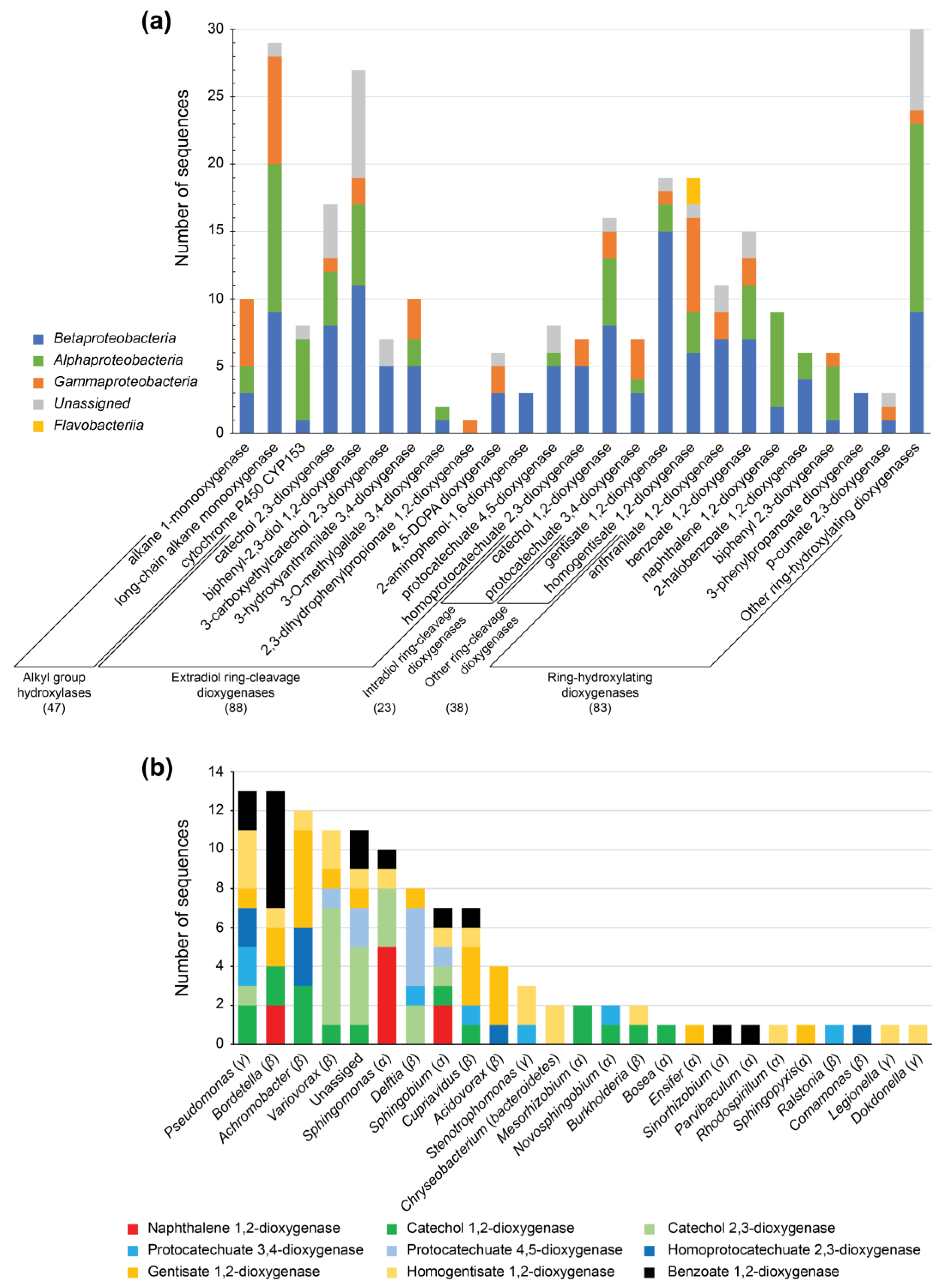
3.4. Identification of Alkane-Degrading CDSs
3.5. Identification of PAH-Degrading CDSs and Central Aromatic Metabolism CDSs
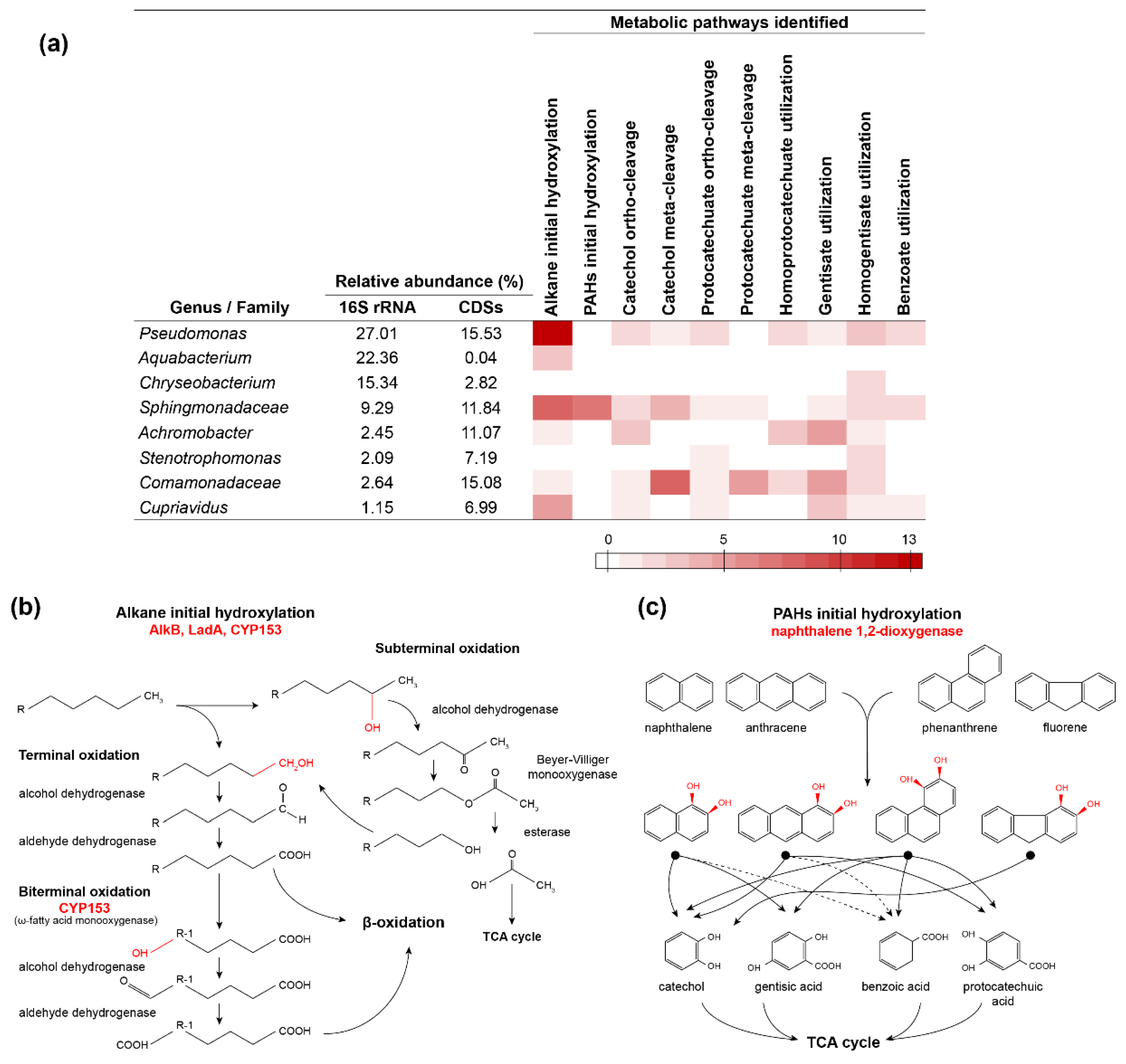
3.6. Metabolic Roles of Specific Populations in the Diesel-Degrading Consortium
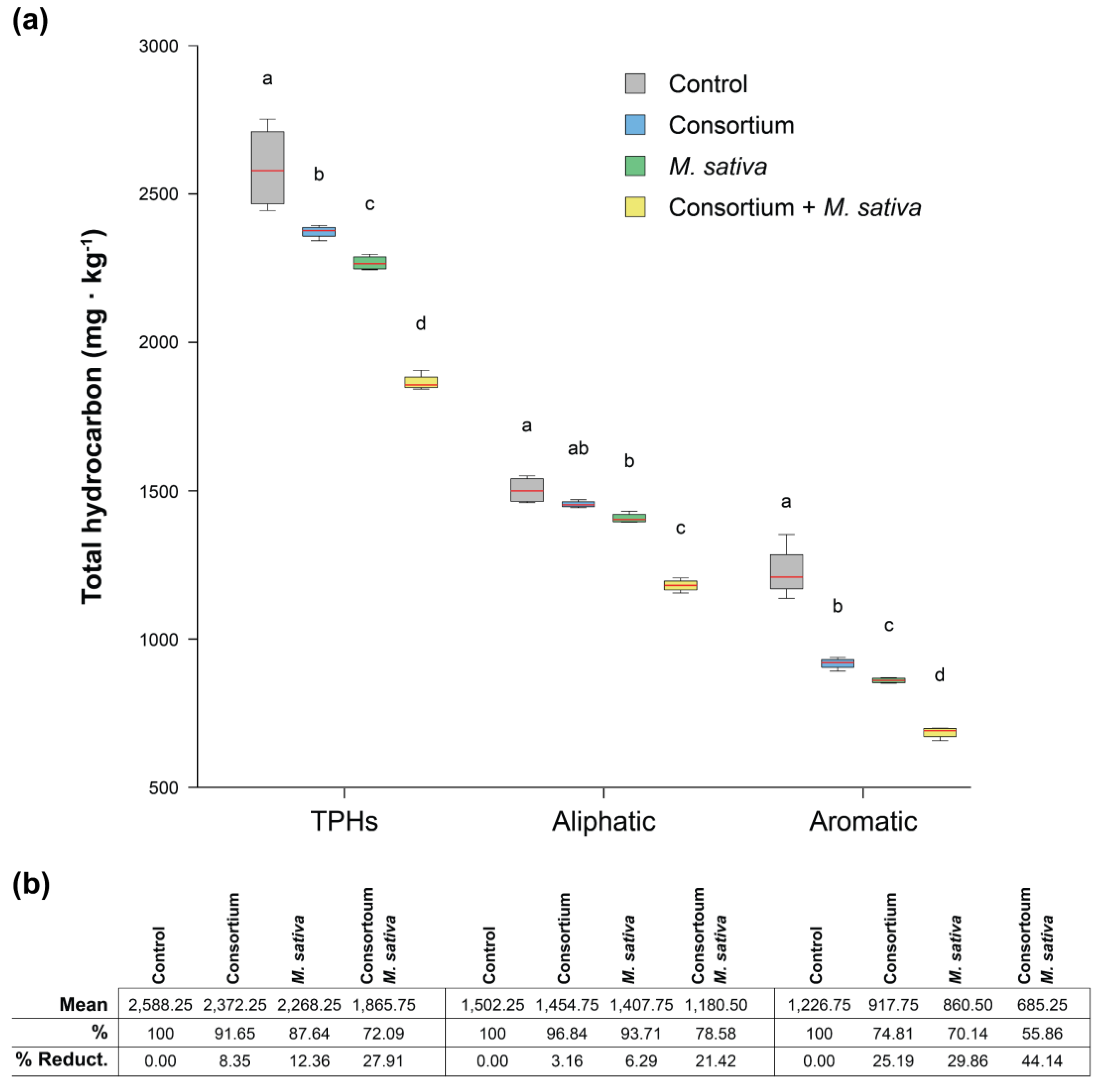
3.7. Rhizoremediation Assays in Diesel-Polluted Soil Microcosms
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Risher, J.; Rhodes, S. Toxicological Profile for Fuel Oils; U.S. Department of Health and Human Services, Public Health Service, Agency for Toxic Substances and Disease Registry: Atlanta, GA, USA, 1995.
- Nessel, C.S. A comprehensive evaluation of the carcinogenic potential of middle distillate fuels. Drug Chem. Toxicol. 1999, 22, 165–180. [Google Scholar] [CrossRef] [PubMed]
- Hentati, O.; Lachhab, R.; Ayadi, M.; Ksibi, M. Toxicity assessment for petroleum-contaminated soil using terrestrial invertebrates and plant bioassays. Environ. Monit. Assess. 2013, 185, 2989–2998. [Google Scholar] [CrossRef] [PubMed]
- Ramadass, K.; Megharaj, M.; Venkateswarlu, K.; Naidu, R. Ecological implications of motor oil pollution: Earthworm survival and soil health. Soil Biol. Biochem. 2015, 85, 72–81. [Google Scholar] [CrossRef]
- Pulles, T.; van der Gon, H.D.; Appelman, W.; Verheul, M. Emission factors for heavy metals from diesel and petrol used in European vehicles. Atmos. Environ. 2012, 61, 641–651. [Google Scholar] [CrossRef]
- Riis, V.; Babel, W.; Pucci, O.H. Influence of heavy metals on the microbial degradation of diesel fuel. Chemosphere 2002, 49, 559–568. [Google Scholar] [CrossRef]
- Ji, Y.; Mao, G.; Wang, Y.; Bartlam, M. Structural insights into diversity and n-alkane biodegradation mechanisms of alkane hydroxylases. Front. Microbiol. 2013, 4, 58. [Google Scholar] [CrossRef] [PubMed]
- Van Beilen, J.; Li, Z.; Duetz, W.; Smits, T.; Witholt, B. Diversity of alkane hydroxylase systems in the environment. Oil Gas Sci. Technol. 2003, 58, 427–440. [Google Scholar] [CrossRef]
- Feng, L.; Wang, W.; Cheng, J.; Ren, Y.; Zhao, G.; Gao, C.; Tang, Y.; Liu, X.; Han, W.; Peng, X.; et al. Genome and proteome of long-chain alkane degrading Geobacillus thermodenitrificans NG80-2 isolated from a deep-subsurface oil reservoir. Proc. Natl. Acad. Sci. USA 2007, 104, 5602–5607. [Google Scholar] [CrossRef]
- Kotani, T.; Yamamoto, T.; Yurimoto, H.; Sakai, Y.; Kato, N. Propane monooxygenase and NAD+-dependent secondary alcohol dehydrogenase in propane metabolism by Gordonia sp. strain TY-5. J. Bacteriol. 2003, 185, 7120–7128. [Google Scholar] [CrossRef]
- Throne-Holst, M.; Wentzel, A.; Ellingsen, T.E.; Kotlar, H.K.; Zotchev, S.B. Identification of novel genes involved in long-chain n-alkane degradation by Acinetobacter sp. strain DSM 17874. Appl. Environ. Microbiol. 2007, 73, 3327–3332. [Google Scholar] [CrossRef]
- Coon, M.J. Omega oxygenases: Nonheme-iron enzymes and P450 cytochromes. Biochem. Biophys. Res. Commun. 2005, 338, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Van Beilen, J.B.; Wubbolts, M.G.; Witholt, B. Genetics of alkane oxidation by Pseudomonas oleovorans. Biodegradation 1994, 5, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Hamamura, N.; Yeager, C.M.; Arp, D.J. Two Distinct Monooxygenases for Alkane Oxidation in Nocardioides sp. Strain CF8. Appl. Environ. Microbiol. 2001, 67, 4992–4998. [Google Scholar] [CrossRef] [PubMed]
- Bihari, Z.; Szvetnik, A.; Szabó, Z.; Blastyák, A.; Zombori, Z.; Balázs, M.; Kiss, I. Functional analysis of long-chain n-alkane degradation by Dietzia spp. FEMS Microbiol. Lett. 2011, 316, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liu, X.; Yang, W.; Xu, F.; Wang, W.; Feng, L.; Bartlam, M.; Wang, L.; Rao, Z. Crystal structure of long-chain alkane monooxygenase (LadA) in complex with coenzyme FMN: Unveiling the long-chain alkane hydroxylase. J. Mol. Biol. 2008, 376, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Scheps, D.; Malca, S.H.; Hoffmann, H.; Nestl, B.M.; Hauer, B. Regioselective omega-hydroxylation of medium-chain n-alkanes and primary alcohols by CYP153 enzymes from Mycobacterium marinum and Polaromonas sp. strain JS666. Org. Biomol. Chem. 2011, 9, 6727–6733. [Google Scholar] [CrossRef]
- Cerniglia, C.E. Biodegradation of polycyclic aromatic hydrocarbons. Curr. Opin. Biotechnol. 1993, 4, 331–338. [Google Scholar] [CrossRef]
- Samanta, S.K.; Singh, O.V.; Jain, R.K. Polycyclic aromatic hydrocarbons: Environmental pollution and bioremediation. Trends Biotechnol. 2002, 20, 243–248. [Google Scholar] [CrossRef]
- Mallick, S.; Chakraborty, J.; Dutta, T.K. Role of oxygenases in guiding diverse metabolic pathways in the bacterial degradation of low-molecular-weight polycyclic aromatic hydrocarbons: A review. Crit. Rev. Microbiol. 2011, 37, 64–90. [Google Scholar] [CrossRef]
- Ensley, B.; Gibson, D. Naphthalene dioxygenase: Purification and properties of a terminal oxygenase component. J. Bacteriol. 1983, 155, 505–511. [Google Scholar]
- Jerina, D.M.; Selander, H.; Yagi, H.; Wells, M.C.; Davey, J.F.; Mahadevan, V.; Gibson, D.T. Dihydrodiols from anthracene and phenanthrene. J. Am. Chem. Soc. 1976, 98, 5988–5996. [Google Scholar] [CrossRef]
- Bouchez, M.; Blanchet, D.; Vandecasteele, J. Degradation of polycyclic aromatic hydrocarbons by pure strains and by defined strain associations: Inhibition phenomena and cometabolism. Appl. Microbiol. Biotechnol. 1995, 43, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Cunliffe, M.; Kertesz, M.A. Effect of Sphingobium yanoikuyae B1 inoculation on bacterial community dynamics and polycyclic aromatic hydrocarbon degradation in aged and freshly PAH-contaminated soils. Environ. Pollut. 2006, 144, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Dandie, C.E.; Weber, J.; Aleer, S.; Adetutu, E.M.; Ball, A.S.; Juhasz, A.L. Assessment of five bioaccessibility assays for predicting the efficacy of petroleum hydrocarbon biodegradation in aged contaminated soils. Chemosphere 2010, 81, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Ranc, B.; Faure, P.; Croze, V.; Simonnot, M.O. Selection of oxidant doses for in situ chemical oxidation of soils contaminated by polycyclic aromatic hydrocarbons (PAHs): A review. J. Hazard. Mater. 2016, 312, 280–297. [Google Scholar] [CrossRef] [PubMed]
- Solano-Serena, F.; Marchal, R.; Vandecasteele, J. Biodegradation of gasoline in the environment: From overall assessment to the case of recalcitrant hydrocarbons. Oil Gas Sci. Technol. 2001, 56, 479–498. [Google Scholar] [CrossRef]
- Gallego, J.L.R.; Sierra, C.; Permanyer, A.; Peláez, A.I.; Menéndez-Vega, D.; Sánchez, J. Full-scale remediation of a jet fuel-contaminated soil: Assessment of biodegradation, volatilization, and bioavailability. Water Air Soil Pollut. 2011, 217, 197–211. [Google Scholar] [CrossRef]
- Richard, J.; Vogel, T. Characterization of a soil bacterial consortium capable of degrading diesel fuel. Int. Biodeterior. Biodegrad. 1999, 44, 93–100. [Google Scholar] [CrossRef]
- Dos Santos, H.F.; Cury, J.C.; do Carmo, F.L.; dos Santos, A.L.; Tiedje, J.; van Elsas, J.D.; Rosado, A.S.; Peixoto, R.S. Mangrove bacterial diversity and the impact of oil contamination revealed by pyrosequencing: Bacterial proxies for oil pollution. PLoS ONE 2011, 6, e16943. [Google Scholar] [CrossRef]
- Margesin, R.; Hammerle, M.; Tscherko, D. Microbial activity and community composition during bioremediation of diesel-oil-contaminated soil: Effects of hydrocarbon concentration, fertilizers, and incubation time. Microb. Ecol. 2007, 53, 259–269. [Google Scholar] [CrossRef]
- Sutton, N.B.; Maphosa, F.; Morillo, J.A.; Al-Soud, W.A.; Langenhoff, A.A.; Grotenhuis, T.; Rijnaarts, H.H.; Smidt, H. Impact of long term diesel contamination on soil microbial community structure. Appl. Environ. Microbiol. 2013, 79, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Sanz, D.; Manzano, J.; Martin, M.; Redondo-Nieto, M.; Rivilla, R. Metagenomic Analysis of a Biphenyl-Degrading Soil Bacterial Consortium Reveals the Metabolic Roles of Specific Populations. Front. Microbiol. 2018, 9, 232. [Google Scholar] [CrossRef] [PubMed]
- Yergeau, E.; Sanschagrin, S.; Beaumier, D.; Greer, C.W. Metagenomic analysis of the bioremediation of diesel-contaminated Canadian high arctic soils. PLoS ONE 2012, 7, e30058. [Google Scholar] [CrossRef] [PubMed]
- Miya, R.K.; Firestone, M.K. Enhanced phenanthrene biodegradation in soil by slender oat root exudates and root debris. J. Environ. Qual. 2001, 30, 1911–1918. [Google Scholar] [CrossRef] [PubMed]
- Chaineau, C.; Morel, J.; Oudot, J. Biodegradation of fuel oil hydrocarbons in the rhizosphere of maize. J. Environ. Qual. 2000, 29, 569–578. [Google Scholar] [CrossRef]
- Kuiper, I.; Lagendijk, E.L.; Bloemberg, G.V.; Lugtenberg, B.J. Rhizoremediation: A beneficial plant-microbe interaction. Mol. Plant-Microbe Interact. 2004, 17, 6–15. [Google Scholar] [CrossRef]
- Liste, H.-H.; Felgentreu, D. Crop growth, culturable bacteria, and degradation of petrol hydrocarbons (PHCs) in a long-term contaminated field soil. Appl. Soil Ecol. 2006, 31, 43–52. [Google Scholar] [CrossRef]
- Wu, M.; Li, W.; Dick, W.A.; Ye, X.; Chen, K.; Kost, D.; Chen, L. Bioremediation of hydrocarbon degradation in a petroleum-contaminated soil and microbial population and activity determination. Chemosphere 2017, 169, 124–130. [Google Scholar] [CrossRef]
- Brazil, G.M.; Kenefick, L.; Callanan, M.; Haro, A.; de Lorenzo, V.; Dowling, D.N.; O’Gara, F. Construction of a rhizosphere pseudomonad with potential to degrade polychlorinated biphenyls and detection of bph gene expression in the rhizosphere. Appl. Environ. Microbiol. 1995, 61, 1946–1952. [Google Scholar]
- Bedard, D.L.; Unterman, R.; Bopp, L.H.; Brennan, M.J.; Haberl, M.L.; Johnson, C. Rapid assay for screening and characterizing microorganisms for the ability to degrade polychlorinated biphenyls. Appl. Environ. Microbiol. 1986, 51, 761–768. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. J. Comput. Mol. Cell Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F. QIIME 2: Reproducible, interactive, scalable, and extensible microbiome data science. PeerJ Preprints 2018, 6, e27295v2. [Google Scholar]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Caporaso, J.G. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2′s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Fåhraeus, G. The infection of clover root hairs by nodule bacteria studied by a simple glass slide technique. Microbiology 1957, 16, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, O.P.; Pastor, R.M.P.; Segovia, O.E. An analytical method for quantifying petroleum hydrocarbon fractions in soils, and its associated uncertainties. Anal. Methods 2014, 6, 5527–5536. [Google Scholar] [CrossRef]
- García-Alonso, S.; Pérez-Pastor, R.; García-Frutos, F. An evaluation of analytical quality for selected PAH measurements in a fuel-contaminated soil. Accredit. Qual. Assur. 2011, 16, 369–377. [Google Scholar] [CrossRef]
- Leahy, J.G.; Colwell, R.R. Microbial degradation of hydrocarbons in the environment. Microbiol. Rev. 1990, 54, 305–315. [Google Scholar]
- Whyte, L.G.; Schultz, A.; Beilen, J.B.; Luz, A.P.; Pellizari, V.; Labbe, D.; Greer, C.W. Prevalence of alkane monooxygenase genes in Arctic and Antarctic hydrocarbon-contaminated and pristine soils. FEMS Microbiol. Ecol. 2002, 41, 141–150. [Google Scholar] [CrossRef]
- Gontikaki, E.; Potts, L.D.; Anderson, J.A.; Witte, U. Hydrocarbon-degrading bacteria in deep-water subarctic sediments (Faroe-Shetland Channel). J. Appl. Microbiol. 2018, 125, 1040–1053. [Google Scholar] [CrossRef]
- Viggor, S.; Joesaar, M.; Vedler, E.; Kiiker, R.; Parnpuu, L.; Heinaru, A. Occurrence of diverse alkane hydroxylase alkB genes in indigenous oil-degrading bacteria of Baltic Sea surface water. Mar. Pollut. Bull. 2015, 101, 507–516. [Google Scholar] [CrossRef]
- Wald, J.; Hroudova, M.; Jansa, J.; Vrchotova, B.; Macek, T.; Uhlik, O. Pseudomonads Rule Degradation of Polyaromatic Hydrocarbons in Aerated Sediment. Front. Microbiol. 2015, 6, 1268. [Google Scholar] [CrossRef]
- Rentz, J.A.; Alvarez, P.J.; Schnoor, J.L. Benzo[a]pyrene degradation by Sphingomonas yanoikuyae JAR02. Environ. Pollut. 2008, 151, 669–677. [Google Scholar] [CrossRef]
- Van Herwijnen, R.; Wattiau, P.; Bastiaens, L.; Daal, L.; Jonker, L.; Springael, D.; Govers, H.A.; Parsons, J.R. Elucidation of the metabolic pathway of fluorene and cometabolic pathways of phenanthrene, fluoranthene, anthracene and dibenzothiophene by Sphingomonas sp. LB126. Res. Microbiol. 2003, 154, 199–206. [Google Scholar] [CrossRef]
- Kotani, T.; Yurimoto, H.; Kato, N.; Sakai, Y. Novel acetone metabolism in a propane-utilizing bacterium, Gordonia sp. strain TY-5. J. Bacteriol. 2007, 189, 886–893. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, J.; ICHIKAWA, Y.; Sagae, H.; Komura, I.; Kanou, H.; Yamada, K. Isolation and identification of n-butane-assimilating bacterium. Agric. Biol. Chem. 1980, 44, 1835–1840. [Google Scholar] [CrossRef][Green Version]
- Hauben, L.; Vauterin, L.; Moore, E.R.; Hoste, B.; Swings, J. Genomic diversity of the genus Stenotrophomonas. Int. J. Syst. Bacteriol. 1999, 49 Pt 4, 1749–1760. [Google Scholar] [CrossRef]
- Zanaroli, G.; Di Toro, S.; Todaro, D.; Varese, G.C.; Bertolotto, A.; Fava, F. Characterization of two diesel fuel degrading microbial consortia enriched from a non acclimated, complex source of microorganisms. Microb. Cell Fact. 2010, 9, 10. [Google Scholar] [CrossRef] [PubMed]
- Kok, M.; Oldenhuis, R.; van der Linden, M.P.; Raatjes, P.; Kingma, J.; van Lelyveld, P.H.; Witholt, B. The Pseudomonas oleovorans alkane hydroxylase gene. Sequence and expression. J. Biol. Chem. 1989, 264, 5435–5441. [Google Scholar] [PubMed]
- Maier, T.; Forster, H.H.; Asperger, O.; Hahn, U. Molecular characterization of the 56-kDa CYP153 from Acinetobacter sp. EB104. Biochem. Biophys. Res. Commun. 2001, 286, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Resnick, S.; Lee, K.; Gibson, D. Diverse reactions catalyzed by naphthalene dioxygenase from Pseudomonas sp strain NCIB 9816. J. Ind. Microbiol. 1996, 17, 438–457. [Google Scholar]
- Ferraro, D.J.; Okerlund, A.; Brown, E.; Ramaswamy, S. One enzyme, many reactions: Structural basis for the various reactions catalyzed by naphthalene 1,2-dioxygenase. IUCrJ 2017, 4, 648–656. [Google Scholar] [CrossRef]
- Selifonov, S.A.; Grifoll, M.; Eaton, R.W.; Chapman, P.J. Oxidation of naphthenoaromatic and methyl-substituted aromatic compounds by naphthalene 1,2-dioxygenase. Appl. Environ. Microbiol. 1996, 62, 507–514. [Google Scholar]
- Pilon-Smits, E. Phytoremediation. Annu. Rev. Plant Biol. 2005, 56, 15–39. [Google Scholar] [CrossRef] [PubMed]
- Del Reino, S.; Rodríguez-Rastrero, M.; Escolano, O.; Welte, L.; Bueno, J.; Fernández, J.; Schmid, T.; Millán, R. In Situ Chemical Oxidation Based on Hydrogen Peroxide: Optimization of Its Application to an Hydrocarbon Polluted Site. In Environment, Energy and Climate Change I; Springer: Berlin, Germany, 2014; pp. 207–228. [Google Scholar]
- Huang, X.-D.; El-Alawi, Y.; Gurska, J.; Glick, B.R.; Greenberg, B.M. A multi-process phytoremediation system for decontamination of persistent total petroleum hydrocarbons (TPHs) from soils. Microchem. J. 2005, 81, 139–147. [Google Scholar] [CrossRef]
- Singer, A.C.; Crowley, D.E.; Thompson, I.P. Secondary plant metabolites in phytoremediation and biotransformation. Trends Biotechnol. 2003, 21, 123–130. [Google Scholar] [CrossRef]
- Nichols, T.; Wolf, D.; Rogers, H.; Beyrouty, C.; Reynolds, C. Rhizosphere microbial populations in contaminated soils. Water Air Soil Pollut. 1997, 95, 165–178. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TPH Fraction | Diesel Oil (mg·mL−1) | Soil (µg·g−1) |
|---|---|---|
| Aliphatic hydrocarbons | ||
| >C10–C12 | 82 ± 1 | 3.5 ± 0.5 |
| >C12–C16 | 257 ± 7 | 151 ± 4 |
| >C16–C21 | 283 ± 8 | 563 ± 28 |
| >C21–C35 | 55 ± 4 | 1086 ± 73 |
| >C35 | 0.05 ± 0.001 | 116 ± 16 |
| Aromatic hydrocarbons | ||
| >EC10–C12 | 17 ± 1 | 11 ± 5 |
| >EC12–C16 | 13 ± 1 | 8 ± 1 |
| >EC16–C21 | 57 ± 3 | 484 ± 48 |
| >EC21–C35 | 2 ± 0.1 | 530 ± 70 |
| >EC35 | 0.1 ± 0.004 | 22 ± 4 |
| TPHs | 764 ± 7 | 2974 ± 143 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garrido-Sanz, D.; Redondo-Nieto, M.; Guirado, M.; Pindado Jiménez, O.; Millán, R.; Martin, M.; Rivilla, R. Metagenomic Insights into the Bacterial Functions of a Diesel-Degrading Consortium for the Rhizoremediation of Diesel-Polluted Soil. Genes 2019, 10, 456. https://doi.org/10.3390/genes10060456
Garrido-Sanz D, Redondo-Nieto M, Guirado M, Pindado Jiménez O, Millán R, Martin M, Rivilla R. Metagenomic Insights into the Bacterial Functions of a Diesel-Degrading Consortium for the Rhizoremediation of Diesel-Polluted Soil. Genes. 2019; 10(6):456. https://doi.org/10.3390/genes10060456
Chicago/Turabian StyleGarrido-Sanz, Daniel, Miguel Redondo-Nieto, María Guirado, Oscar Pindado Jiménez, Rocío Millán, Marta Martin, and Rafael Rivilla. 2019. "Metagenomic Insights into the Bacterial Functions of a Diesel-Degrading Consortium for the Rhizoremediation of Diesel-Polluted Soil" Genes 10, no. 6: 456. https://doi.org/10.3390/genes10060456
APA StyleGarrido-Sanz, D., Redondo-Nieto, M., Guirado, M., Pindado Jiménez, O., Millán, R., Martin, M., & Rivilla, R. (2019). Metagenomic Insights into the Bacterial Functions of a Diesel-Degrading Consortium for the Rhizoremediation of Diesel-Polluted Soil. Genes, 10(6), 456. https://doi.org/10.3390/genes10060456