Histone Methylation Participates in Gene Expression Control during the Early Development of the Pacific Oyster Crassostrea gigas

Abstract
1. Introduction
2. Materials and Methods
2.1. Animals, Fecundation Assays, and Early Development in the Presence of the Jumonji Histone Demethylase Inhibitor, Methylstat
2.2. Histones Extraction
2.3. Histone Methylation Quantification
2.4. Scanning Electron Microscopy
2.5. Microarrays
2.5.1. RNA Amplification, Labeling, and Hybridization
2.5.2. Correction and Normalization
2.5.3. Data Analysis
2.5.4. Gene Ontology Analyses
2.5.5. RT-qPCR Validation of Microarray Analysis
3. Results
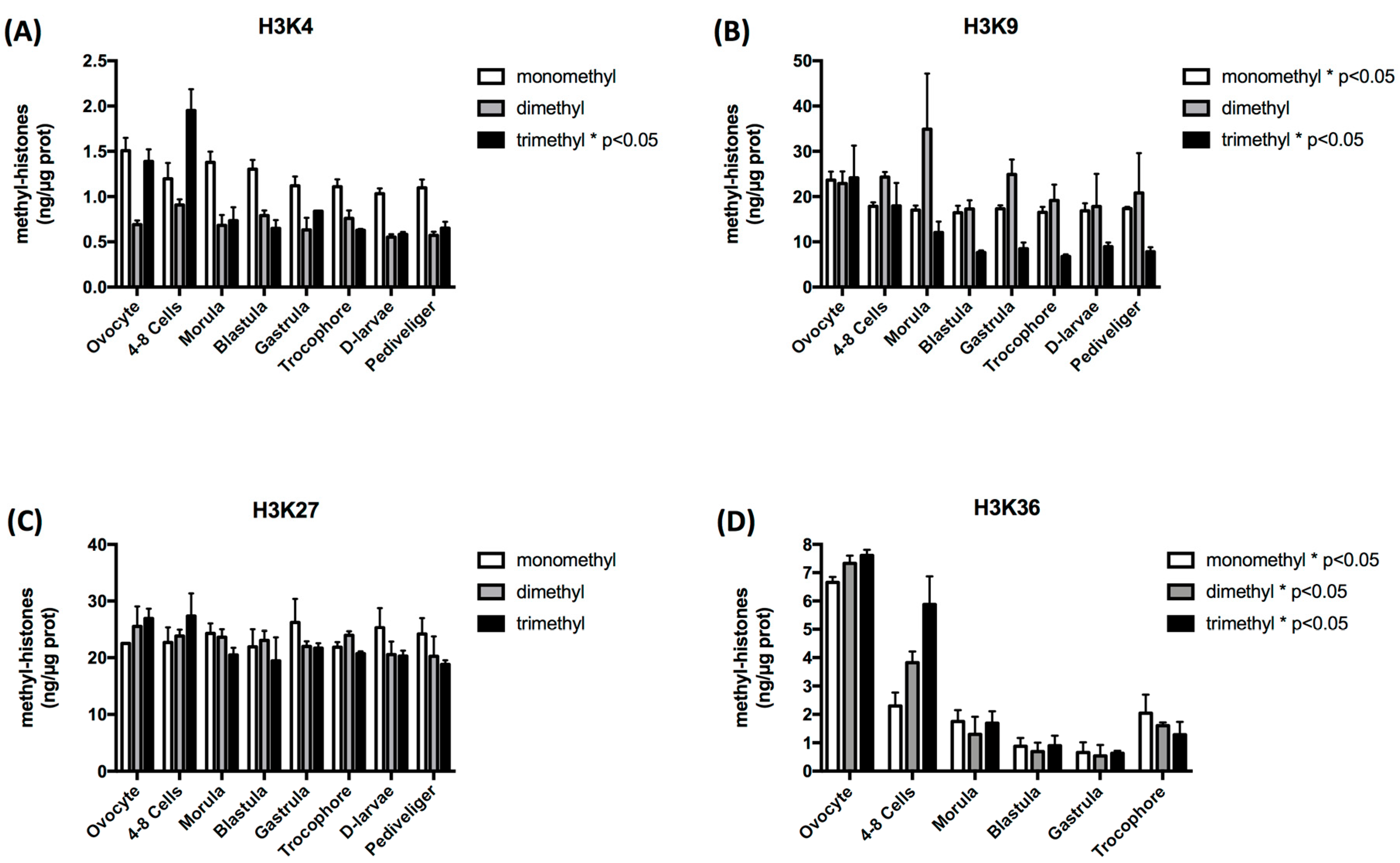
3.1. Histone Methylation Exhibits Stage-Specific Patterns during Oyster Early Development
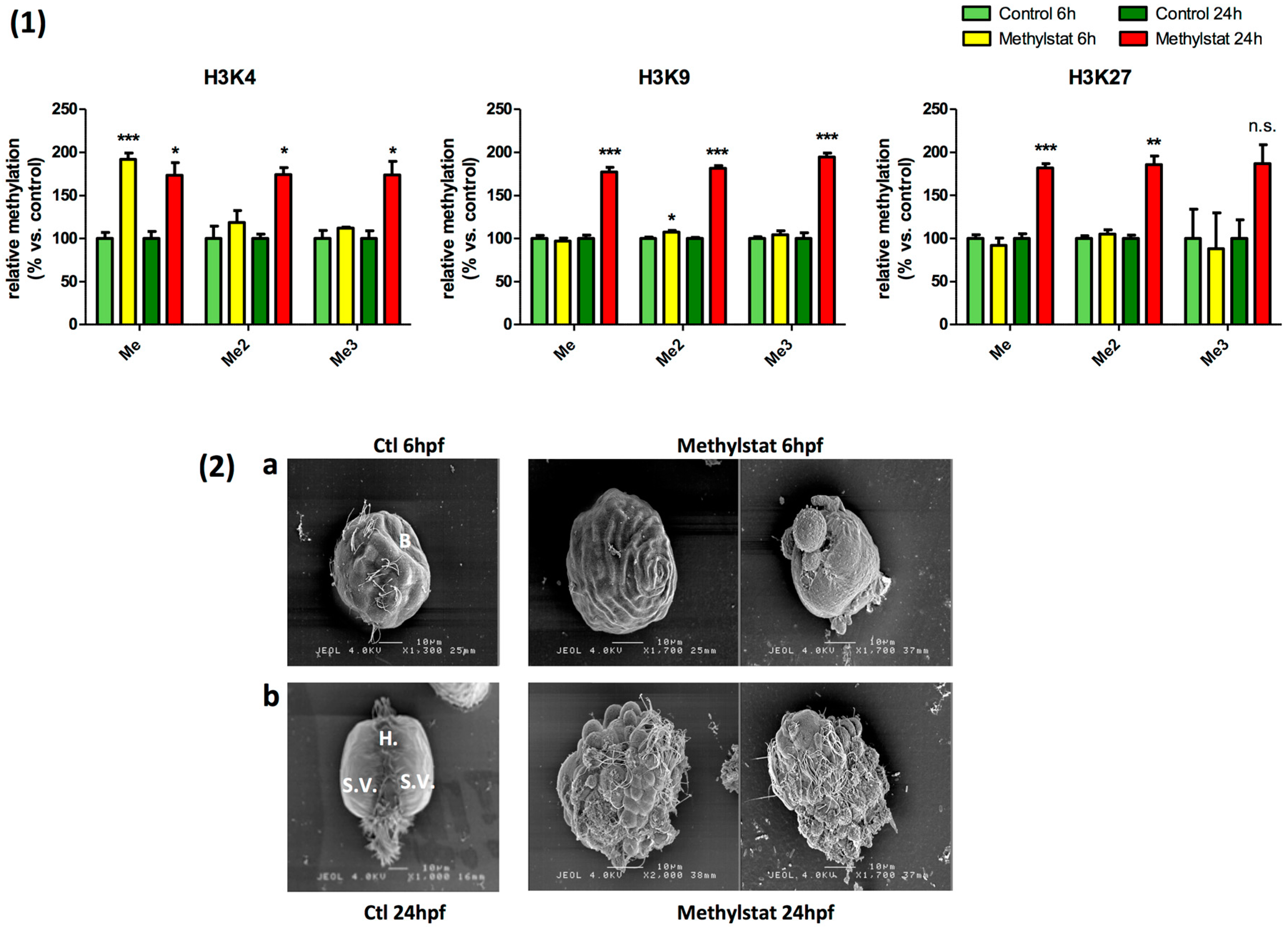
3.2. Methylstat-Induced Histone Hypermethylation is Correlated to Severe Developmental Alterations
3.3. Methylstat Treatment Induces Transcriptomic Variations
3.4. Identification of the Putative Metabolic Pathways Implicating the Differentially Expressed Genes in Larvae Exposed to Methylstat
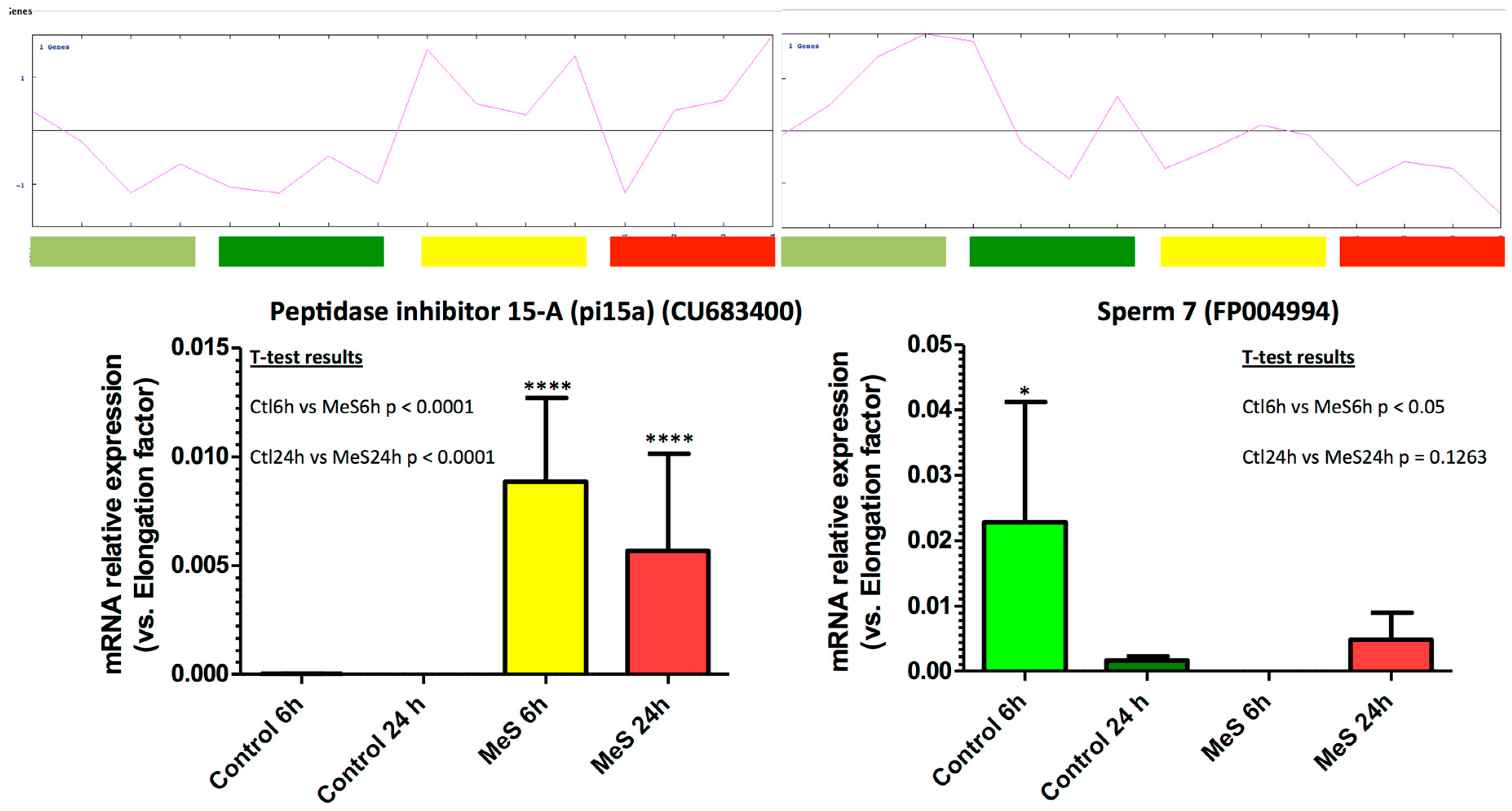
3.5. Selected Marker Genes RT-qPCR Measurements Confirm Microarray Signals
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Black, J.C.; Van Rechem, C.; Whetstine, J.R. Histone Lysine Methylation Dynamics: Establishment, Regulation, and Biological Impact. Mol. Cell 2013, 48, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Zhang, Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 2005, 6, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Vastenhouw, N.L.; Schier, A.F. Bivalent Histone Modifications in Early Embryogenesis. Curr. Opin. Cell Biol. 2012, 24, 374–386. [Google Scholar] [CrossRef] [PubMed]
- Ooi, S.K.T.; Qiu, C.; Bernstein, E.; Li, K.; Jia, D.; Yang, Z.; Tempst, P.; Lin, S.-P.; Allis, C.D.; Cheng, X.; et al. DNMT3L connects unmethylated lysine 4 of histone H3 to denovo methylation of DNA. Nature 2007, 448, 714–717. [Google Scholar] [CrossRef] [PubMed]
- Agger, K.; Christensen, J.; Cloos, P.A.; Helin, K. The emerging functions of histone demethylases. Curr. Opin. Genet. Dev. 2008, 18, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Feldman, N.; Gerson, A.; Fang, J.; Li, E.; Zhang, Y.; Shinkai, Y.; Cedar, H.; Bergman, Y. G9a-mediated irreversible epigenetic inactivation of Oct-3/4 during early embryogenesis. Nature 2006, 8, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, G.; Chung, A.C.-K.; Jackson, K.J.; Hummelke, G.; Baniahmad, A.; Sutter, J.; Sylvester, I.; Scholer, H.R.; Cooney, A.J. Mouse Germline Restriction of Oct4 Expression by Germ Cell Nuclear Factor. Dev. Cell 2001, 1, 377–387. [Google Scholar] [CrossRef]
- Li, J.; Ahn, J.H.; Wang, G.G. Understanding histone H3 lysine 36 methylation and its deregulation in disease. Cell. Mol. Life Sci. 2019, 76, 2899–2916. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Chen, X. Dynamics of histone H3 lysine 27 trimethylation in Plant Development. Curr. Opin. Plant Biol. 2011, 14, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-E. Bookmarking by histone methylation ensures chromosomal integrity during mitosis. Arch. Pharmacal Res. 2019, 42, 466–480. [Google Scholar] [CrossRef]
- Klose, R.J.; Kallin, E.M.; Zhang, Y. JmjC-domain-containing proteins and histone demethylation. Nat. Rev. Genet. 2006, 7, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Sun, L.; Li, Q.; Liang, J.; Yu, W.; Yi, X.; Yang, X.; Li, Y.; Han, X.; Zhang, Y.; et al. Histone demethylase JMJD2B coordinates H3K4/H3K9 methylation and promotes hormonally responsive breast carcinogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 7541–7546. [Google Scholar] [CrossRef] [PubMed]
- Skvortsova, K.; Iovino, N.; Bogdanović, O. Functions and mechanisms of epigenetic inheritance in animals. Nat. Rev. Mol. Cell Biol. 2018, 19, 1. [Google Scholar] [CrossRef] [PubMed]
- Eirin-lopez, J.M.; Putnam, H.M. Marine Environmental Epigenetics. Ann. Rev. Mar. Sci. 2018, 11, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Fellous, A.; Favrel, P.; Rivière, G. Temperature influences histone methylation and mRNA expression of the Jmj-C histone-demethylase orthologues during the early development of the oyster Crassostrea gigas. Mar. Genom. 2015, 19, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.; Hart, C.; Pierce, R.; Lloyd, V. Ontogenetic Survey of Histone Modifications in an Annelid. Genet. Res. Int. 2012, 2012, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Glastad, K.M.; Hunt, B.G.; Goodisman, M.A. Epigenetics in Insects: Genome Regulation and the Generation of Phenotypic Diversity. Annu. Rev. Èntomol. 2019, 64, 185–203. [Google Scholar] [CrossRef] [PubMed]
- Cosseau, C.; Wolkenhauer, O.; Padalino, G.; Geyer, K.K.; Hoffmann, K.F.; Grunau, C. (Epi)genetic Inheritance in Schistosoma mansoni: A Systems Approach. Trends Parasitol. 2017, 33, 285–294. [Google Scholar] [CrossRef]
- Fabrizio, P.; Garvis, S.; Palladino, F. Histone Methylation and Memory of Environmental Stress. Cells 2019, 8, 339. [Google Scholar] [CrossRef] [PubMed]
- Raj, A.; Oudenaarden, A. Van Review Nature, Nurture, or Chance: Stochastic Gene Expression and Its Consequences. Cell 2008, 135, 216–226. [Google Scholar] [CrossRef] [PubMed]
- LaForge, B.; Guez, D.; Martinez, M.; Kupiec, J.-J. Modeling embryogenesis and cancer: An approach based on an equilibrium between the autostabilization of stochastic gene expression and the interdependence of cells for proliferation. Prog. Biophys. Mol. Biol. 2005, 89, 93–120. [Google Scholar] [CrossRef] [PubMed]
- Andersen, I.S.; Lindeman, L.C.; Reiner, A.H.; Østrup, O.; Aanes, H.; Aleström, P.; Collas, P. Epigenetic Marking of the Zebrafish Developmental Program. In Current Topics in Developmental Biology; Elsevier BV: Amsterdam, The Netherlands, 2013; Volume 104, pp. 85–112. [Google Scholar]
- Gavery, M.R.; Roberts, S.B. DNA methylation patterns provide insight into epigenetic regulation in the Pacific oyster (Crassostrea gigas). BMC Genom. 2010, 11, 483. [Google Scholar] [CrossRef] [PubMed]
- Olson, C.E.; Roberts, S.B. Genome-wide profiling of DNA methylation and gene expression in Crassostrea gigas male gametes. Front. Physiol. 2014, 5, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Riviere, G.; Wu, G.-C.; Fellous, A.; Goux, D.; Sourdaine, P.; Favrel, P. DNA Methylation is Crucial for the Early Development in the Oyster C. gigas. Mar. Biotechnol. 2013, 15, 739–753. [Google Scholar] [CrossRef] [PubMed]
- Riviere, G. Epigenetic features in the oyster Crassostrea gigas suggestive of functionally relevant promoter DNA methylation in invertebrates. Front. Physiol. 2014, 5, 1–7. [Google Scholar]
- Rivière, G.; He, Y.; Tecchio, S.; Crowell, E.; Gras, M.; Sourdaine, P.; Guo, X.; Favrel, P. Dynamics of DNA methylomes underlie oyster development. PLoS Genet. 2017, 13, e1006807. [Google Scholar] [CrossRef] [PubMed]
- Saint-Carlier, E.; Riviere, G. Regulation of HOX orthologues in the oyster Crassostrea gigas evidences a functional role for promoter DNA methylation in an invertebrate. FEBS Lett. 2015, 589, 1459–1466. [Google Scholar] [CrossRef] [PubMed]
- Fellous, A.; Favrel, P.; Guo, X.; Rivière, G. The Jumonji gene family in Crassostrea gigas suggests evolutionary conservation of Jmj-C histone demethylases orthologues in the oyster gametogenesis and development. Gene 2014, 538, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Dheilly, N.M.; Lelong, C.; Huvet, A.; Favrel, P. Development of a Pacific oyster (Crassostrea gigas) 31, 918-feature microarray: Identification of reference genes and tissue-enriched expression patterns Development of a Pacific oyster (Crassostrea gigas) 31, 918-feature microarray: Identificatio. BMC Genom. 2011, 12, 468. [Google Scholar] [CrossRef] [PubMed]
- Režen, T.; Juvan, P.; Tacer, K.F.; Kuzman, D.; Roth, A.; Pompon, D.; Aggerbeck, L.P.; A Meyer, U.; Rozman, D. The Sterolgene v0 cDNA microarray: A systemic approach to studies of cholesterol homeostasis and drug metabolism. BMC Genom. 2008, 9, 76. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.; Sharov, V.; White, J.; Li, J.; Liang, W.; Bhagabati, N.; Braisted, J.; Klapa, M.; Currier, T.; Thiagarajan, M.; et al. TM4: A Free, Open-Source System for Microarray Data Management and Analysis. Biotechniques 2003, 34, 374–378. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.I.; Bhagabati, N.K.; Braisted, J.C.; Liang, W.; Sharov, V.; Howe, E.A.; Li, J.; Thiagarajan, M.; White, J.A.; Quackenbush, J. TM4 Microarray Software Suite. Methods Enzymol. 2006, 411, 99–119. [Google Scholar]
- Conesa, A.; Götz, S.; García-gómez, J.M.; Terol, J.; Talón, M.; Genómica, D.; Valenciano, I.; Agrarias, D.I.; Valencia, U.P. De Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinform. Appl. Notes 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Fleury, E.; Huvet, A. Microarray Analysis Highlights Immune Response of Pacific Oysters as a Determinant of Resistance to Summer Mortality. Mar. Biotechnol. 2012, 14, 203–217. [Google Scholar] [CrossRef] [PubMed]
- Riviere, G.; Klopp, C.; Ibouniyamine, N.; Huvet, A.; Boudry, P.; Favrel, P. GigaTON: An extensive publicly searchable database providing a new reference transcriptome in the pacific oyster Crassostrea gigas. BMC Bioinform. 2015, 16, 1. [Google Scholar] [CrossRef] [PubMed]
- Cock, P.J.A.; Chilton, J.M.; Grüning, B.; Johnson, J.E.; Soranzo, N. NCBI BLAST + integrated into Galaxy. Gigascience 2015, 4, 39. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST +: Architecture and applications. BMC Bioinform. 2009, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Open Access METHOD Gene ontology analysis for RNA-seq: Accounting for selection bias GOseq GOseq is a method for GO analysis of RNA-seq data that takes into account the length bias inherent in RNA-seq. Genome Biol. 2010, 11, 14. [Google Scholar] [CrossRef]
- Rivière, G.; Fellous, A.; Franco, A.; Bernay, B.; Favrel, P. A Crucial Role in Fertility for the Oyster Angiotensin-Converting Enzyme Orthologue CgACE. PLoS ONE 2011, 6, e27833. [Google Scholar] [CrossRef]
- Biscotti, M.A.; Canapa, A.; Forconi, M.; Barucca, M. HOX and ParaHox Genes: A Review on Molluscs. Genesis 2014, 52, 935–945. [Google Scholar] [CrossRef]
- Liu, C.; Ma, Y.; Shang, Y.; Huo, R.; Li, W. Post-translational regulation of the maternal-to-zygotic transition. Cell. Mol. Life Sci. 2018, 75, 1707–1722. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wu, X.; Zhang, W.; Shen, W.; Sun, Q.; Liu, K.; Zhang, Y.; Wang, Q.; Li, Y.; Meng, A.; et al. Widespread Enhancer Dememorization and Promoter Priming during Parental-to-Zygotic Transition. Mol. Cell 2018, 72, 673–686. [Google Scholar] [CrossRef] [PubMed]
- Murphy, P.J.; Wu, S.F.; James, C.R.; Wike, C.L.; Cairns, B.R. Placeholder Nucleosomes Underlie Germline-to-Embryo DNA Methylation Reprogramming. Cell 2018, 172, 993–1006. [Google Scholar] [CrossRef] [PubMed]
- Horsfield, J.A. Packaging development: How chromatin controls transcription in zebrafish embryogenesis. Biochem. Soc. Trans. 2019, 47, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, L.; Li, Y.; Li, W.; Guo, Z.; Li, R.; Hu, X.; Bao, Z.; Wang, S. Dynamics of DNA Methylation and DNMT Expression During Gametogenesis and Early Development of Scallop Patinopecten yessoensis. Mar. Biotechnol. 2019, 21, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Eckersley-Maslin, M.A.; Alda-Catalinas, C.; Reik, W. Dynamics of the epigenetic landscape during the maternal-to-zygotic transition. Nat. Rev. Mol. Cell Biol. 2018, 19, 436–450. [Google Scholar] [CrossRef] [PubMed]
- Fellous, A.; Labed-Veydert, T.; Locrel, M.; Voisin, A.-S.; Earley, R.L.; Silvestre, F. DNA methylation in adults and during development of the self-fertilizing mangrove rivulus, Kryptolebias marmoratus. Ecol. Evol. 2018, 8, 6016–6033. [Google Scholar] [CrossRef] [PubMed]
- Vastenhouw, N.L.; Cao, W.X.; Lipshitz, H.D. The maternal-to-zygotic transition revisited. Development 2019, 146, dev161471. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A Bivalent Chromatin Structure Marks Key Developmental Genes in Embryonic Stem Cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef]
- Lepikhov, K.; Walter, J. Differential dynamics of histone H3 methylation at positions K4 and K9 in the mouse zygote. BMC Dev. Biol. 2004, 4, 12. [Google Scholar] [CrossRef]
- Rose, N.R.; Klose, R.J. Understanding the relationship between DNA methylation and histone lysine methylation. Biochim. Biophys. Acta (BBA) Bioenerg. 2014, 1839, 1362–1372. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Liu, Y.; Kubicek, S.; Myllyharju, J.; Tumber, A.; Ng, S.; Che, K.H.; Podoll, J.; Heightman, T.D.; Oppermann, U.; et al. A Selective Inhibitor and Probe of the Cellular Functions of Jumonji C Domain-Containing Histone Demethylases. J. Am. Chem. Soc. 2011, 133, 9451–9456. [Google Scholar] [CrossRef] [PubMed]
- Tsurumi, A.; Xue, S.; Zhang, L.; Li, J.; Li, W.X. Genome-wide Kdm4 histone demethylase transcriptional regulation in Drosophila. Mol. Genet. Genom. 2019. [Google Scholar] [CrossRef] [PubMed]
- Nichol, J.N.; Dupéré-Richer, D.; Ezponda, T.; Licht, J.D.; Miller, W.H. H3K27 Methylation: A Focal Point of Epigenetic Deregulation in Cancer. Adv. Breast Cancer Res. 2016, 131, 59–95. [Google Scholar]
- Pedersen, M.T.; Kooistra, S.M.; Radzisheuskaya, A.; Laugesen, A.; Johansen, J.V.; Hayward, D.G.; Nilsson, J.; Agger, K.; Helin, K. Continual removal of H3K9 promoter methylation by Jmjd2 demethylases is vital for ESC self-renewal and early development. EMBO J. 2016, 35, 1550–1564. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GO-ID | GO-Term | GO-Class | p-Value | |
|---|---|---|---|---|
| Cluster 1 | 0005890 | Sodium:potassium-exchanging ATPase complex | CC | 0.0027 |
| 0044183 | Protein binding involved in protein folding | MF | 0.0027 | |
| 0006814 | Sodium ion transport | BP | 0.0031 | |
| 0004126 | Cytidine deaminase activity | MF | 0.0054 | |
| 0009972 | Cytidine deamination | BP | 0.0054 | |
| 0080019 | Fatty-acyl-CoA reductase (alcohol-forming) activity | MF | 0.0054 | |
| 0030126 | COPI vesicle coat | CC | 0.0080 | |
| Cluster 2 | 0005882 | Intermediate filament | CC | 0.000045 |
| 0004952 | Dopamine neurotransmitter receptor activity | MF | 0.0015 | |
| 0005198 | Structural molecule activity | MF | 0.0022 | |
| 0050829 | Defense response to Gram-negative bacterium | BP | 0.0030 | |
| 0050830 | Defense response to Gram-positive bacterium | BP | 0.0030 | |
| 0000087 | Mitotic M phase | BP | 0.0045 | |
| 0008250 | Oligosaccharyltransferase complex | CC | 0.0045 | |
| 0004579 | Dolichyl-diphosphooligosaccharide-protein glycontransferase | MF | 0.0060 | |
| 0005887 | Integral component of plasma membrane | CC | 0.0089 | |
| 0016641 | Oxidoreductase activity, acting on the CH-NH2 group of donors, oxygen as acceptor | MF | 0.0089 | |
| Cluster 3 | 0006511 | Ubiquitin-dependent protein catabolic process | BP | 0.0020 |
| 0003868 | 4-hydroxyphenylpyruvate dioxygenase activity | MF | 0.0031 | |
| 0003922 | GMP synthase (glutamine-hydrolizing) activity | MF | 0.0031 | |
| 0006177 | GMP biosynthetic process | BP | 0.0031 | |
| 0004879 | RNA polymerase II transcription factor activity, ligand-activated sequence-specific DNA binding | MF | 0.0062 | |
| 0010309 | Acireductone dioxygenase (iron(II)-requiring) activity | MF | 0.0062 | |
| 0016701 | Oxydoreductase activity, acting on single donors with incorporation of molecular oxygen | MF | 0.0062 | |
| 0030833 | Regulation of actin filament polymerization | BP | 0.0062 | |
| 0000220 | Vacuolar proton-transporting V-type ATPase, V0 domain | CC | 0.0092 | |
| 0004427 | Inorganic diphosphatase activity | MF | 0.0092 | |
| 0006206 | Pyrimidine nucleobase metabolic process | BP | 0.0092 | |
| 0006796 | Phosphate-containing compound metabolic process | BP | 0.0092 | |
| 0016154 | Pyrimidine-nucleoside phosorylase activity | MF | 0.0092 | |
| 0016462 | Pyrophosphatase activity | MF | 0.0092 | |
| 0019509 | L-methionine biosynthetic process from methylthioadenosine | BP | 0.0092 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fellous, A.; Le Franc, L.; Jouaux, A.; Goux, D.; Favrel, P.; Rivière, G. Histone Methylation Participates in Gene Expression Control during the Early Development of the Pacific Oyster Crassostrea gigas. Genes 2019, 10, 695. https://doi.org/10.3390/genes10090695
Fellous A, Le Franc L, Jouaux A, Goux D, Favrel P, Rivière G. Histone Methylation Participates in Gene Expression Control during the Early Development of the Pacific Oyster Crassostrea gigas. Genes. 2019; 10(9):695. https://doi.org/10.3390/genes10090695
Chicago/Turabian StyleFellous, Alexandre, Lorane Le Franc, Aude Jouaux, Didier Goux, Pascal Favrel, and Guillaume Rivière. 2019. "Histone Methylation Participates in Gene Expression Control during the Early Development of the Pacific Oyster Crassostrea gigas" Genes 10, no. 9: 695. https://doi.org/10.3390/genes10090695
APA StyleFellous, A., Le Franc, L., Jouaux, A., Goux, D., Favrel, P., & Rivière, G. (2019). Histone Methylation Participates in Gene Expression Control during the Early Development of the Pacific Oyster Crassostrea gigas. Genes, 10(9), 695. https://doi.org/10.3390/genes10090695