Temporal Dynamics of DNA Methylation Patterns in Response to Rearing Juvenile Steelhead (Oncorhynchus mykiss) in a Hatchery versus Simulated Stream Environment

 ,
,
Abstract
1. Introduction
2. Materials and Methods
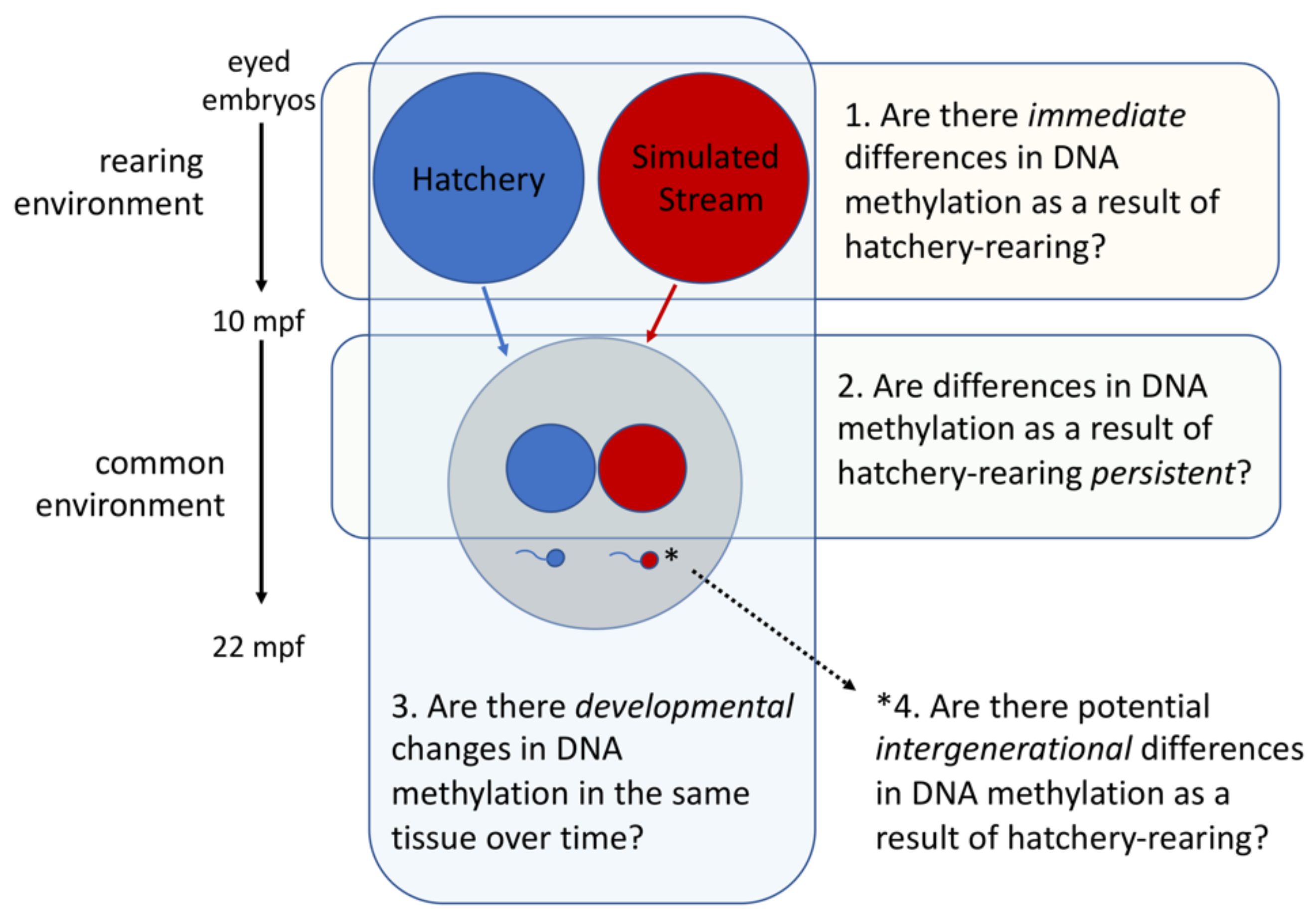
2.1. Experimental Design
2.1.1. Overview
2.1.2. Study Population and Crosses
2.1.3. Stream Rearing Treatment
2.1.4. Hatchery Rearing Treatment
2.1.5. Testing for Immediate Effects of Rearing Environment
2.1.6. Testing for Persistent Effects of Early Rearing Environment
2.1.7. Testing for Potential Intergenerational Effects of Early Rearing Environment
2.2. DNA Based Assays to Identify Genetic Sex and Family
2.2.1. Genetic Sex
2.2.2. Parentage Analysis
2.3. RRBS Library Preparation and Data Analysis
2.3.1. DNA Isolation: Liver
2.3.2. DNA Isolation: Sperm
2.3.3. RRBS Library Preparation
2.3.4. Sequence Trimming and Mapping
2.3.5. Identifying Differences in DNA Methylation
3. Results
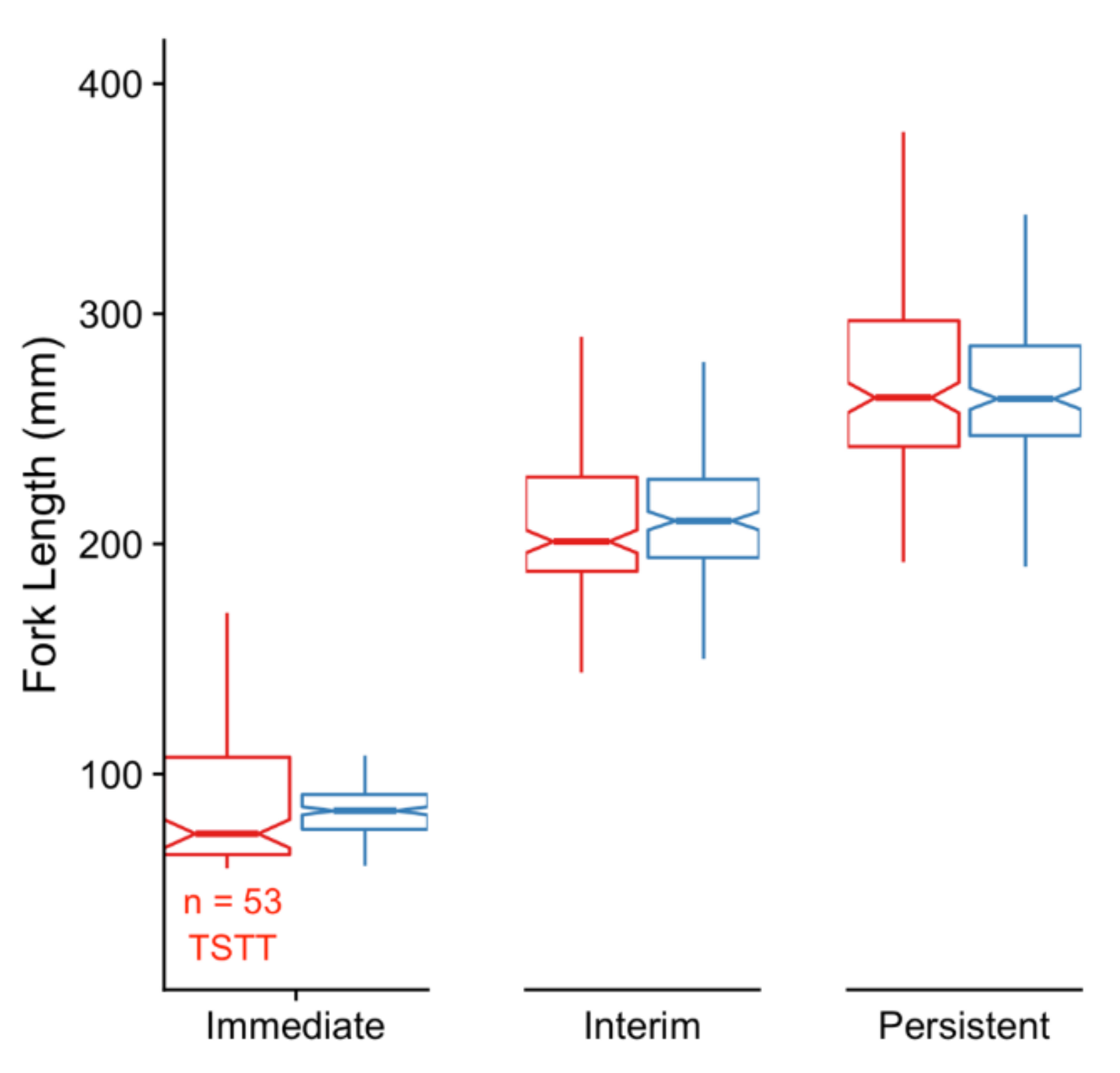
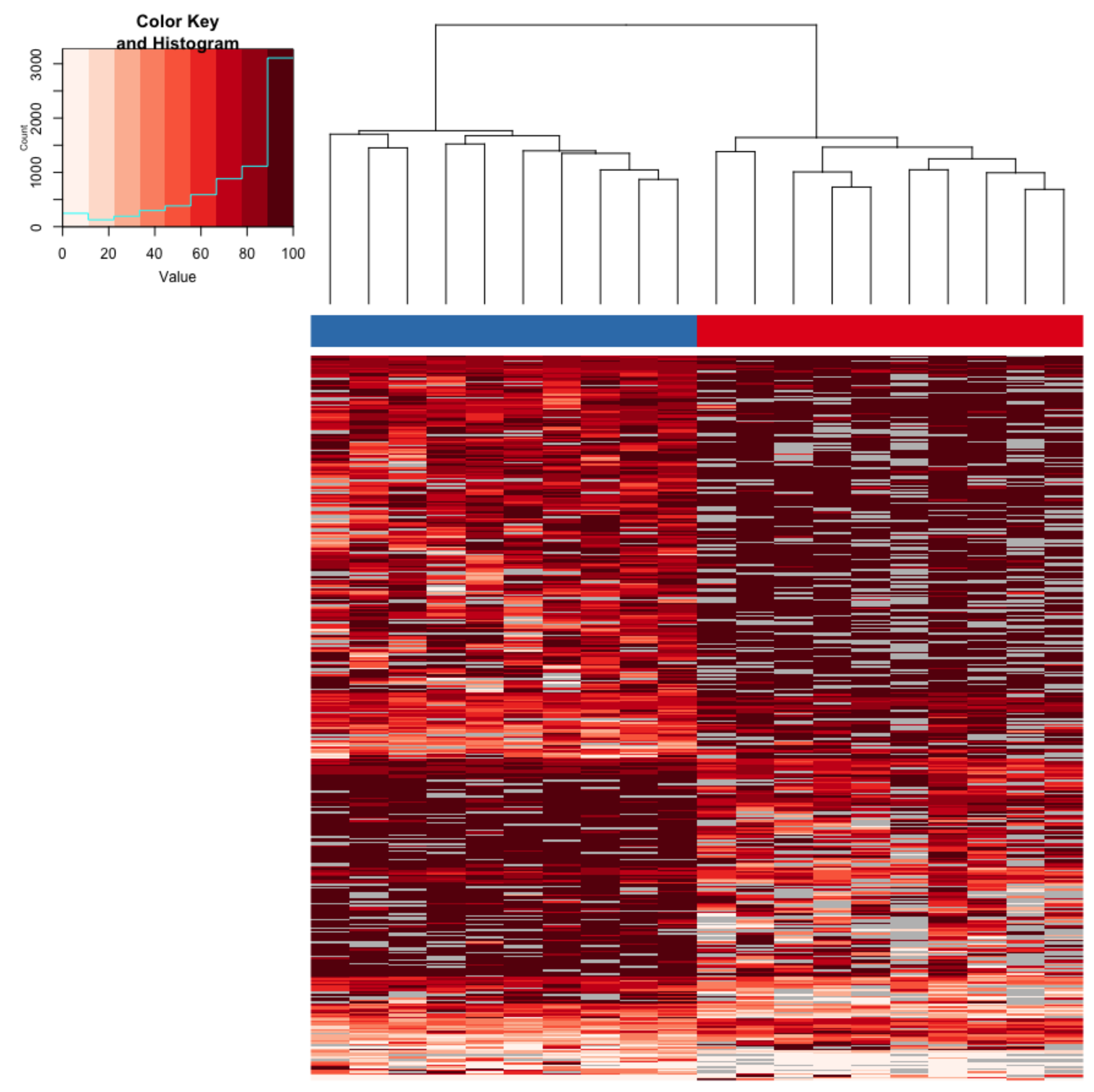
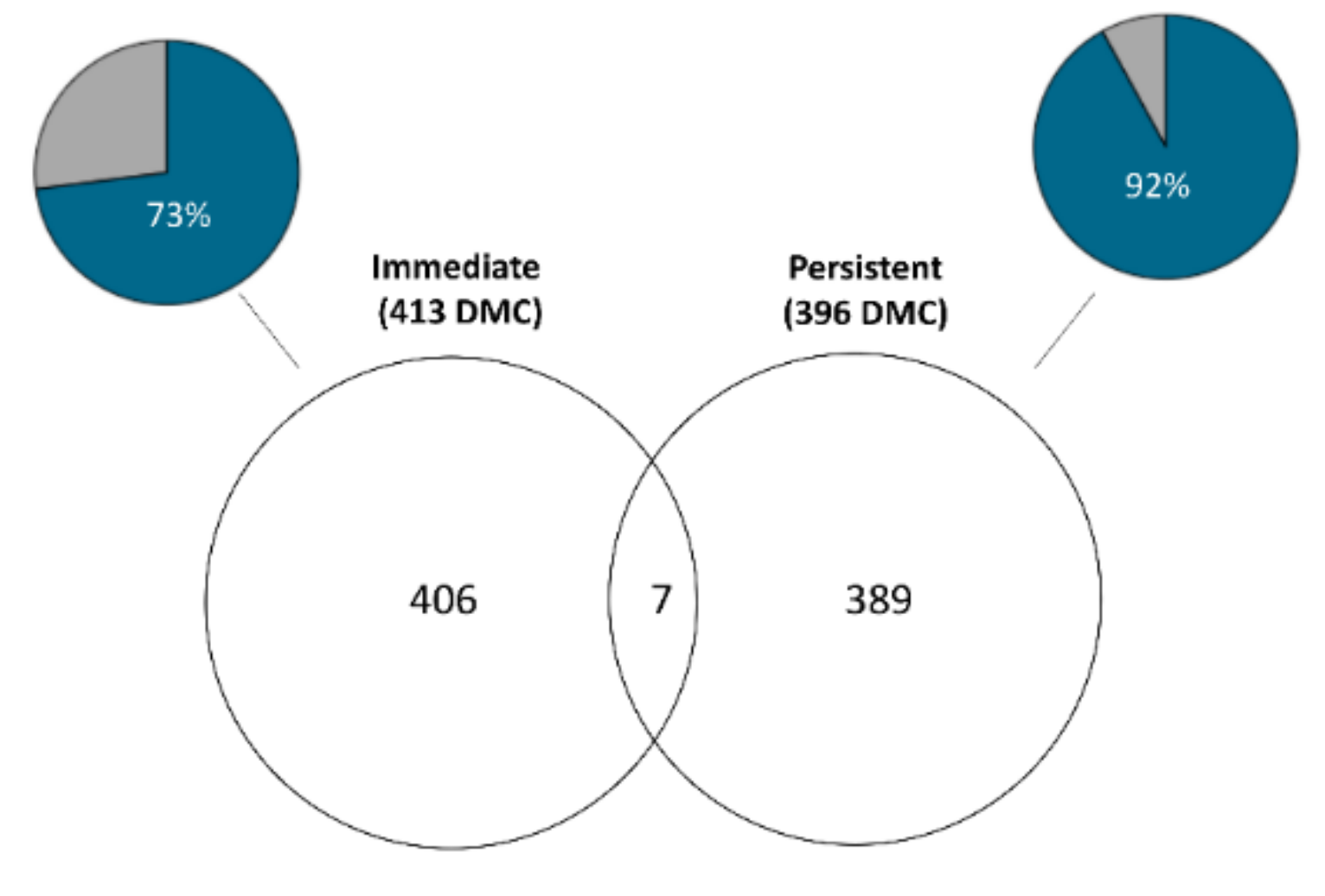
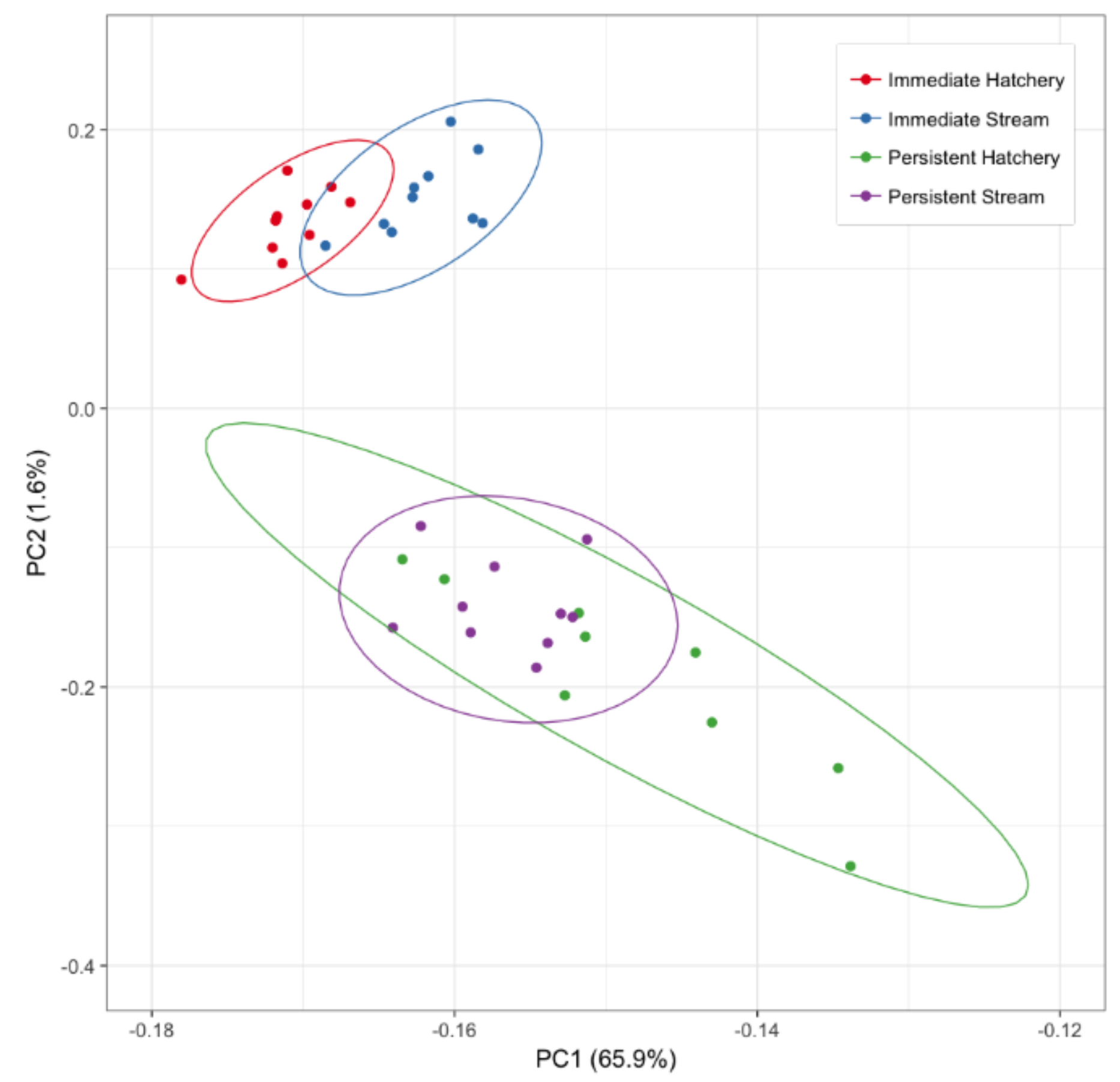
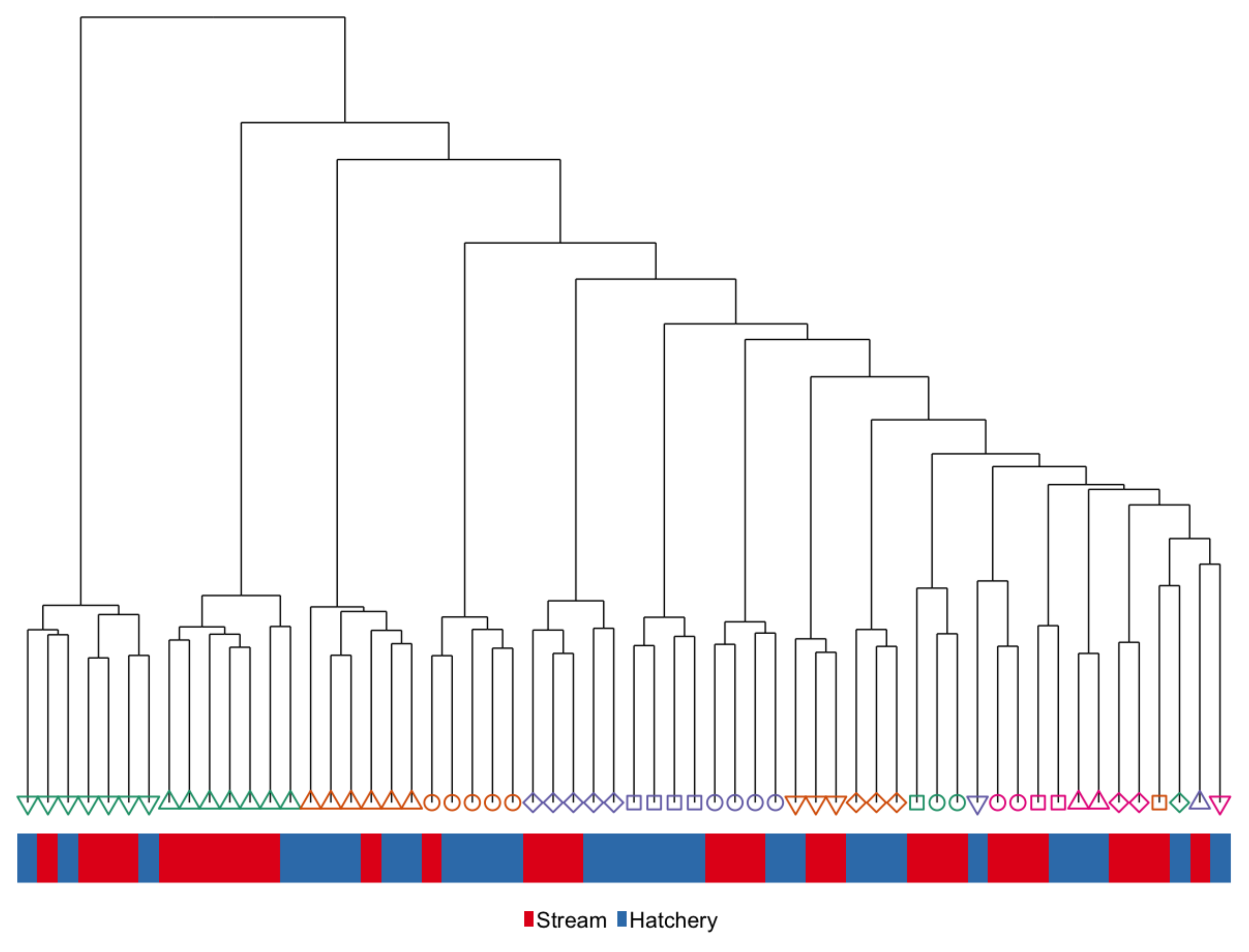
3.1. Immediate Effects: Differences in DNA Methylation in the Liver of Hatchery vs. Stream-Reared Males (Age-1 Fish)
3.2. Persistent Effects: DNA Methylation Differences in the Liver of Hatchery vs. Stream Reared Males After One Year in a Common Environment (Age-2 Fish)
3.3. Developmental Effects: Differences in DNA Methylation in the Liver of Age-1 vs. Age-2 Males
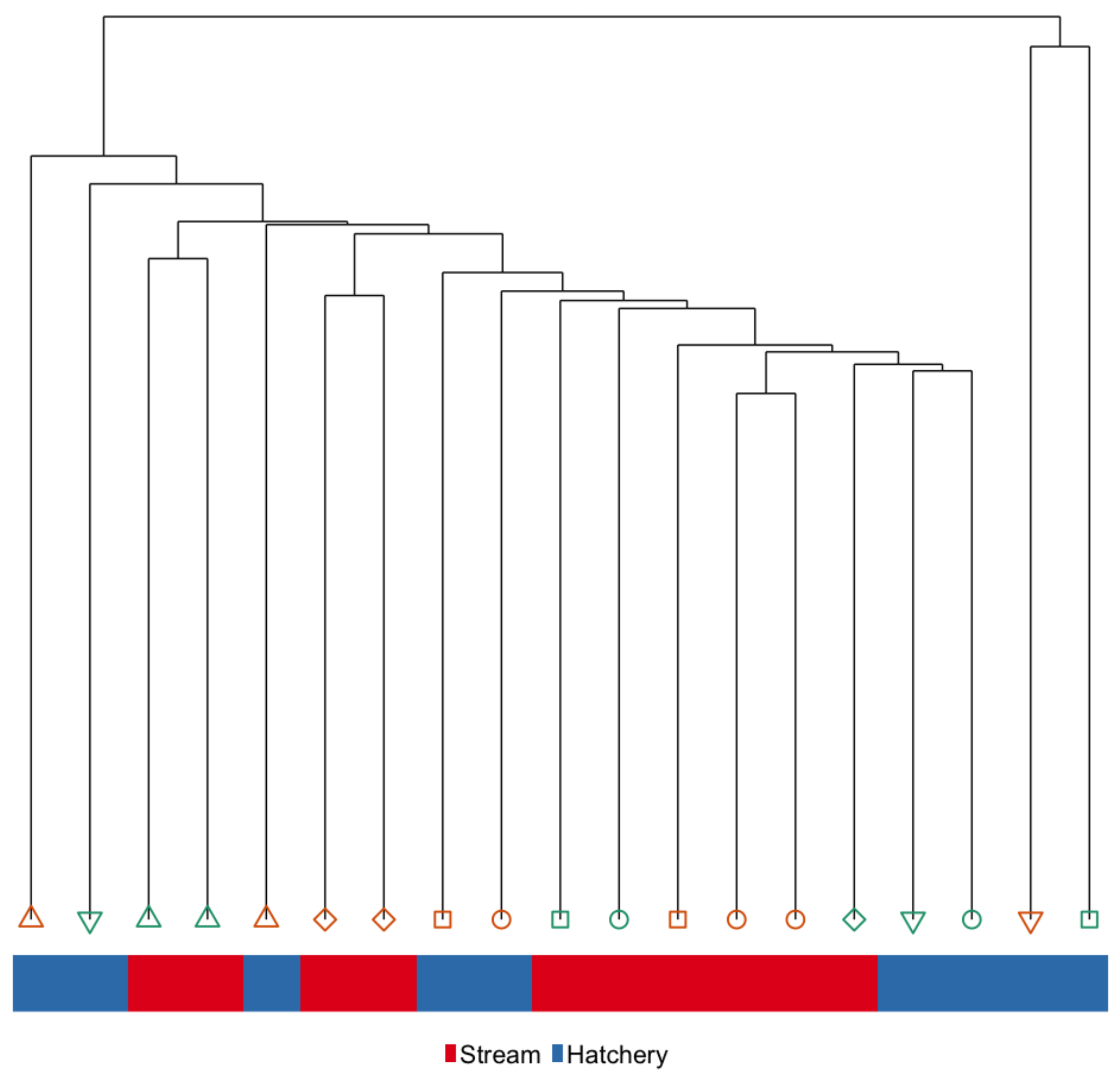
3.4. Intergenerational Effects: Differences in DNA Methylation in the Sperm of Hatchery vs. Stream-Reared Reared Fish After One Year in a Common Hatchery Environment
4. Discussion
4.1. Immediate
4.2. Persistent
4.3. Developmental
4.4. Intergenerational
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ford, M.J. Selection in captivity during supportive breeding may reduce fitness in the wild. Conserv. Biol. 2002, 16, 815–825. [Google Scholar] [CrossRef]
- Frankham, R. Genetic adaptation to captivity in species conservation programs. Mol. Ecol. 2008, 17, 325–333. [Google Scholar] [CrossRef]
- Fraser, D.J. How well can captive breeding programs conserve biodiversity? A review of salmonids. Evol. Appl. 2008, 1, 535–586. [Google Scholar] [CrossRef] [PubMed]
- Christie, M.R.; Marine, M.L.; Fox, S.E.; French, R.A.; Blouin, M.S. A single generation of domestication heritably alters the expression of hundreds of genes. Nat. Commun. 2016, 7, 10676. [Google Scholar] [CrossRef]
- Ford, M.J.; Murdoch, A.R.; Hughes, M.S.; Seamons, T.R.; LaHood, E.S. Broodstock History Strongly Influences Natural Spawning Success in Hatchery Steelhead (Oncorhynchus mykiss). PLoS ONE 2016, 11, e0164801. [Google Scholar] [CrossRef]
- Araki, H.; Ardren, W.R.; Olsen, E.; Cooper, B.; Blouin, M.S. Reproductive success of captive-bred steelhead trout in the wild: evaluation of three hatchery programs in the Hood river. Conserv. Biol. 2007, 21, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Araki, H.; Cooper, B.; Blouin, M.S. Genetic effects of captive breeding cause a rapid, cumulative fitness decline in the wild. Science 2007, 318, 100–103. [Google Scholar] [CrossRef]
- Bateson, P.; Barker, D.; Clutton-Brock, T.; Deb, D.; D’Udine, B.; Foley, R.A.; Gluckman, P.; Godfrey, K.; Kirkwood, T.; Lahr, M.M.; et al. Developmental plasticity and human health. Nature 2004, 430, 419–421. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, B.; Jonsson, N. Early environment influences later performance in fishes. J. Fish Biol. 2014, 85, 151–188. [Google Scholar] [CrossRef] [PubMed]
- Lea, A.J.; Tung, J.; Archie, E.A.; Alberts, S.C. Developmental plasticity: Bridging research in evolution and human health. Evol Med Public Health 2017, 2017, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.C.K. Environmental quality, developmental plasticity and the thrifty phenotype: a review of evolutionary models. Evol. Bioinform. Online 2007, 3, 109–120. [Google Scholar] [CrossRef]
- Berejikian, B.A.; Larsen, D.A.; Swanson, P.; Moore, M.E.; Tatara, C.P.; Gale, W.L.; Pasley, C.R.; Beckman, B.R. Development of natural growth regimes for hatchery-reared steelhead to reduce residualism, fitness loss, and negative ecological interactions. Environ. Biol. Fishes 2012, 94, 29–44. [Google Scholar] [CrossRef]
- Jonsson, B.; Jonsson, N. Egg incubation temperature affects the timing of the Atlantic salmon Salmo salar homing migration. J. Fish Biol. 2018, 93, 1016–1020. [Google Scholar] [CrossRef] [PubMed]
- Gavery, M.R.; Roberts, S.B. Epigenetic considerations in aquaculture. PeerJ 2017, 5, e4147. [Google Scholar] [CrossRef] [PubMed]
- Lea, A.J.; Vilgalys, T.P.; Durst, P.A.P.; Tung, J. Maximizing ecological and evolutionary insight in bisulfite sequencing data sets. Nat. Ecol. Evol. 2017, 1, 1074–1083. [Google Scholar] [CrossRef]
- Houston, R.D.; Macqueen, D.J. Atlantic salmon (Salmo salar L.) genetics in the 21st century: taking leaps forward in aquaculture and biological understanding. Anim. Genet. 2018. [Google Scholar] [CrossRef]
- Jablonka, E.; Lamb, M.J. The changing concept of epigenetics. Ann. N. Y. Acad. Sci. 2002, 981, 82–96. [Google Scholar] [CrossRef]
- Feil, R.; Fraga, M.F. Epigenetics and the environment: emerging patterns and implications. Nat. Rev. Genet. 2012, 13, 97–109. [Google Scholar] [CrossRef]
- Szyf, M.; Bick, J. DNA methylation: A mechanism for embedding early life experiences in the genome. Child Dev. 2013, 84, 49–57. [Google Scholar] [CrossRef]
- El-Heis, S.; Godfrey, K. Developmental origins of health and disease. Obstet. Gynaecol. Reprod. Med. 2015, 25, 236–238. [Google Scholar]
- Navarro-Martín, L.; Viñas, J.; Ribas, L.; Díaz, N.; Gutiérrez, A.; Di Croce, L.; Piferrer, F. DNA Methylation of the Gonadal Aromatase (cyp19a) Promoter Is Involved in Temperature-Dependent Sex Ratio Shifts in the European Sea Bass. PLoS Genet. 2011, 7, e1002447. [Google Scholar] [CrossRef] [PubMed]
- Geurden, I.; Borchert, P.; Balasubramanian, M.N.; Schrama, J.W.; Dupont-Nivet, M.; Quillet, E.; Kaushik, S.J.; Panserat, S.; Médale, F. The Positive Impact of the Early-Feeding of a Plant-Based Diet on Its Future Acceptance and Utilisation in Rainbow Trout. PLoS ONE 2013, 8, e83162. [Google Scholar] [CrossRef]
- Balasubramanian, M.N.; Panserat, S.; Dupont-Nivet, M.; Quillet, E.; Montfort, J.; Le Cam, A.; Medale, F.; Kaushik, S.J.; Geurden, I. Molecular pathways associated with the nutritional programming of plant-based diet acceptance in rainbow trout following an early feeding exposure. BMC Genomics 2016, 17, 449. [Google Scholar] [CrossRef] [PubMed]
- Burgerhout, E.; Mommens, M.; Johnsen, H.; Aunsmo, A.; Santi, N.; Andersen, Ø. Genetic background and embryonic temperature affect DNA methylation and expression of myogenin and muscle development in Atlantic salmon (Salmo salar). PLoS ONE 2017, 12, e0179918. [Google Scholar] [CrossRef]
- Moghadam, H.K.; Johnsen, H.; Robinson, N.; Andersen, Ø.; H Jørgensen, E.; Johnsen, H.K.; Bæhr, V.J.; Tveiten, H. Impacts of Early Life Stress on the Methylome and Transcriptome of Atlantic Salmon. Sci. Rep. 2017, 7, 5023. [Google Scholar] [CrossRef] [PubMed]
- Le Luyer, J.; Laporte, M.; Beacham, T.D.; Kaukinen, K.H.; Withler, R.E.; Leong, J.S.; Rondeau, E.B.; Koop, B.F.; Bernatchez, L. Parallel epigenetic modifications induced by hatchery rearing in a Pacific salmon. Proc. Natl. Acad. Sci. USA 2017, 114, 12964–12969. [Google Scholar] [CrossRef] [PubMed]
- Herb, B.R.; Wolschin, F.; Hansen, K.D.; Aryee, M.J.; Langmead, B.; Irizarry, R.; Amdam, G.V.; Feinberg, A.P. Reversible switching between epigenetic states in honeybee behavioral subcastes. Nat. Neurosci. 2012, 15, 1371–1373. [Google Scholar] [CrossRef] [PubMed]
- Burggren, W.W. Dynamics of epigenetic phenomena: intergenerational and intragenerational phenotype “washout”. J. Exp. Biol. 2015, 218, 80–87. [Google Scholar] [CrossRef]
- Metzger, D.C.H.; Schulte, P.M. Persistent and plastic effects of temperature on DNA methylation across the genome of threespine stickleback (Gasterosteus aculeatus). Proc. R. Soc. B Biol. Sci. 2017, 284, 20171667. [Google Scholar] [CrossRef]
- Gavery, M.R.; Nichols, K.M.; Goetz, G.W.; Middleton, M.A.; Swanson, P. Characterization of Genetic and Epigenetic Variation in Sperm and Red Blood Cells from Adult Hatchery and Natural-Origin Steelhead, Oncorhynchus mykiss. G3 2018, 8, 3723–3736. [Google Scholar] [CrossRef] [PubMed]
- Berejikian, B.A.; Tezak, E.P.; LaRae, A.L. Female mate choice and spawning behavior of Chinook salmon (Oncorhynchus tshawytscha) under experimental conditions. J Fish Biol. 2000, 57, 647–661. [Google Scholar] [CrossRef]
- Yano, A.; Nicol, B.; Jouanno, E.; Quillet, E.; Fostier, A.; Guyomard, R.; Guiguen, Y. The sexually dimorphic on the Y-chromosome gene (sdY) is a conserved male-specific Y-chromosome sequence in many salmonids. Evol. Appl. 2013, 6, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Tatara, C.P.; Larsen, D.A.; Cooper, M.R.; Swanson, P.; Middleton, M.A.; Dickey, J.T.; Humling, M.; Pasley, C.R.; Berejikian, B.A. Age at release, size, and maturation status influence residualism in hatchery steelhead. N. Am. J. Fish. Manage. 2019. [Google Scholar] [CrossRef]
- Steele, C.A.; Anderson, E.C.; Ackerman, M.W.; Hess, M.A.; Campbell, N.R.; Narum, S.R.; Campbell, M.R. A validation of parentage-based tagging using hatchery steelhead in the Snake River basin. Can. J. Fish. Aquat. Sci. 2013, 70, 1046–1054. [Google Scholar] [CrossRef]
- Riester, M.; Stadler, P.F.; Klemm, K. FRANz: reconstruction of wild multi-generation pedigrees. Bioinformatics 2009, 25, 2134–2139. [Google Scholar] [CrossRef] [PubMed]
- Boyle, P.; Clement, K.; Gu, H.; Smith, Z.D.; Ziller, M.; Fostel, J.L.; Holmes, L.; Meldrim, J.; Kelley, F.; Gnirke, A.; et al. Gel-free multiplexed reduced representation bisulfite sequencing for large-scale DNA methylation profiling. Genome Biol. 2012, 13, R92. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 1 January 2018).
- Pearse, D.E.; Barson, N.J.; Nome, T.; Gao, G.; Campbell, M.A.; Abadía-Cardoso, A.; Anderson, E.C.; Rundio, D.E.; Williams, T.H.; Naish, K.A.; et al. Sex-dependent dominance maintains migration supergene in rainbow trout. bioRxiv 2018, 504621. [Google Scholar]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Zou, D.; Mao, L.; Liu, H.; Song, P.; Chen, Y.; Zhao, S.; Gao, C.; Li, X.; Gao, Z.; et al. BS-SNPer: SNP calling in bisulfite-seq data. Bioinformatics 2015, 31, 4006–4008. [Google Scholar] [CrossRef]
- Lea, A.J.; Tung, J.; Zhou, X. A Flexible, Efficient Binomial Mixed Model for Identifying Differential DNA Methylation in Bisulfite Sequencing Data. PLoS Genet. 2015, 11, e1005650. [Google Scholar] [CrossRef] [PubMed]
- Pembleton, L.W.; Cogan, N.O.I.; Forster, J.W. StAMPP: an R package for calculation of genetic differentiation and structure of mixed-ploidy level populations. Mol. Ecol. Resour. 2013, 13, 946–952. [Google Scholar] [CrossRef] [PubMed]
- Storey, J.D. A direct approach to false discovery rates. J. R. Stat. Soc. Series B Stat. Methodol. 2002, 64, 479–498. [Google Scholar] [CrossRef]
- Lea, A.J.; Altmann, J.; Alberts, S.C.; Tung, J. Resource base influences genome-wide DNA methylation levels in wild baboons (Papio cynocephalus). Mol. Ecol. 2016, 25, 1681–1696. [Google Scholar] [CrossRef]
- Wickham, H.; François, R.; Henry, L.; Müller, K. dplyr: A Grammar of Data Manipulation. R package version _0.7.7. 2018. Available online: https://cran.r-project.org/package=dplyr (accessed on 1 January 2018).
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.-M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Birshtein, B.K. Epigenetic Regulation of Individual Modules of the immunoglobulin heavy chain locus 3′ Regulatory Region. Front. Immunol. 2014, 5, 163. [Google Scholar] [CrossRef]
- Libertini, E.; Lebreton, A.; Lakisic, G.; Dillies, M.-A.; Beck, S.; Coppée, J.-Y.; Cossart, P.; Bierne, H. Overexpression of the Heterochromatinization Factor BAHD1 in HEK293 Cells Differentially Reshapes the DNA Methylome on Autosomes and X Chromosome. Front. Genet. 2015, 6, 339. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Robertson, F.M.; Gundappa, M.K.; Grammes, F.; Hvidsten, T.R.; Redmond, A.K.; Lien, S.; Martin, S.A.M.; Holland, P.W.H.; Sandve, S.R.; Macqueen, D.J. Lineage-specific rediploidization is a mechanism to explain time-lags between genome duplication and evolutionary diversification. Genome Biol. 2017, 18, 111. [Google Scholar] [CrossRef]
- Campbell, M.A.; Hale, M.C.; McKinney, G.J.; Nichols, K.M.; Pearse, D.E. Long-Term Conservation of Ohnologs Through Partial Tetrasomy Following Whole-Genome Duplication in Salmonidae. G3 2019. [Google Scholar] [CrossRef]
- Taggart, A.K.P.; Fisher, T.S.; Pugh, B.F. The TATA-binding protein and associated factors are components of pol III transcription factor TFIIIB. Cell 1992, 71, 1015–1028. [Google Scholar] [CrossRef]
- Qiu, X.-B.; Shao, Y.-M.; Miao, S.; Wang, L. The diversity of the DnaJ/Hsp40 family, the crucial partners for Hsp70 chaperones. Cell. Mol. Life Sci. 2006, 63, 2560–2570. [Google Scholar] [CrossRef]
- Berasain, C.; Avila, M.A. The EGFR signalling system in the liver: from hepatoprotection to hepatocarcinogenesis. J. Gastroenterol. 2014, 49, 9–23. [Google Scholar] [CrossRef]
- Larsen, D.A.; Harstad, D.L.; Strom, C.R.; Johnston, M.V.; Knudsen, C.M.; Fast, D.E.; Pearsons, T.N.; Beckman, B.R. Early Life History Variation in Hatchery- and Natural-Origin Spring Chinook Salmon in the Yakima River, Washington. Trans. Am. Fish. Soc. 2013, 142, 540–555. [Google Scholar] [CrossRef]
- Kass, S.U.; Pruss, D.; Wolffe, A.P. How does DNA methylation repress transcription? Trends Genet. 1997, 13, 444–449. [Google Scholar] [CrossRef]
- Shao, C.; Li, Q.; Chen, S.; Zhang, P.; Lian, J.; Hu, Q.; Sun, B.; Jin, L.; Liu, S.; Wang, Z.; et al. Epigenetic modification and inheritance in sexual reversal of fish. Genome Res. 2014, 24, 604–615. [Google Scholar] [CrossRef]
- Zhang, N. Epigenetic modulation of DNA methylation by nutrition and its mechanisms in animals. Anim. Nutr. 2015, 1, 144–151. [Google Scholar] [CrossRef]
- Kostow, K.E. Differences in juvenile phenotypes and survival between hatchery stocks and a natural population provide evidence for modified selection due to captive breeding. Can. J. Fish. Aquat. Sci. 2004, 61, 577–589. [Google Scholar] [CrossRef]
- Tipping, J.M. Management Briefs: Heritability of Age at Maturity in Steelhead. N. Am. J. Fish. Manage. 1991, 11, 105–108. [Google Scholar] [CrossRef]
- Blouin, M.S.; Thuillier, V.; Cooper, B.; Amarasinghe, V.; Cluzel, L.; Araki, H.; Grunau, C. No evidence for large differences in genomic methylation between wild and hatchery steelhead (Oncorhynchus mykiss). Can. J. Fish. Aquat. Sci. 2010, 67, 217–224. [Google Scholar] [CrossRef]
- Panserat, S.; Hortopan, G.A.; Plagnes-Juan, E.; Kolditz, C.; Lansard, M.; Skiba-Cassy, S.; Esquerré, D.; Geurden, I.; Médale, F.; Kaushik, S.; et al. Differential gene expression after total replacement of dietary fish meal and fish oil by plant products in rainbow trout (Oncorhynchus mykiss) liver. Aquaculture 2009, 294, 123–131. [Google Scholar] [CrossRef]
- Vickers, M.H. Early life nutrition, epigenetics and programming of later life disease. Nutrients 2014, 6, 2165–2178. [Google Scholar] [CrossRef]
- Vindas, M.A.; Madaro, A.; Fraser, T.W.K.; Höglund, E.; Olsen, R.E.; Øverli, Ø.; Kristiansen, T.S. Coping with a changing environment: the effects of early life stress. R. Soc. Open. Sci. 2016, 3, 160382. [Google Scholar] [CrossRef] [PubMed]
- Tatara, C.P.; Cooper, M.R.; Gale, W.; Kennedy, B.M.; Pasley, C.R.; Berejikian, B.A. Age and Method of Release Affect Migratory Performance of Hatchery Steelhead. N. Am. J. Fish. Manag. 2017, 37, 700–713. [Google Scholar] [CrossRef]
- Potok, M.E.; Nix, D.A.; Parnell, T.J.; Cairns, B.R. Reprogramming the maternal zebrafish genome after fertilization to match the paternal methylation pattern. Cell 2013, 153, 759–772. [Google Scholar] [CrossRef]
- Boks, M.P.; Derks, E.M.; Weisenberger, D.J.; Strengman, E.; Janson, E.; Sommer, I.E.; Kahn, R.S.; Ophoff, R.A. The relationship of DNA methylation with age, gender and genotype in twins and healthy controls. PLoS ONE 2009, 4, e6767. [Google Scholar] [CrossRef]
- Horvath, S.; Zhang, Y.; Langfelder, P.; Kahn, R.S.; Boks, M.P.M.; van Eijk, K.; van den Berg, L.H.; Ophoff, R.A. Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biol. 2012, 13, R97. [Google Scholar] [CrossRef]
- Folmar, L.C.; Dickhoff, W.W. The parr—Smolt transformation (smoltification) and seawater adaptation in salmonids: A review of selected literature. Aquaculture 1980, 21, 1–37. [Google Scholar] [CrossRef]
- Folmar, L.C.; Dickhoff, W.W. Evaluation of some physiological parameters as predictive indices of smoltification. Aquaculture 1981, 23, 309–324. [Google Scholar] [CrossRef]
- Hoar, W.S. 4 The Physiology of Smolting Salmonids. In Fish Physiology; Hoar, W.S., Randall, D.J., Eds.; Academic Press: San Deigo, CA, USA, 1988; Volume 11, pp. 275–343. [Google Scholar]
- Nichols, K.M.; Edo, A.F.; Wheeler, P.A.; Thorgaard, G.H. The genetic basis of smoltification-related traits in Oncorhynchus mykiss. Genetics 2008, 179, 1559–1575. [Google Scholar] [CrossRef]
- Hecht, B.C.; Campbell, N.R.; Holecek, D.E.; Narum, S.R. Genome-wide association reveals genetic basis for the propensity to migrate in wild populations of rainbow and steelhead trout. Mol. Ecol. 2013, 22, 3061–3076. [Google Scholar] [CrossRef]
- Hale, M.C.; McKinney, G.J.; Thrower, F.P.; Nichols, K.M. RNA-seq reveals differential gene expression in the brains of juvenile resident and migratory smolt rainbow trout (Oncorhynchus mykiss). Comp. Biochem. Physiol. Part D Genomics Proteomics 2016, 20, 136–150. [Google Scholar] [CrossRef]
- Baerwald, M.R.; Meek, M.H.; Stephens, M.R.; Nagarajan, R.P.; Goodbla, A.M.; Tomalty, K.M.H.; Thorgaard, G.H.; May, B.; Nichols, K.M. Migration-related phenotypic divergence is associated with epigenetic modifications in rainbow trout. Mol. Ecol. 2016, 25, 1785–1800. [Google Scholar] [CrossRef] [PubMed]
- Kendall, N.W.; McMillan, J.R.; Sloat, M.R.; Buehrens, T.W.; Quinn, T.P.; Pess, G.R.; Kuzishchin, K.V.; McClure, M.M.; Zabel, R.W. Anadromy and residency in steelhead and rainbow trout (Oncorhynchus mykiss): A review of the processes and patterns. Can. J. Fish. Aquat. Sci. 2015, 72, 319–342. [Google Scholar] [CrossRef]
- Berejikian, B.A.; Hard, J.J.; Tatara, C.P.; Van Doornik, D.M.; Swanson, P.; Larsen, D.A. Rearing strategies alter patterns of size-selective mortality and heritable size variation in steelhead trout (Oncorhynchus mykiss). Can. J. Fish. Aquat. Sci. 2016, 74, 273–283. [Google Scholar] [CrossRef]
- Dickhoff, W.W.; Folmar, L.C.; Mighell, J.L.; Mahnken, C.V.W. Plasma thyroid hormones during smoltification of yearling and underyearling coho salmon and yearling chinook salmon and steelhead trout. Aquaculture 1982, 28, 39–48. [Google Scholar] [CrossRef]
- Bell, A.C.; Felsenfeld, G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 2000, 405, 482–485. [Google Scholar] [CrossRef]
- Jones, P.A. The DNA methylation paradox. Trends Genet. 1999, 15, 34–37. [Google Scholar] [CrossRef]
- Wilson, V.L.; Smith, R.A.; Ma, S.; Cutler, R.G. Genomic 5-methyldeoxycytidine decreases with age. J. Biol. Chem. 1987, 262, 9948–9951. [Google Scholar]
- Unnikrishnan, A.; Freeman, W.M.; Jackson, J.; Wren, J.D.; Porter, H.; Richardson, A. The role of DNA methylation in epigenetics of aging. Pharmacol. Ther. 2018. [Google Scholar] [CrossRef]
- De Paoli-Iseppi, R.; Deagle, B.E.; McMahon, C.R.; Hindell, M.A.; Dickinson, J.L.; Jarman, S.N. Measuring Animal Age with DNA Methylation: From Humans to Wild Animals. Front. Genet. 2017, 8, 106. [Google Scholar] [CrossRef] [PubMed]
- Hampton, J.A.; Lantz, R.C.; Hinton, D.E. Functional units in rainbow trout (Salmo gairdneri, Richardson) liver: III. Morphometric analysis of parenchyma, stroma, and component cell types. Am. J. Anat. 1989, 185, 58–73. [Google Scholar] [CrossRef]
- McCormick, S.D. 5—Smolt Physiology and Endocrinology. In Fish Physiology; McCormick, S.D., Farrell, A.P., Brauner, C.J., Eds.; Academic Press: San Diego, CA, USA, 2012; Volume 32, pp. 199–251. [Google Scholar]
- Björnsson, B.T.; Stefansson, S.O.; McCormick, S.D. Environmental endocrinology of salmon smoltification. Gen. Comp. Endocrinol. 2011, 170, 290–298. [Google Scholar] [CrossRef]
- Sheridan, M.A. Alterations in lipid metabolism accompanying smoltification and seawater adaptation of salmonid fish. Aquaculture 1989, 82, 191–203. [Google Scholar] [CrossRef]
- Harada, M.; Yoshinaga, T.; Ojima, D.; Iwata, M. cDNA cloning and expression analysis of thyroid hormone receptor in the coho salmon Oncorhynchus kisutch during smoltification. Gen. Comp. Endocrinol. 2008, 155, 658–667. [Google Scholar] [CrossRef]
- Stefansson, S.O.; Haugland, M.; Björnsson, B.T.; McCormick, S.D.; Holm, M.; Ebbesson, L.O.E.; Holst, J.C.; Nilsen, T.O. Growth, osmoregulation and endocrine changes in wild Atlantic salmon smolts and post-smolts during marine migration. Aquaculture 2012, 362–363, 127–136. [Google Scholar] [CrossRef]
- Beckman, B.R.; Larsen, D.A.; Moriyama, S.; Lee-Pawlak, B.; Dickhoff, W.W. Insulin-like Growth Factor-I and Environmental Modulation of Growth during Smoltification of Spring Chinook Salmon (Oncorhynchus tshawytscha). Gen. Comp. Endocrinol. 1998, 109, 325–335. [Google Scholar] [CrossRef]
- Poulsen, L.; Siersbæk, M.; Mandrup, S. PPARs: fatty acid sensors controlling metabolism. Semin. Cell Dev. Biol. 2012, 23, 631–639. [Google Scholar] [CrossRef]
- Varga, T.; Czimmerer, Z.; Nagy, L. PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. Biochim. Biophys. Acta 2011, 1812, 1007–1022. [Google Scholar] [CrossRef] [PubMed]
- Houde, A.L.S.; Schulze, A.D.; Kaukinen, K.H.; Strohm, J.; Patterson, D.A.; Beacham, T.D.; Farrell, A.P.; Hinch, S.G.; Miller, K.M. Transcriptional shifts during juvenile Coho salmon (Oncorhynchus kisutch) life stage changes in freshwater and early marine environments. Comp. Biochem. Physiol. Part D Genomics Proteomics 2018, 29, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Ly, L.; Chan, D.; Trasler, J.M. Developmental windows of susceptibility for epigenetic inheritance through the male germline. Semin. Cell Dev. Biol. 2015, 43, 96–105. [Google Scholar] [CrossRef]
- Billard, R. Reproduction in rainbow trout: sex differentiation, dynamics of gametogenesis, biology and preservation of gametes. Aquaculture 1992, 100, 263–298. [Google Scholar] [CrossRef]
- Molaro, A.; Hodges, E.; Fang, F.; Song, Q.; McCombie, W.R.; Hannon, G.J.; Smith, A.D. Sperm methylation profiles reveal features of epigenetic inheritance and evolution in primates. Cell 2011, 146, 1029–1041. [Google Scholar] [CrossRef]
- Jiang, L.; Zhang, J.; Wang, J.-J.; Wang, L.; Zhang, L.; Li, G.; Yang, X.; Ma, X.; Sun, X.; Cai, J.; et al. Sperm, but not oocyte, DNA methylome is inherited by zebrafish early embryos. Cell 2013, 153, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Maclean, J.A., 2nd; Wilkinson, M.F. Gene regulation in spermatogenesis. Curr. Top. Dev. Biol. 2005, 71, 131–197. [Google Scholar]
- Kobayashi, N.; Miyauchi, N.; Tatsuta, N.; Kitamura, A.; Okae, H.; Hiura, H.; Sato, A.; Utsunomiya, T.; Yaegashi, N.; Nakai, K.; et al. Factors associated with aberrant imprint methylation and oligozoospermia. Sci. Rep. 2017, 7, 42336. [Google Scholar] [CrossRef]
- Woods, L.C., III; Li, Y.; Ding, Y.; Liu, J.; Reading, B.J.; Fuller, S.A.; Song, J. DNA methylation profiles correlated to striped bass sperm fertility. BMC Genomics 2018, 19, 244. [Google Scholar] [CrossRef]
- Laity, J.H.; Lee, B.M.; Wright, P.E. Zinc finger proteins: New insights into structural and functional diversity. Curr. Opin. Struct. Biol. 2001, 11, 39–46. [Google Scholar] [CrossRef]
- Castillo, J.; Amaral, A.; Oliva, R. Sperm nuclear proteome and its epigenetic potential. Andrology 2014, 2, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Olson, C.E.; Roberts, S.B. Indication of family-specific DNA methylation patterns in developing oysters. bioRxiv 2015, 012831. [Google Scholar]
- Metzger, D.C.H.; Schulte, P.M. The DNA Methylation Landscape of Stickleback Reveals Patterns of Sex Chromosome Evolution and Effects of Environmental Salinity. Genome Biol. Evol. 2018, 10, 775–785. [Google Scholar] [CrossRef] [PubMed]
- Lienert, F.; Wirbelauer, C.; Som, I.; Dean, A.; Mohn, F.; Schübeler, D. Identification of genetic elements that autonomously determine DNA methylation states. Nat. Genet. 2011, 43, 1091–1097. [Google Scholar] [CrossRef] [PubMed]
- Gertz, J.; Varley, K.E.; Reddy, T.E.; Bowling, K.M.; Pauli, F.; Parker, S.L.; Kucera, K.S.; Willard, H.F.; Myers, R.M. Analysis of DNA methylation in a three-generation family reveals widespread genetic influence on epigenetic regulation. PLoS Genet. 2011, 7, e1002228. [Google Scholar] [CrossRef] [PubMed]
- Teh, A.L.; Pan, H.; Chen, L.; Ong, M.-L.; Dogra, S.; Wong, J.; MacIsaac, J.L.; Mah, S.M.; McEwen, L.M.; Saw, S.-M.; et al. The effect of genotype and in utero environment on interindividual variation in neonate DNA methylomes. Genome Res. 2014, 24, 1064–1074. [Google Scholar] [CrossRef] [PubMed]
- Tang, A.; Huang, Y.; Li, Z.; Wan, S.; Mou, L.; Yin, G.; Li, N.; Xie, J.; Xia, Y.; Li, X.; et al. Analysis of a four generation family reveals the widespread sequence-dependent maintenance of allelic DNA methylation in somatic and germ cells. Sci. Rep. 2016, 6, 19260. [Google Scholar] [CrossRef] [PubMed]
- Gonseth, S.; de Smith, A.J.; Roy, R.; Zhou, M.; Lee, S.-T.; Shao, X.; Ohja, J.; Wrensch, M.R.; Walsh, K.M.; Metayer, C.; et al. Genetic contribution to variation in DNA methylation at maternal smoking-sensitive loci in exposed neonates. Epigenetics 2016, 11, 664–673. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Contrast | Immediate | Persistent | Developmental | Intergenerational |
|---|---|---|---|---|
| Tissue | liver | liver | liver | sperm |
| Sample size | 20 | 19 | 39 | 60 |
| CG covered across half of samples at > 10x coverage | 1,449,887 | 1,251,548 | 1,354,166 | 1,494,268 |
| CG remaining after filtering for low variation across samples | 1,377,392 | 1,183,061 | 1,286,457 | 1,419,554 |
| CG remaining after filtering for hypo-methylation | 1,318,452 | 1,117,756 | 1,236,987 | 1,359,452 |
| CG remaining after filtering for hyper-methylation | 559,680 | 443,280 | 492,583 | 84,661 |
| CG remaining after filtering for CG SNPs | 532,017 | 418,800 | 465,391 | 67,661 |
| Ingenuity Canonical Pathways | P-Value | Ratio |
|---|---|---|
| Axonal Guidance Signaling | 6.31 × 10−11 | 266/421 |
| CREB Signaling in Neurons | 2.40 × 10−10 | 138/198 |
| Role of NFAT in Cardiac Hypertrophy | 3.55 × 10−10 | 137/197 |
| G-Protein Coupled Receptor Signaling | 1.23 × 10−9 | 169/255 |
| GNRH Signaling | 3.24 × 10−8 | 107/154 |
| Netrin Signaling | 5.75 × 10−8 | 49/60 |
| Molecular Mechanisms of Cancer | 7.08 × 10−8 | 217/352 |
| Opioid Signaling Pathway | 8.51 × 10−8 | 142/217 |
| Neuropathic Pain Signaling in Dorsal Horn Neurons | 1.48 × 10−7 | 80/111 |
| PPARα/RXRα Activation | 2.82 × 10−7 | 104/153 |
| cAMP-mediated signaling | 2.82 × 10−7 | 128/195 |
| Synaptic Long-Term Depression | 3.31 × 10−7 | 109/162 |
| Adrenomedullin signaling pathway | 6.46 × 10−7 | 119/181 |
| nNOS Signaling in Skeletal Muscle Cells | 1.00 × 10−6 | 32/37 |
| GPCR-Mediated Nutrient Sensing in Enteroendocrine Cells | 1.51 × 10−6 | 70/98 |
| G Beta Gamma Signaling | 1.70 × 10−6 | 77/110 |
| Hepatic Fibrosis/Hepatic Stellate Cell Activation | 1.70 × 10−6 | 95/141 |
| Leukocyte Extravasation Signaling | 2.00 × 10−6 | 113/173 |
| Synaptic Long-Term Potentiation | 2.34 × 10−6 | 79/114 |
| Protein Kinase A Signaling | 3.24 × 10−6 | 194/322 |
| Dopamine-DARPP32 Feedback in cAMP Signaling | 4.37 × 10−6 | 95/143 |
| Regulation of the Epithelial-Mesenchymal Transition Pathway | 5.89 × 10−6 | 116/181 |
| PTEN Signaling | 6.61 × 10−6 | 79/116 |
| GABA Receptor Signaling | 6.92 × 10−6 | 59/82 |
| Corticotropin Releasing Hormone Signaling | 6.92 × 10−6 | 87/130 |
| Neuregulin Signaling | 8.13 × 10−6 | 63/89 |
| Signaling by Rho Family GTPases | 8.51 × 10−6 | 137/220 |
| Cellular Effects of Sildenafil (Viagra) | 9.33 × 10−6 | 74/108 |
| RAR Activation | 1.05 × 10−5 | 104/161 |
| Glutamate Receptor Signaling | 1.12 × 10−5 | 39/50 |
| DMR_ID (chr.start.stop) | Number of CG | Methylation Difference | Relationship DMR to Gene | Gene |
|---|---|---|---|---|
| NC_035079.1.50470786.50470912 | 8 | −60.7 | 0 | plakophilin-4 |
| NC_035086.1.43986683.43986897 | 13 | −58.4 | 0 | coagulation factor IX |
| NC_035090.1.20320098.20320139 | 3 | −55.9 | 0 | thyroid hormone receptor beta |
| NC_035079.1.54857805.54857941 | 7 | −55.5 | 0 | aryl hydrocarbon receptor |
| NC_035094.1.42211884.42211949 | 4 | −53.9 | 0 | thyroid hormone receptor beta |
| NC_035098.1.39974903.39974975 | 6 | −51.9 | −4743 | activin receptor type-2A |
| NC_035103.1.6240302.6240419 | 8 | −51.0 | 0 | protein phosphatase 1 regulatory subunit 37 |
| NC_035077.1.65859491.65859615 | 5 | −48.6 | 0 | tetratricopeptide repeat protein 27 |
| NC_035104.1.2409783.2409794 | 3 | −45.6 | 0 | kinesin-1 heavy chain |
| NC_035092.1.14183000.14183073 | 3 | −45.1 | 0 | transcription factor 7-like 2 |
| NC_035079.1.70446509.70446615 | 9 | −44.6 | 0 | B-cell CLL/lymphoma 9 protein |
| NW_018557253.1.30187.30270 | 6 | −44.2 | 0 | thrombospondin-2 |
| NC_035096.1.18319228.18319305 | 5 | −44.1 | 0 | nuclear factor erythroid 2-related factor 1 |
| NC_035089.1.22399625.22399721 | 6 | −43.9 | 0 | interferon regulatory factor 2-binding protein 2-B |
| NC_035084.1.6294684.6294737 | 3 | −42.5 | 0 | cholesterol 24-hydroxylase |
| NC_035080.1.55253415.55253602 | 3 | −42.4 | 4013 | 5-hydroxytryptamine receptor 1E |
| NC_035078.1.62045061.62045113 | 3 | −42.0 | 0 | NLR family CARD domain-containing protein 3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gavery, M.R.; Nichols, K.M.; Berejikian, B.A.; Tatara, C.P.; Goetz, G.W.; Dickey, J.T.; Van Doornik, D.M.; Swanson, P. Temporal Dynamics of DNA Methylation Patterns in Response to Rearing Juvenile Steelhead (Oncorhynchus mykiss) in a Hatchery versus Simulated Stream Environment. Genes 2019, 10, 356. https://doi.org/10.3390/genes10050356
Gavery MR, Nichols KM, Berejikian BA, Tatara CP, Goetz GW, Dickey JT, Van Doornik DM, Swanson P. Temporal Dynamics of DNA Methylation Patterns in Response to Rearing Juvenile Steelhead (Oncorhynchus mykiss) in a Hatchery versus Simulated Stream Environment. Genes. 2019; 10(5):356. https://doi.org/10.3390/genes10050356
Chicago/Turabian StyleGavery, Mackenzie R., Krista M. Nichols, Barry A. Berejikian, Christopher P. Tatara, Giles W. Goetz, Jon T. Dickey, Donald M. Van Doornik, and Penny Swanson. 2019. "Temporal Dynamics of DNA Methylation Patterns in Response to Rearing Juvenile Steelhead (Oncorhynchus mykiss) in a Hatchery versus Simulated Stream Environment" Genes 10, no. 5: 356. https://doi.org/10.3390/genes10050356
APA StyleGavery, M. R., Nichols, K. M., Berejikian, B. A., Tatara, C. P., Goetz, G. W., Dickey, J. T., Van Doornik, D. M., & Swanson, P. (2019). Temporal Dynamics of DNA Methylation Patterns in Response to Rearing Juvenile Steelhead (Oncorhynchus mykiss) in a Hatchery versus Simulated Stream Environment. Genes, 10(5), 356. https://doi.org/10.3390/genes10050356