DNA Methylation Analysis of the Citrullus lanatus Response to Cucumber Green Mottle Mosaic Virus Infection by Whole-Genome Bisulfite Sequencing


Abstract
:1. Introduction
2. Plants and Materials
2.1. Plant Materials and Cucumber Green Mottle Mosaic Virus Inoculation
2.2. DNA Extraction
2.3. Library Preparation and Illumina HiSeq Sequencing
2.4. Reads Quality Control and Mapping
2.5. Identification of Methylated Cytosine Sites
2.6. Identification of Differentially Methylated Regions and DMR-Associated Genes
2.7. Kyoto Encyclopedia of Genes and Genomes Pathway Analysis of DMR-Associated Genes
2.8. Data Visualization
2.9. RNA Sequencing and Data Analysis
3. Results
3.1. Phenotypic Observation of Watermelon Leaves Before and After Cucumber Green Mottle Mosaic Virus Infection
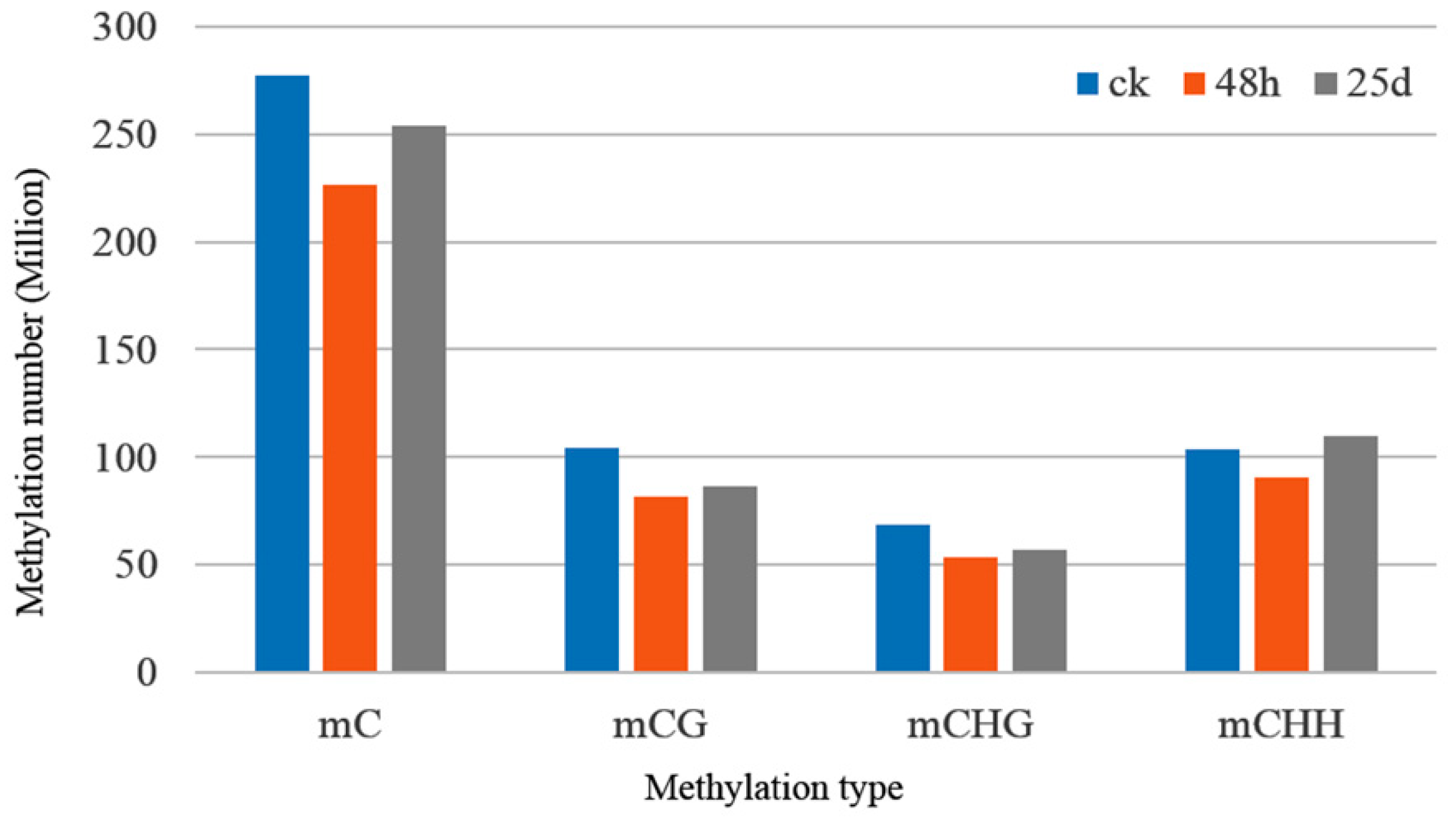
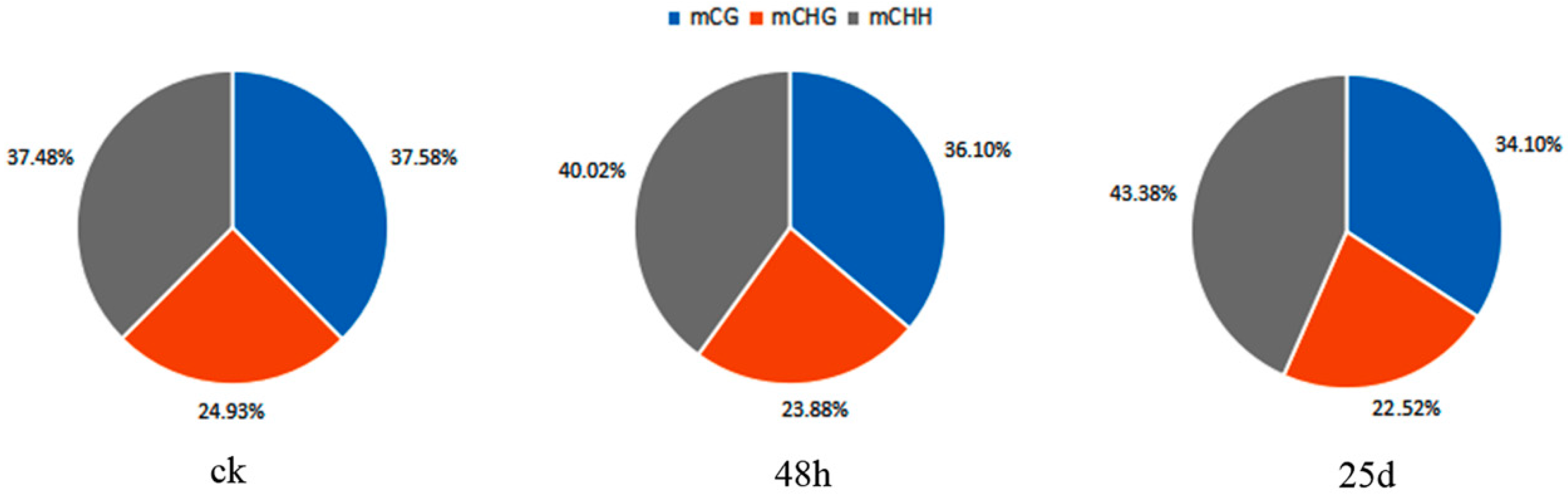
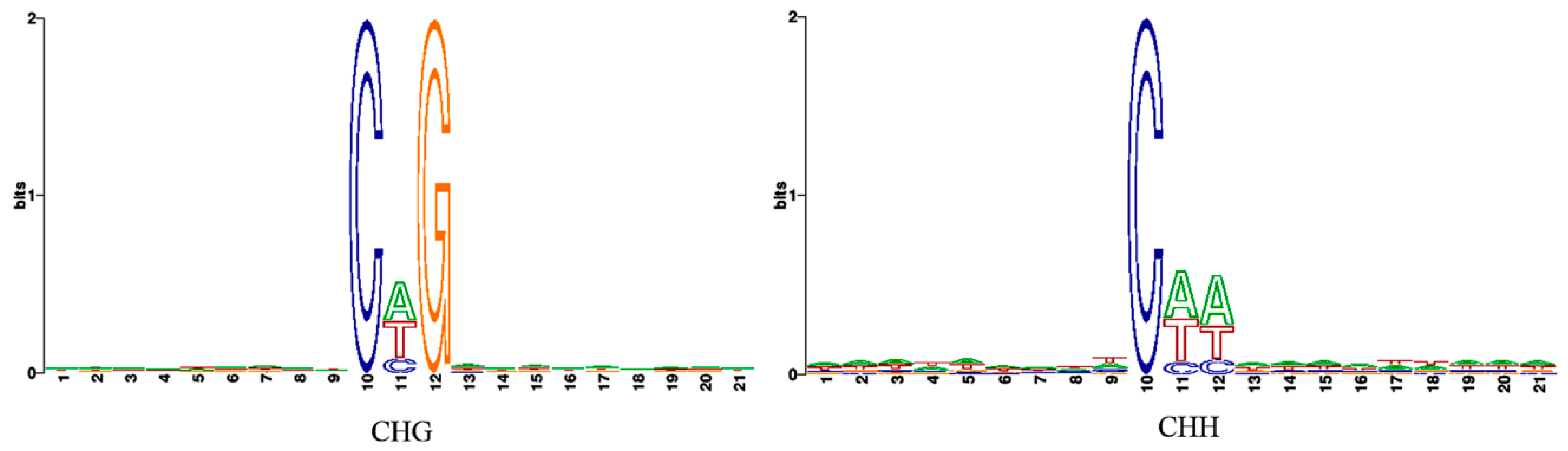
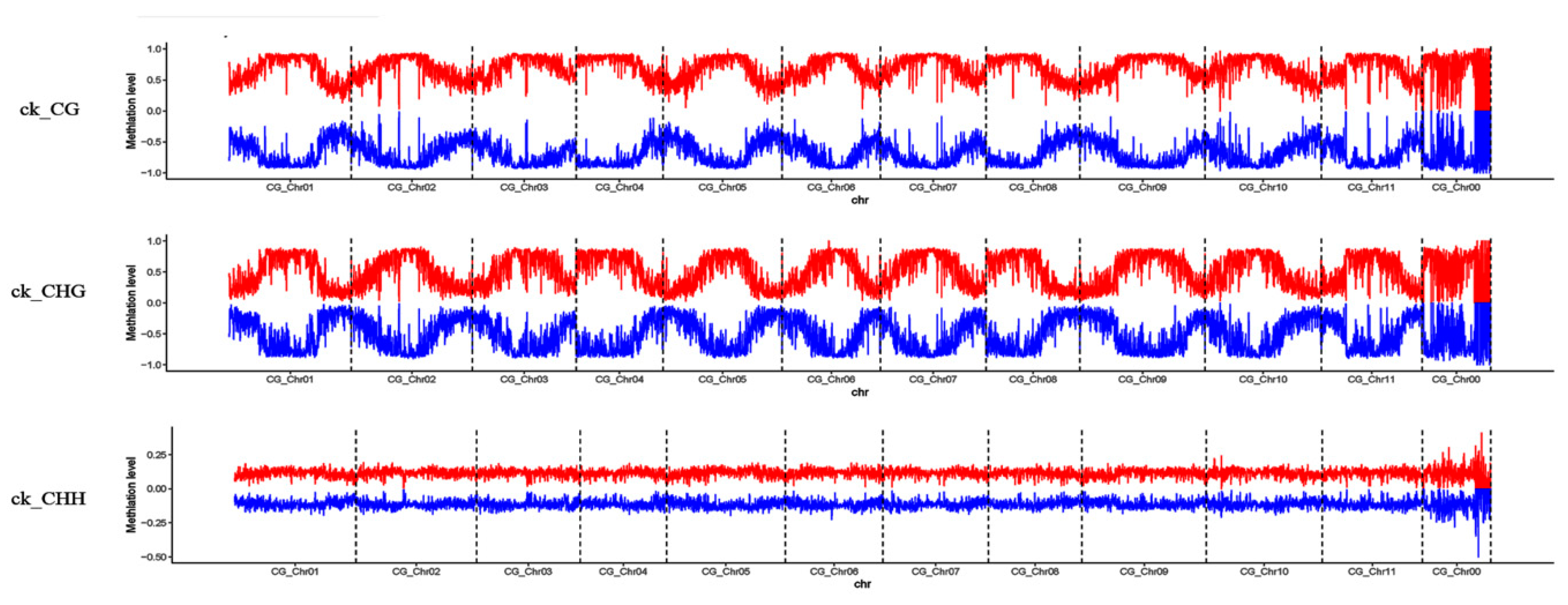
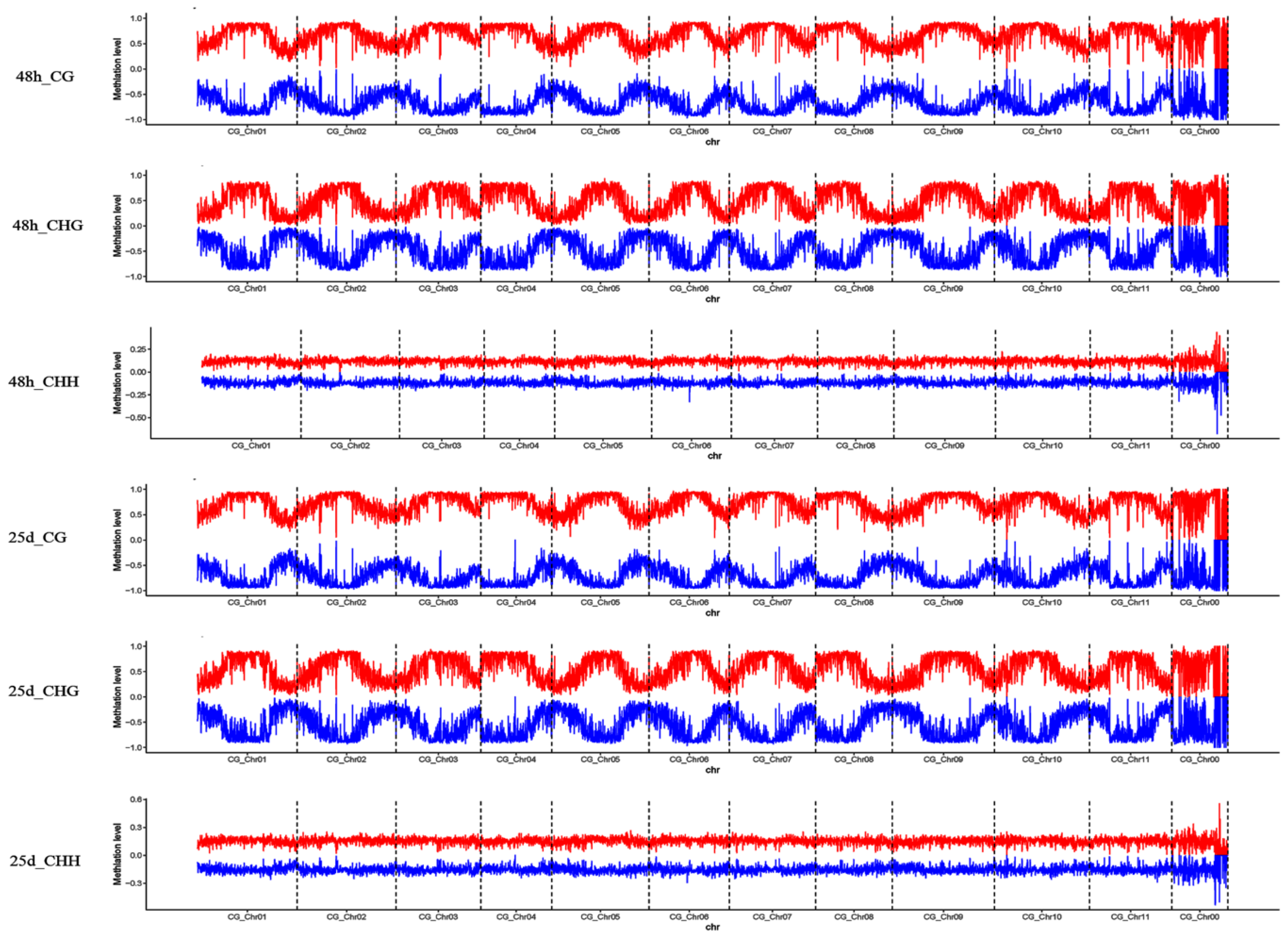
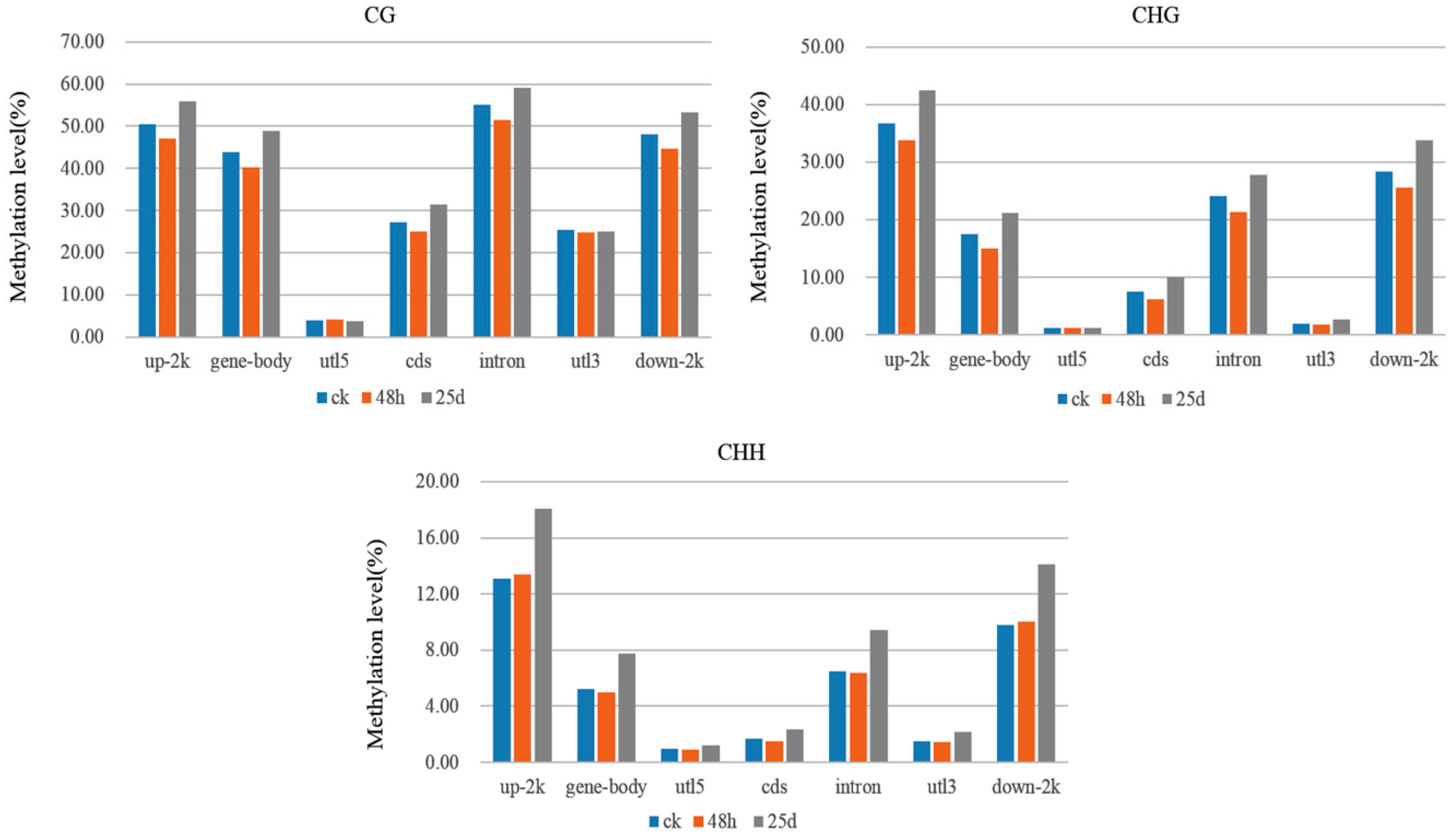
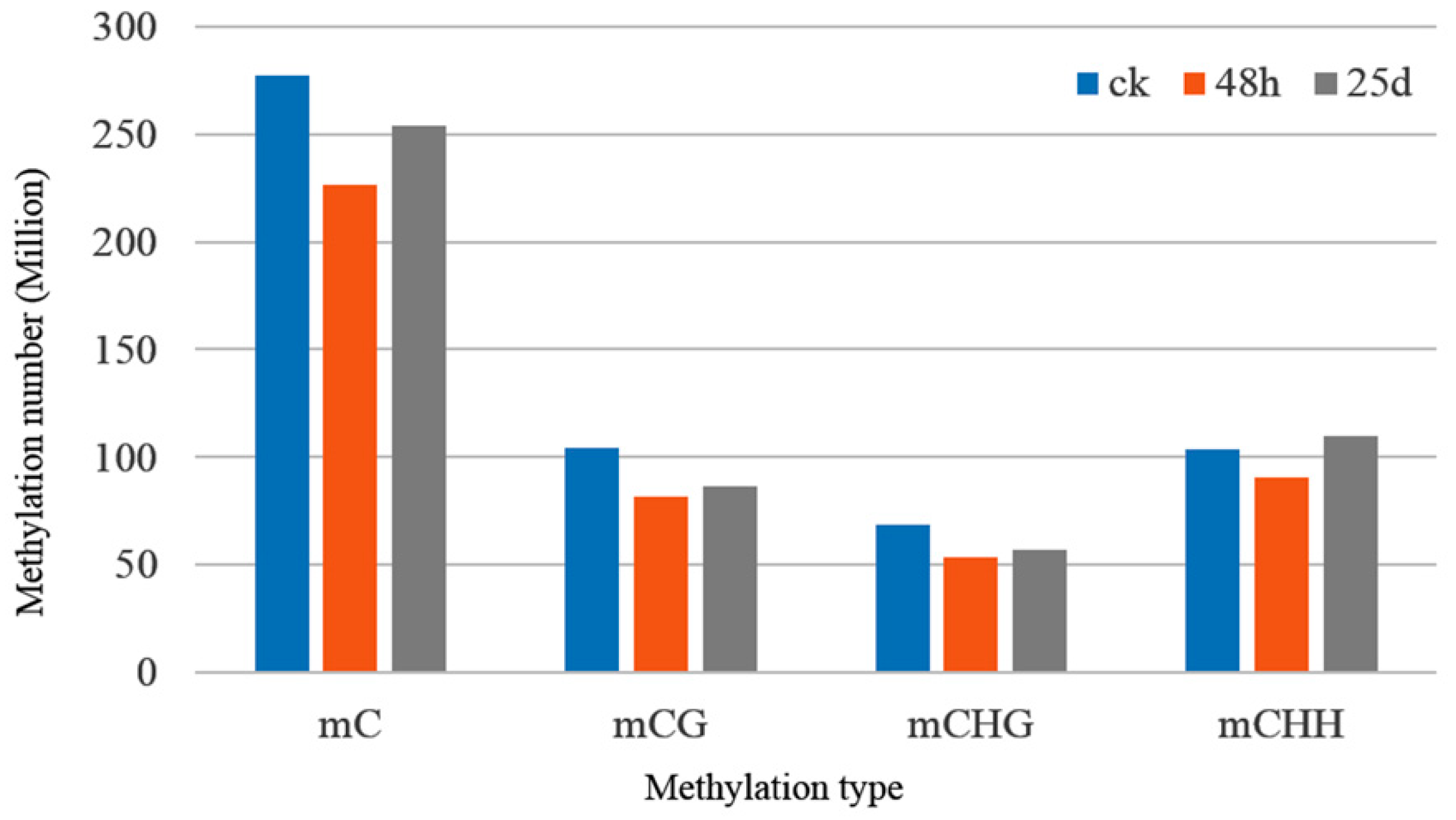
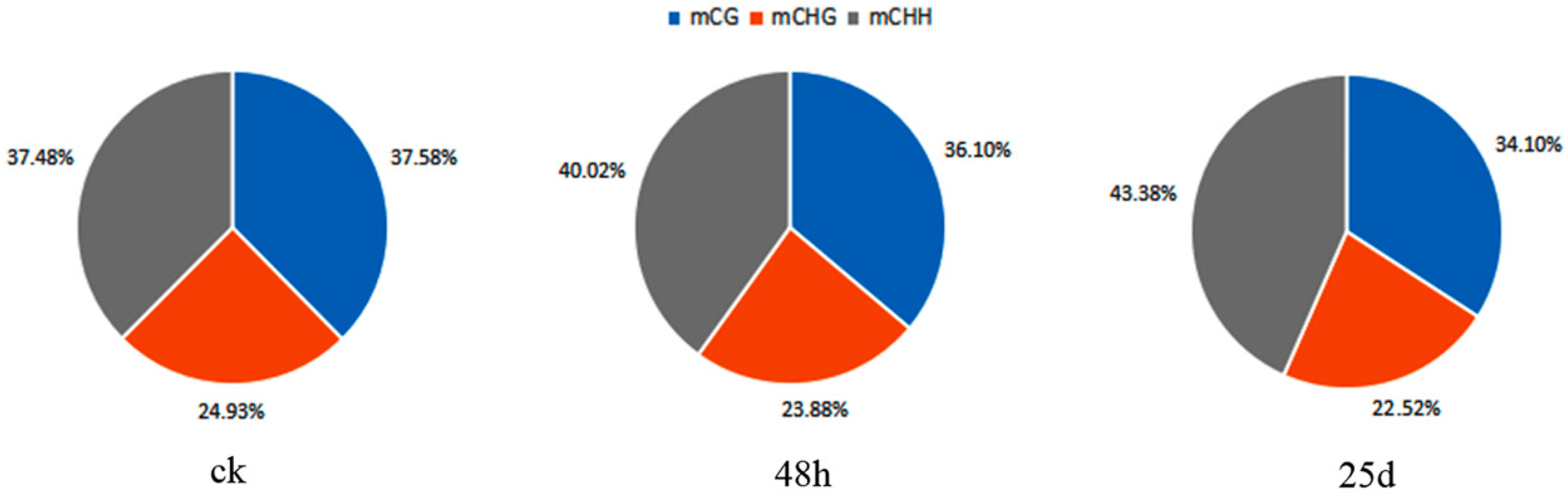
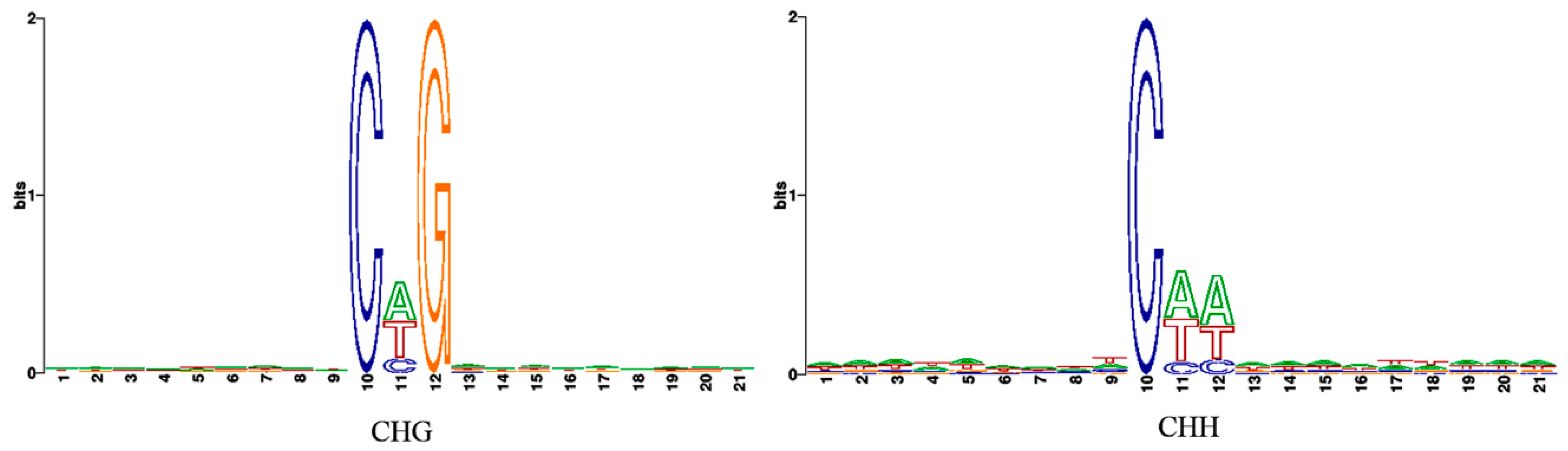
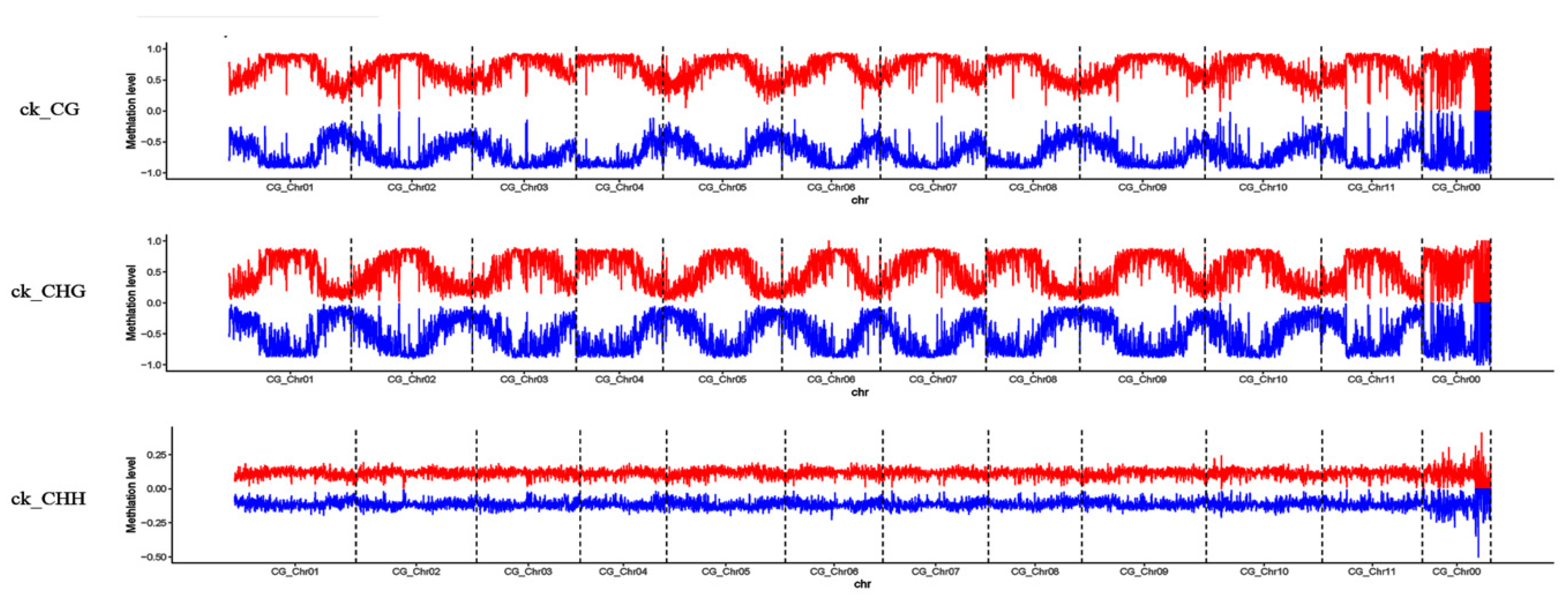
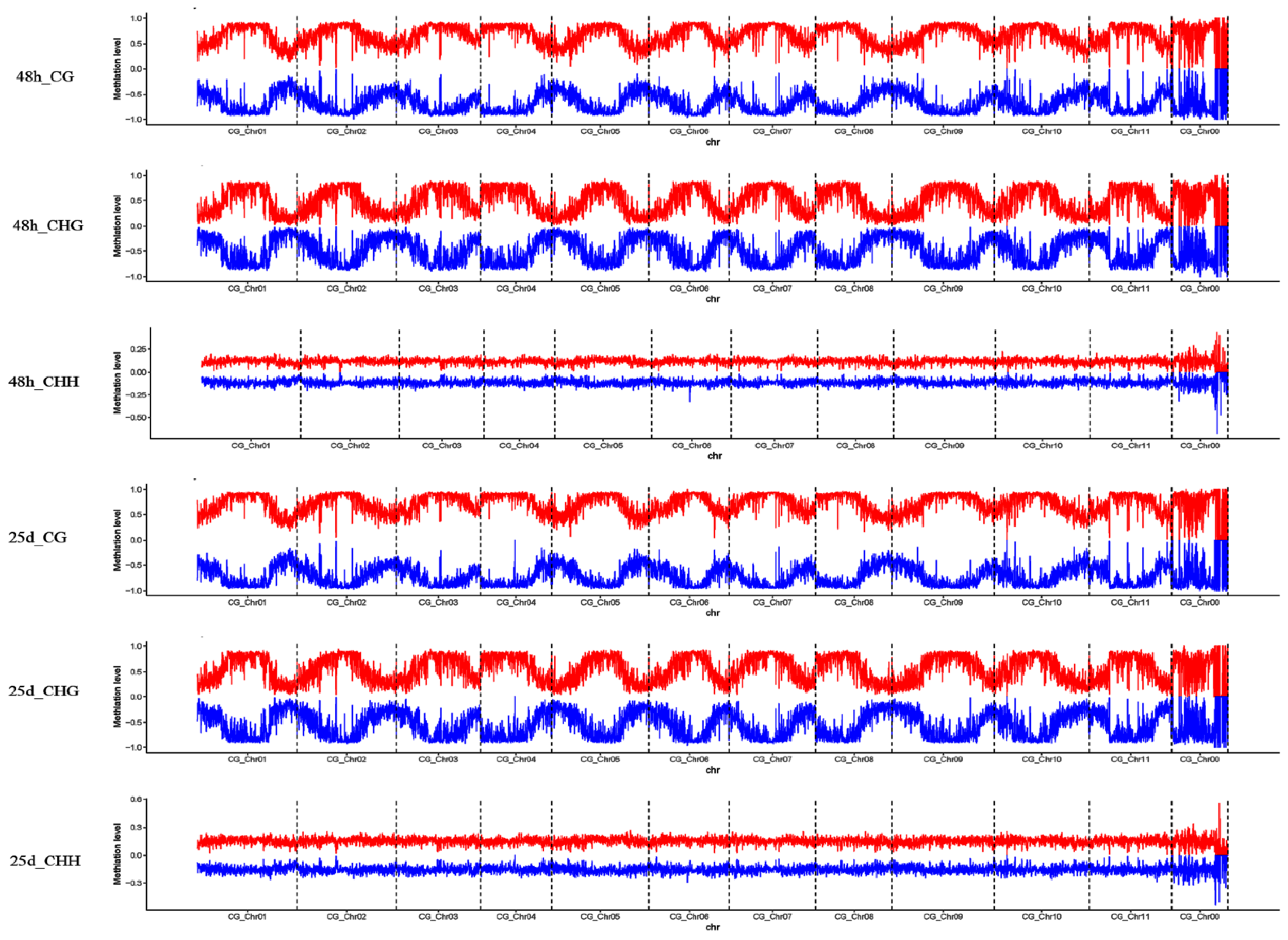
3.2. Profiles of Genome-Wide DNA Methylation in Watermelon Leaves
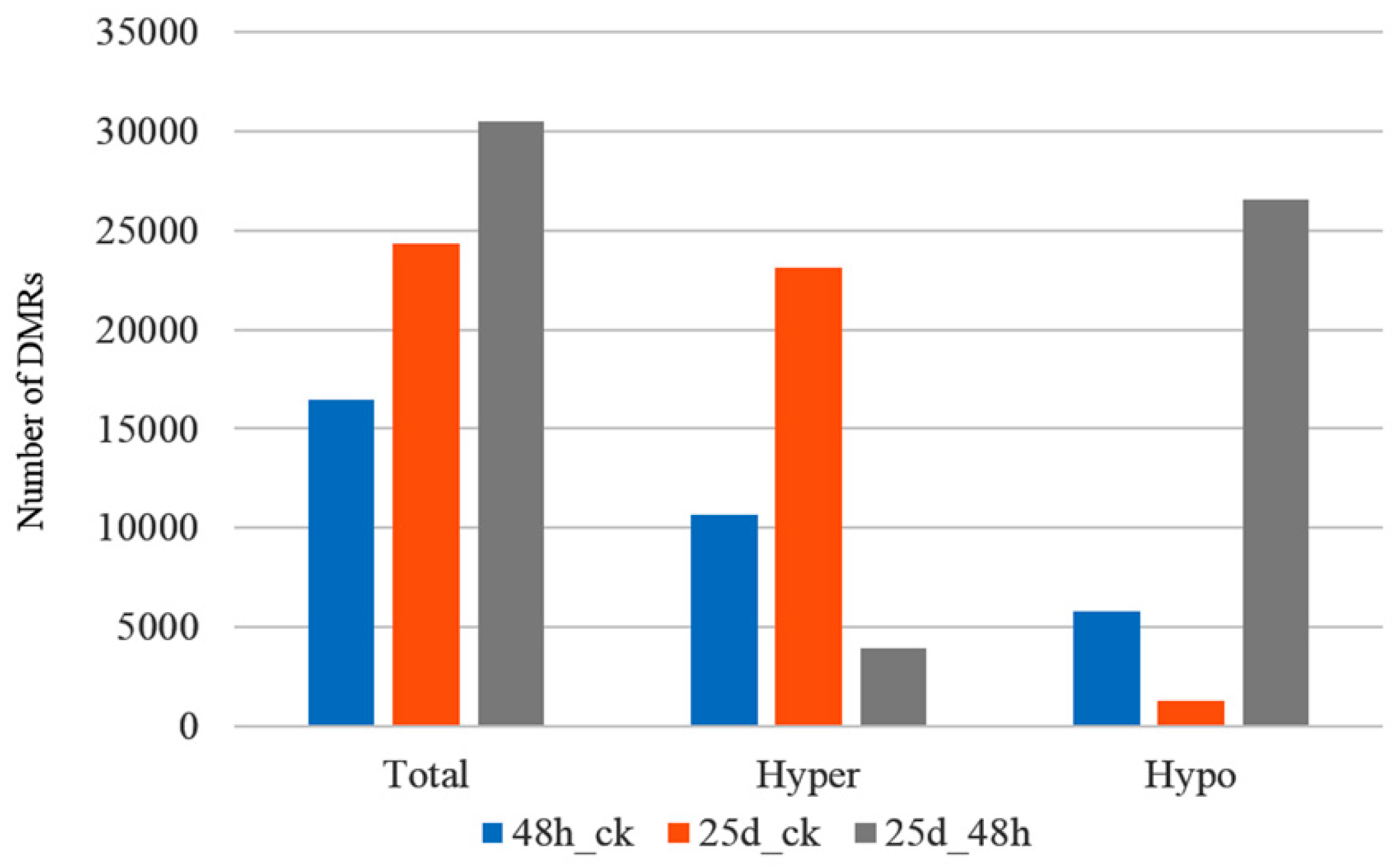
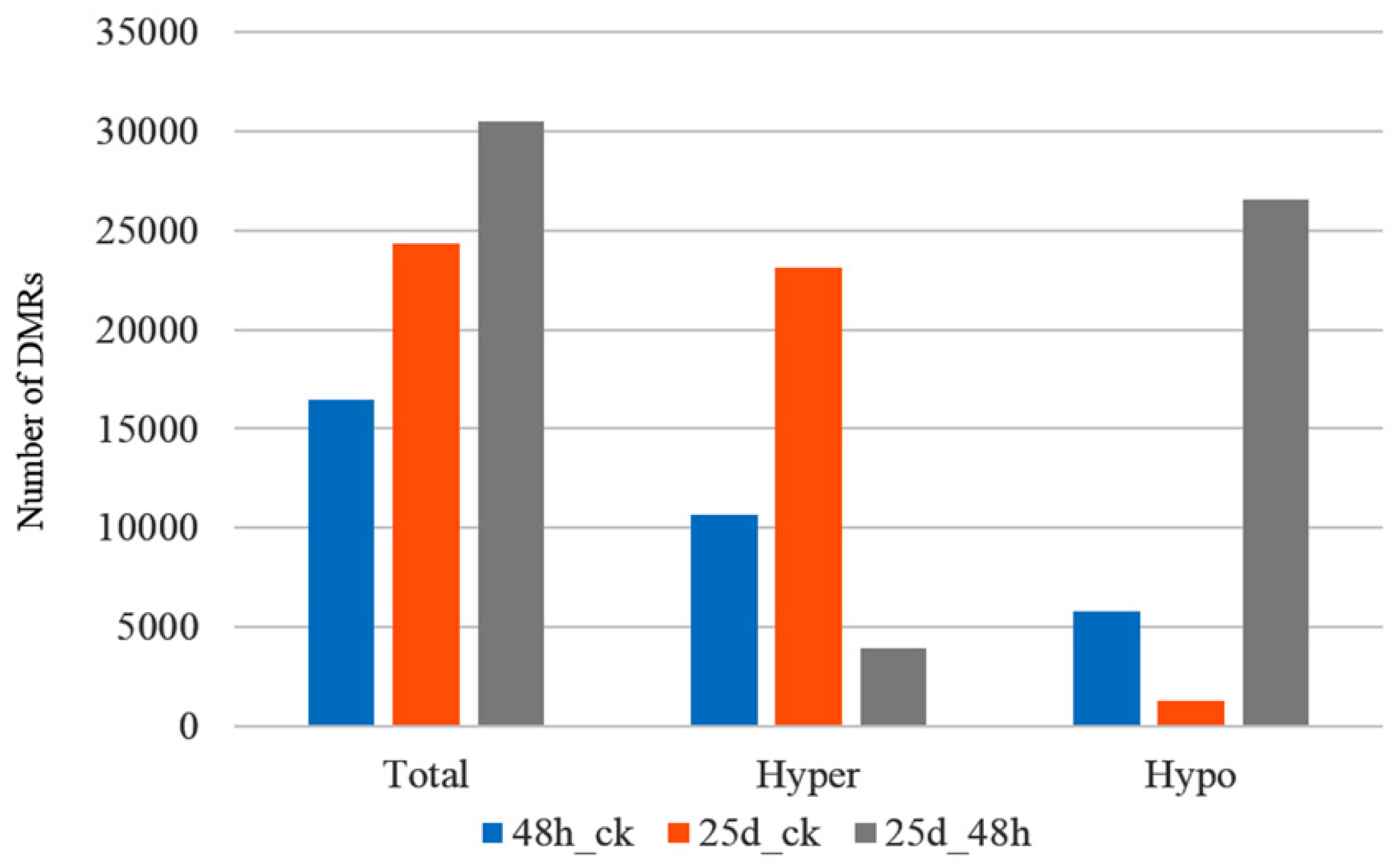
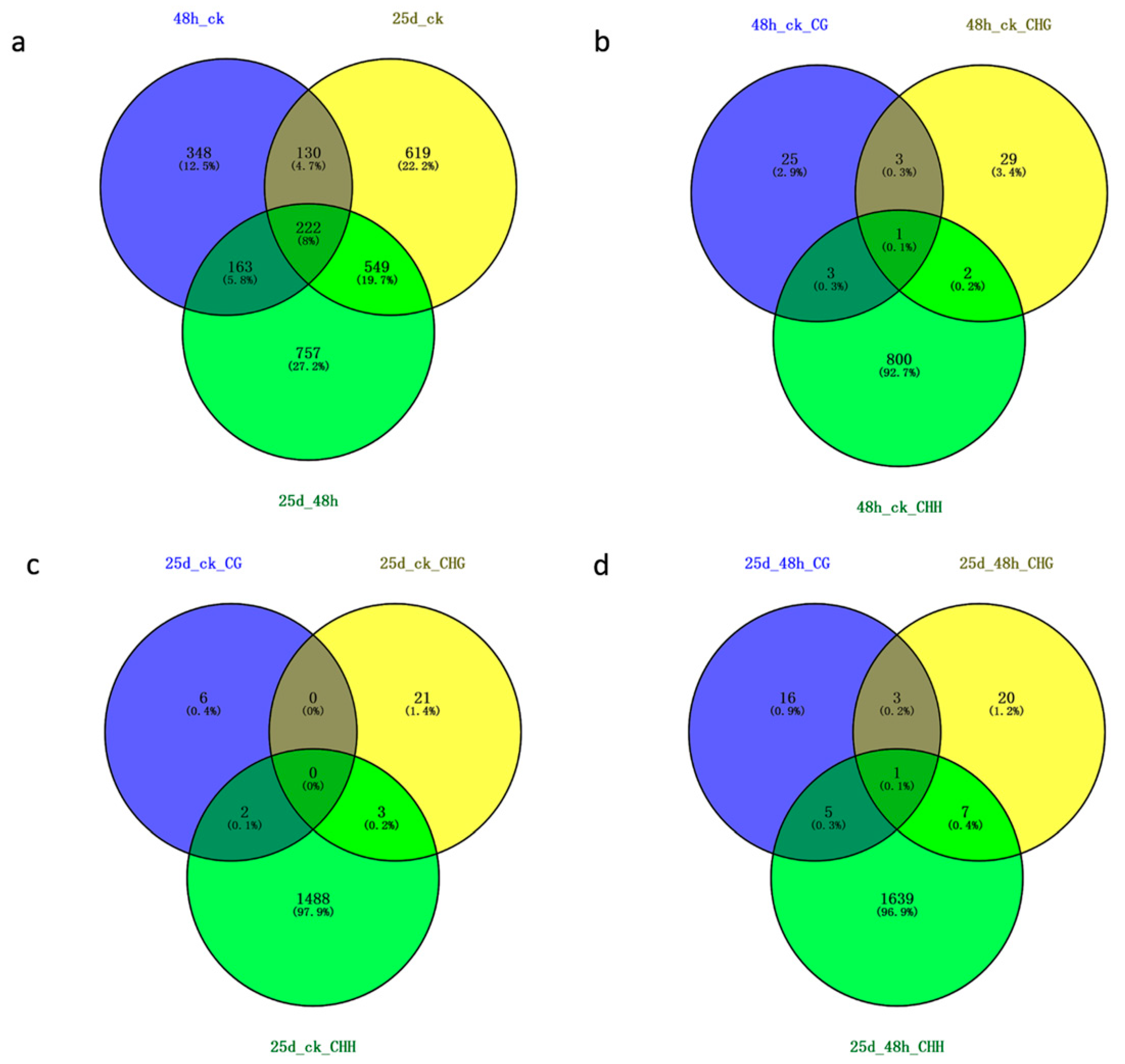
3.3. Detection of Differentially Methylated Regions
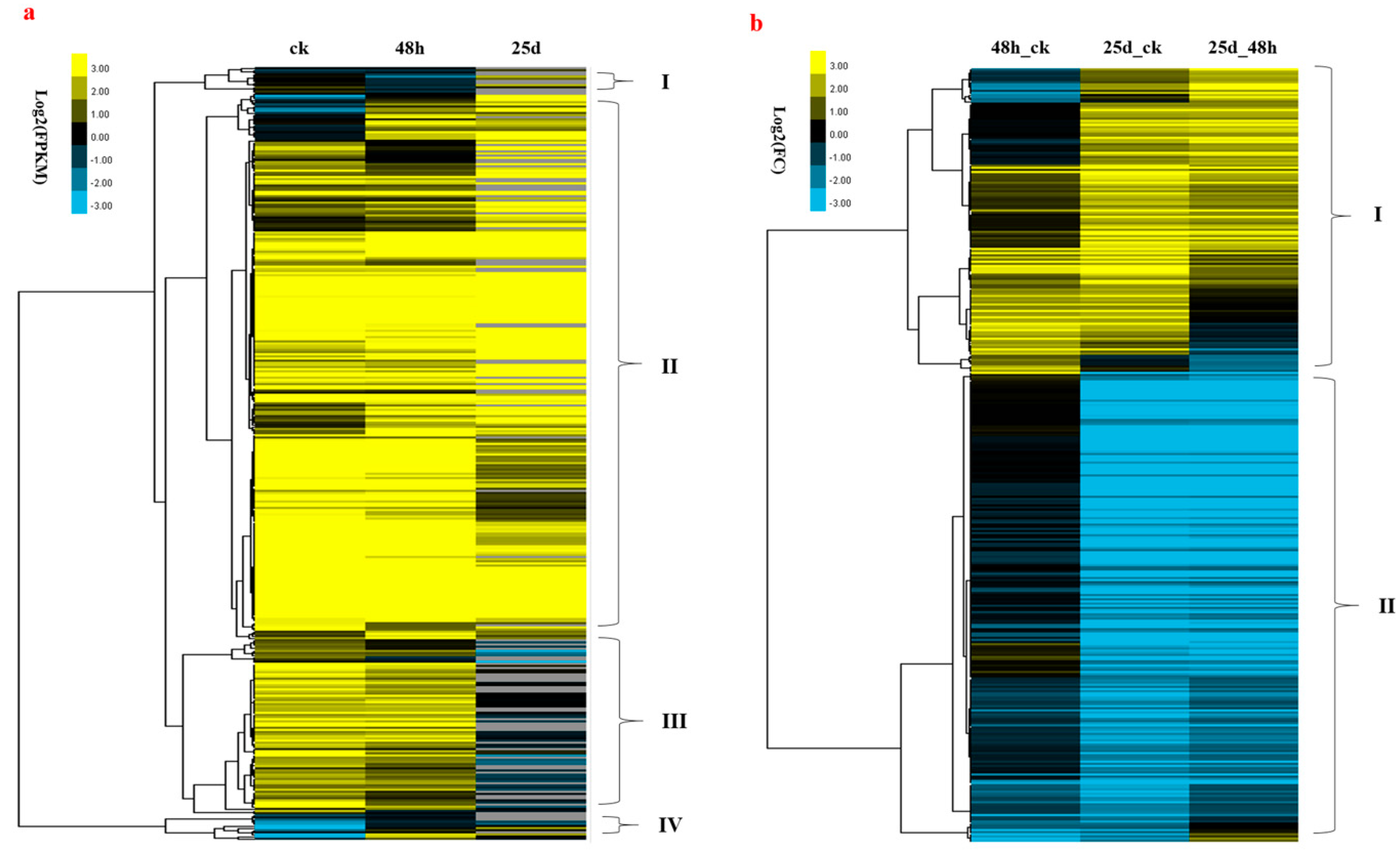
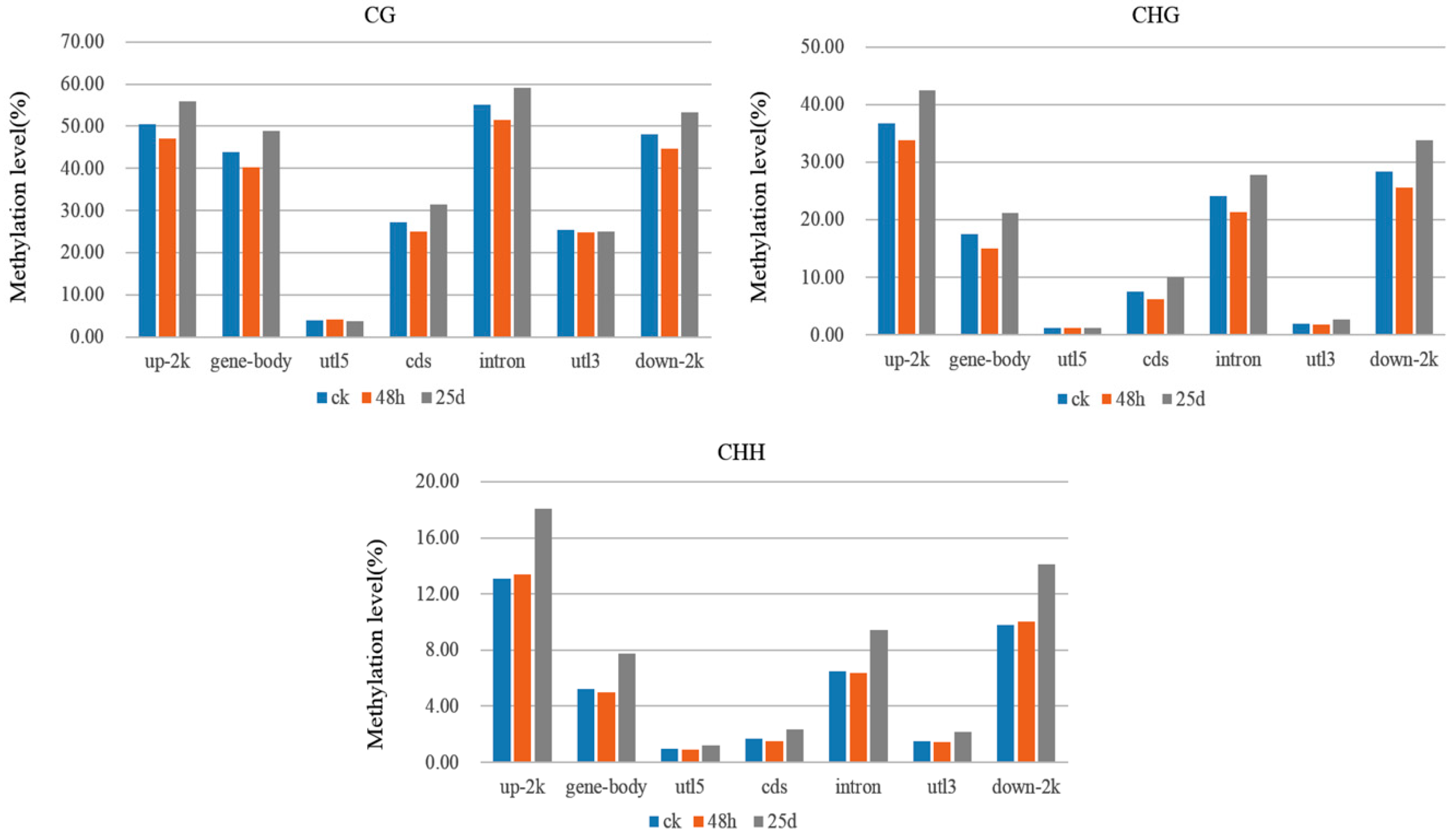
3.4. Detection of DMR-Associated Genes
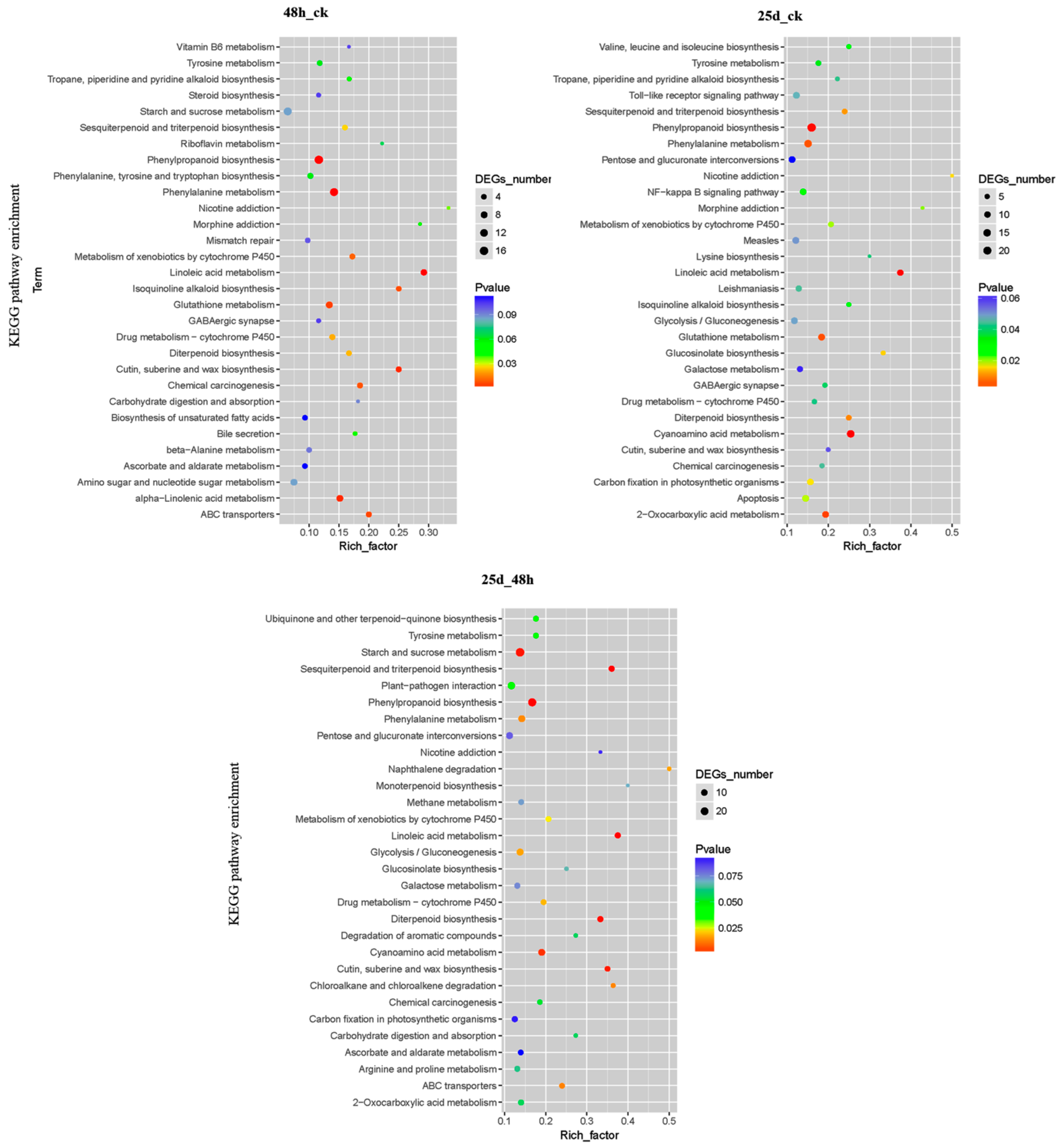
3.5. Functional Analysis of DMR-Associated Genes
3.6. DMR-Associated Genes Related to the Cucumber Green Mottle Mosaic Virus Response in Watermelon Leaves
3.6.1. DMR-Associated Genes Involved in Secondary Biosynthesis and Metabolism
3.6.2. DMR-Associated Genes Involved in Plant–Pathogen Interactions
3.6.3. DMR-Associated Genes Involved in the Toll-like Receptor Signaling Pathway
3.6.4. DMR-Associated Genes Involved in ABC Transporters
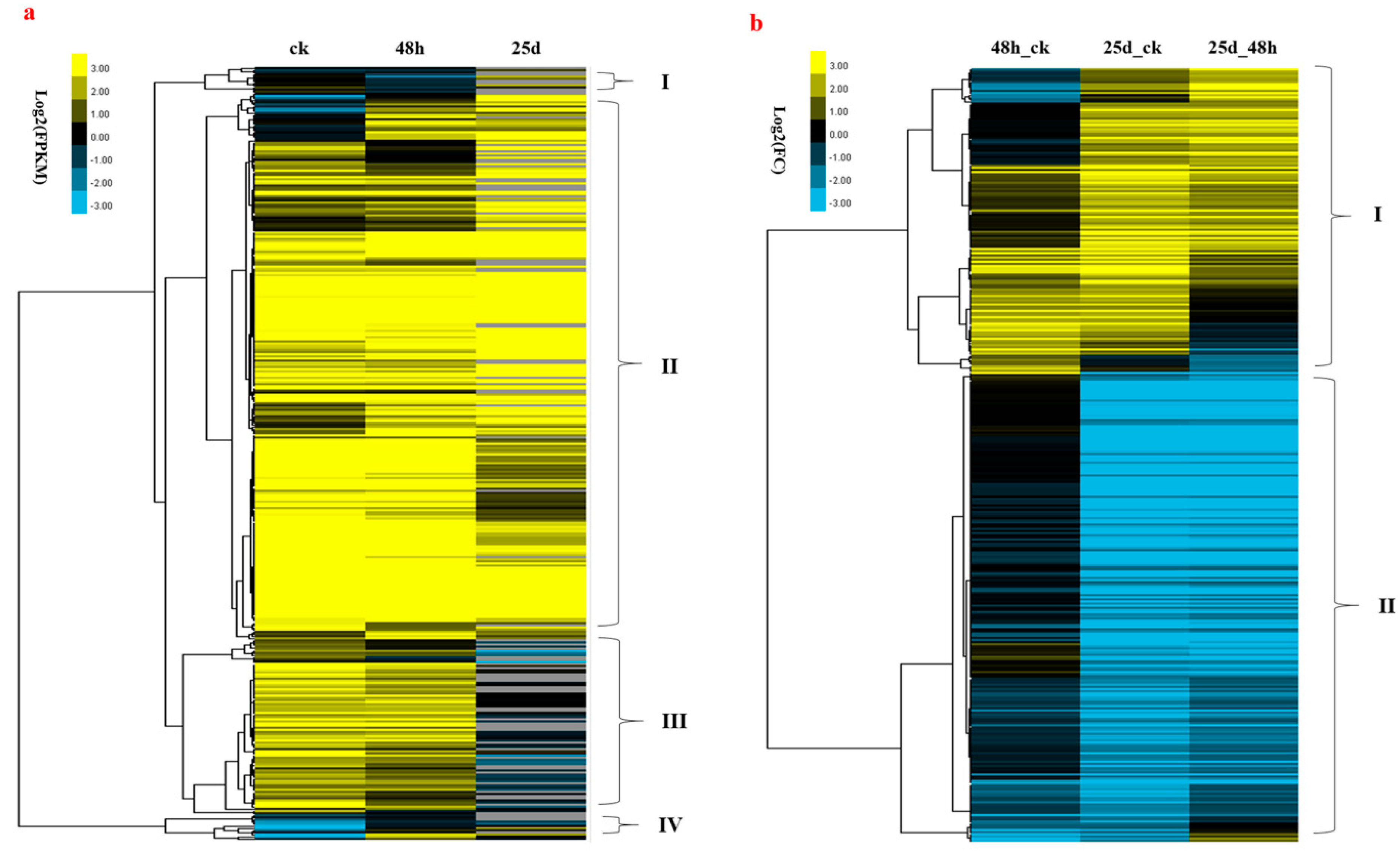
3.7. Association between DNA Methylation and Gene Expression
3.8. Expression of Genes Invovled in the RdDM Pathway and RNA Interference in Watermelon
4. Discussion
4.1. Global DNA Methylation Level and Genomic Distribution of DNA Methylation under Cucumber Green Mottle Mosaic Virus Infection
4.2. DMRs and DMGs Mainly Gathered under the CHH Context During Cucumber Green Mottle Mosaic Virus Infection
4.3. DMR-Associated Genes Enriched in Plant–Pathogen Interactions During Cucumber Green Mottle Mosaic Virus Infection
4.4. DMR-Associated Genes Enriched in Toll-Like Receptor Signaling During Cucumber Green Mottle Mosaic Virus Infection
4.5. DMR-Associated Genes Enriched in ABC Transporters During Cucumber Green Mottle Mosaic Virus Infection
4.6. Relationship between DNA Methylation and Gene Expression
4.7. RNA Interference and RdDM Pathways Associated with Watermelon Antiviral Processes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ainsworth, G.C. Mosaic disease of the cucumber. Ann. Appl. Biol. 1935, 22, 55–67. [Google Scholar] [CrossRef]
- Inoue, T.; Inoue, N.; Asatani, M.; Mitsuhata, K. Studies on cucumber green mottle mosaic virus in Japan. Nogaku Kenkyu 1967, 51, 175–186. [Google Scholar]
- Komuro, Y. Cucumber green mottle mosaic virus on cucumber and watermelon and melon necrotic spot virus on muskmelon. Jpn. Agric. Res. Q. 1971, 6, 41–45. [Google Scholar]
- Vasudeva, R.S.; Raychaudhuri, S.P.; Singh, J. A new strain of Cucumis virus 2. Ind. Phytopathol. 1949, 2, 180–185. [Google Scholar]
- Motoyoshi, F.; Nishiguchi, M. Control of virus diseases by attenuated virus strains, comparison between attenuated strains of cucumber green mottle mosaic virus and tobacco mosaic virus. Gamma Field Symp. 1988, 27, 91–109. [Google Scholar]
- Dombrovsky, A.; Tran-Nguyen, L.T.T.; Jones, R.A.C. Cucumber green mottle mosaic virus: Rapidly increasing global distribution, etiology, epidemiology and management. Ann. Rev. Phytopathol. 2017, 55, 231–256. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Win, N.K.K.; Cho, D.M.; Lee, S.H.; Jung, H.Y. Cucumber green mottle mosaic virus (CGMMV) can induce hair-like tissues on genus Cucumis seeds. Sci. Hortic. 2012, 146, 76–80. [Google Scholar] [CrossRef]
- Li, R.; Zheng, Y.; Fei, Z.; Ling, K.S. First complete genome sequence of an emerging cucumber green mottle mosaic virus isolate in North America. Genome Announc. 2015, 3, e00452-15. [Google Scholar] [CrossRef]
- Tesoriero, L.A.; Chambers, G.; Srivastava, M.; Smith, S.; Conde, B.; Tran-Nguyen, L.T.T. First report of cucumber green mottle mosaic virus in Australia. Australas. Plant Dis. Notes 2016, 11, 1. [Google Scholar] [CrossRef]
- Pop, I.; Jilaveanu, A. Identification of cucumber green mottle virus in Romania. Analele Institutului de Cercetari Pentru Protectia Plantelor 1985, 18, 43–47. [Google Scholar]
- Varveri, C.; Vassilakos, N.; Bem, F. Characterization and detection of Cucumber green mottle mosaic virus in Greece. Phytoparasitica 2002, 30, 493–501. [Google Scholar] [CrossRef]
- Li, X.; An, M.; Xia, Z.; Bai, X.; Wu, Y. Transcriptome analysis of watermelon (Citrullus lanatus) fruits in response to Cucumber green mottle mosaic virus (CGMMV) infection. Sci. Rep. 2017, 7, 16747. [Google Scholar] [CrossRef]
- Sun, Y.; Fan, M.; He, Y. Transcriptome analysis of watermelon leaves reveals candidate genes responsive to Cucumber green mottle mosaic virus infection. Int. J. Mol. Sci. 2019, 20, 610. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.W.; Luo, L.X.; Liang, C.Q.; Jiang, N.; Liu, P.F.; Li, J.Q. High-throughput sequencing identifies novel and conserved cucumber (Cucumis sativus L.) microRNAs in response to Cucumber green mottle mosaic virus infection. PLoS ONE 2015, 10, e0129002. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Niu, X.; Fan, M. Genome-wide identification of cucumber green mottle mosaic virus-responsive microRNAs in watermelon. Arch. Virol. 2017, 162, 2591–2602. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Deng, C.; Shang, Q.; Zhao, X.; Liu, X.; Zhou, Q. Characterization of siRNAs derived from cucumber green mottle mosaic virus in infected cucumber plants. Arch. Virol. 2016, 161, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zheng, H.; Zhang, C.; Han, K.; Wang, S.; Peng, J.; Lu, Y.; Zhao, J.; Xu, P.; Wu, X.; Li, G.; Chen, J.; Yan, F. Different virus-derived siRNAs profiles between leaves and fruits in Cucumber green mottle mosaic virus-infected Lagenaria siceraria plants. Front. Microbiol. 2016, 7, 1797. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Niu, X.; Cui, D.; Fan, M. High-throughput sequencing reveals vsiRNAs derived from Cucumber green mottle mosaic virus-infected watermelon. Horticul. Plant J. 2017, 3, 17–22. [Google Scholar] [CrossRef]
- Kim, M.Y.; Zilberman, D. DNA methylation as a system of plant genomic immunity. Trends Plant Sci. 2014, 19, 320–326. [Google Scholar] [CrossRef]
- Niederhuth, C.E.; Schmitz, R.J. Putting DNA methylation in context: From genomes to gene expression in plants. Biochim. Biophys. Acta 2017, 1860, 149–156. [Google Scholar] [CrossRef]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulfite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef]
- Cao, X.; Jacobsen, S.E. Locus-specific control of asymmetric and CpNpG methylation by the DRM and CMT3 methyltransferase genes. Proc. Natl. Acad. Sci. USA 2002, 99, 16491–16498. [Google Scholar] [CrossRef] [PubMed]
- Stroud, H.; Do, T.; Du, J.; Zhong, X.; Feng, S.; Johnson, L.; Patel, D.J.; Jacobsen, S.E. Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat. Struct. Mol. Boil. 2014, 21, 64–72. [Google Scholar] [CrossRef]
- Matzke, M.A.; Mosher, R.A. RNA-directed DNA methylation: An epigenetic pathway of increasing complexity. Nat. Rev. Genet. 2014, 15, 394–408. [Google Scholar] [CrossRef]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Bewick, A.J.; Schmitz, R.J. Gene body DNA methylation in plants. Curr. Opin. Plant Biol. 2017, 36, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Finnegan, E.J.; Peacock, W.J.; Dennis, E.S. Reduced DNA methylation in Arabidopsis results in abnormal plant development. Proc. Natl. Acad. Sci. USA 1996, 93, 8449–8454. [Google Scholar] [CrossRef]
- Xiao, W.; Custard, K.D.; Brown, R.C.; Lemmon, B.E.; Harada, J.J.; Goldberg, R.B.; Fischer, R.L. DNA methylation is critical for Arabidopsis embryogenesis and seed viability. Plant Cell 2006, 18, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, S.E.; Sakai, H.; Finnegan, E.J.; Cao, X.; Meyerowitz, E.M. Ectopic hypermethylation of flower-specific genes in Arabidopsis. Curr. Biol. 2000, 10, 179–186. [Google Scholar] [CrossRef]
- Kim, M.; Ohr, H.; Lee, J.W.; Hyun, Y.; Fischer, R.L.; Choi, Y. Temporal and spatial downregulation of Arabidopsis MET1 activity results in global DNA hypomethylation and developmental defects. Mol. Cells 2008, 26, 611–615. [Google Scholar]
- Xiao, W.; Brown, R.C.; Lemmon, B.E.; Harada, J.J.; Goldberg, R.B.; Fischer, R.L. Regulation of seed size by hypomethylation of maternal and paternal genomes. Plant Physiol. 2006, 142, 1160–1168. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.S.; Kawakatsu, T.; Kim, K.D.; Zhang, N.; Nguyen, C.T.; Khan, S.M.; Batek, J.M.; Joshi, T.; Schmutz, J.; Grimwood, J.; Schmitz, R.J.; Xu, D.; Jackson, S.A.; Ecker, J.R.; Stacey, G. Divergent cytosine DNA methylation patterns in single-cell, soybean root hairs. New Phytol. 2017, 214, 808–819. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, D.R.; Crisp, P.A.; Eichten, S.R.; Pogson, B.J. The Arabidopsis DNA methylome is stable under transgenerational drought stress. Plant Physiol. 2017, 175, 1893–1912. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, C.; Xu, W.; Zou, J.; Qiu, Y.; Kong, J.; Yang, Y.; Zhang, B.; Zhu, S. Epigenetic changes in the regulation of Nicotiana tabacum response to Cucumber mosaic virus infection and symptom recovery through single-base resolution methylomes. Viruses 2018, 10, 402. [Google Scholar] [CrossRef] [PubMed]
- Geng, S.; Kong, X.; Song, G.; Jia, M.; Guan, J.; Wang, F.; Qin, Z.; Wu, L.; Lan, X.; Li, A.; Mao, L. DNA methylation dynamics during the interaction of wheat progenitor Aegilops tauschii with the obligate biotrophic fungus Blumeria graminis f. sp. tritici. New Phytol. 2019, 221, 1023–1035. [Google Scholar] [CrossRef]
- Le, T.N.; Schumann, U.; Smith, N.A.; Tiwari, S.; Au, P.C.; Zhu, Q.H.; Taylor, J.M.; Kazan, K.; Llewellyn, D.J.; Zhang, R.; Dennis, E.S.; Wang, M. DNA demethylases target promoter transposable elements to positively regulate stress responsive genes in Arabidopsis. Genome Biol. 2014, 15, 458. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.; Lepere, G.; Jay, F.; Wang, J.; Bapaume, L.; Wang, Y.; Abraham, A.L.; Penterman, J.; Fischer, R.L.; Voinnet, O.; Navarro, L. Dynamics and biological relevance of DNA demethylation in Arabidopsis antibacterial defense. Proc. Natl. Acad. Sci. USA 2013, 110, 2389–2394. [Google Scholar] [CrossRef]
- Wang, B.; Yang, X.; Wang, Y.; Xie, Y.; Zhou, X. Tomato yellow leaf curl virus V2 interacts with host histone deacetylase 6 to suppress methylation-mediated transcriptional gene silencing in plants. J. Virol. 2018, 92, e00036-18. [Google Scholar] [CrossRef]
- Butterbach, P.; Verlaan, M.G.; Dullemans, A.; Lohuis, D.; Visser, R.G.; Bai, Y.; Kormelink, R. Tomato yellow leaf curl virus resistance by Ty-1 involves increased cytosine methylation of viral genomes and is compromised by cucumber mosaic virus infection. Proc. Natl. Acad. Sci. USA 2014, 111, 12942–12947. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, B.; Sanfaçon, H. Symptom recovery in virus-infected plants: Revisiting the role of RNA silencing mechanisms. Virology 2015, 479, 167–179. [Google Scholar] [CrossRef]
- Ding, S.W. RNA-based antiviral immunity. Nat. Rev. Immunol. 2010, 10, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Duan, C.G.; Fang, Y.Y.; Zhou, B.J.; Zhao, J.H.; Hou, W.N.; Zhu, H.; Ding, S.W.; Guo, H.S. Suppression of Arabidopsis ARGONAUTE1-mediated slicing, transgene-induced RNA silencing, and DNA methylation by distinct domains of the Cucumber mosaic virus 2b protein. Plant Cell 2012, 24, 259–274. [Google Scholar] [CrossRef] [PubMed]
- Piedra-Aguilera, A.; Jiao, C.; Luna, A.P.; Villanueva, F.; Dabad, M.; Esteve-Codina, A.; Díaz-Pendón, J.A.; Fei, Z.; Bejarano, E.R.; Castillo, A.G. Integrated single-base resolution maps of transcriptome, sRNAome and methylome of Tomato yellow leaf curl virus (TYLCV) in tomato. Sci. Rep. 2019, 9, 2863. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Hermanson, P.J.; Springer, N.M. Detection of DNA methylation by whole-genome bisulfite sequencing. Methods Mol. Biol. 2018, 1676, 185–196. [Google Scholar] [PubMed]
- Wang, Z.; Wu, X.; Wu, Z.; An, H.; Yi, B.; Wen, J.; Ma, C.; Shen, J.; Fu, T.; Tu, J. Genome-Wide DNA methylation comparison between Brassica napus genic male sterile line and restorer line. Int J. Mol. Sci. 2018, 10, 19. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Chang, Y.; Lu, J.; Wu, X.; Liu, Q.; Zhang, W.; Su, X.; Zhang, B. Global methylomic and transcriptomic analyses reveal the broad participation of DNA methylation in daily gene expression regulation of Populus trichocarpa. Front. Plant Sci. 2019, 10, 243. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.J.; Liang, L.X.; Diao, S.; Su, X.H.; Zhang, B.Y. Genome-wide analysis of day/night DNA methylation differences in Populus nigra. PLoS ONE 2018, 13, e0190299. [Google Scholar] [CrossRef]
- Xi, Y.; Li, W. BSMAP: Whole genome bisulfite sequence MAPping program. BMC Biol. 2009, 10, 232. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Fei, Z.; Chen, Y.R.; Zheng, Y.; Huang, M.; Vrebalov, J.; McQuinn, R.; Gapper, N.; Liu, B.; Xiang, J.; Shao, Y.; Giovannoni, J.J. Single-base resolution methylomes of tomato fruit development reveal epigenome modifications associated with ripening. Nat. Biotechnol. 2013, 31, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief Bioinf. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.P.; Fang, Y.Y.; An, C.P.; Dong, L.; Zhang, Z.H.; Chen, H.; Xie, Q.; Guo, H.S. C2-mediated decrease in DNA methylation, accumulation of siRNAs, and increase in expression for genes involved in defense pathways in plants infected with beet severe curly top virus. Plant J. 2013, 73, 910–917. [Google Scholar] [CrossRef] [PubMed]
- Dodds, P.N.; Rathjen, J.P. Plant immunity: Towards an integrated view of plant-pathogen interactions. Nat. Rev. Genet. 2010, 11, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.S.N.; Ali, G.S.; Celesnik, H.; Day, I.S. Coping with stresses: Roles of calcium- and calcium/calmodulin-regulated gene expression. Plant Cell 2011, 23, 2010–2032. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Yang, T.; Puthanveettil, S.V.; Poovaiah, B.W. Decoding of calcium signal through calmodulin: Calmodulin-binding proteins in plants. Signal. Commun. Plants 2011, 10, 177–233. [Google Scholar]
- Liu, Q.; Li, X.; Yan, S.; Yu, T.; Yang, J.; Dong, J.; Zhang, S.; Zhao, J.; Yang, T.; Mao, X.; Zhu, X.; Liu, B. OsWRKY67 positively regulates blast and bacteria blight resistance by direct activation of PR genes in rice. BMC Plant Biol. 2018, 18, 257. [Google Scholar] [CrossRef]
- Peng, Y.; Bartley, L.E.; Chen, X.; Dardick, C.; Chern, M.; Ruan, R.; Canlas, P.E.; Ronald, P.C. OsWRKY62 is a negative regulator of basal and Xa21-mediated defense against Xanthomonas oryzae pv. oryzae in rice. Mol. Plant 2008, 1, 446–458. [Google Scholar] [CrossRef]
- Chujo, T.; Miyamoto, K.; Shimogawa, T.; Shimizu, T.; Otake, Y.; Yokotani, N.; Nishizawa, Y.; Shibuya, N.; Nojiri, H.; Yamane, H.; Minami, E.; Okada, K. OsWRKY28, a PAMP-responsive transrepressor, negatively regulates innate immune responses in rice against rice blast fungus. Plant Mol. Biol. 2013, 82, 23–37. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, Q.; Tang, Y.; Ding, W. NtPR1a regulates resistance to Ralstonia solanacearum in Nicotiana tabacum via activating the defense-related genes. Biochem. Biophys. Res. Commun. 2019, 508, 940–945. [Google Scholar] [CrossRef] [PubMed]
- Antoniw, J.F.; Pierpoint, W.S. Purification of a tobacco leaf protein associated with resistance to virus infection. Biochem. Soc. Trans. 1978, 6, 248–250. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.; Goodman, R.M.; Gut-Rella, M.; Glascock, C.; Weymann, K.; Friedrich, L.; Maddox, D.; Ahl-Goy, P.; Luntz, T.; Ward, E. Increased tolerance to two oomycete pathogens in transgenic tobacco expressing pathogenesis-related protein 1a. Proc. Natl. Acad. Sci. USA 1993, 90, 7327–7331. [Google Scholar] [CrossRef] [PubMed]
- Chin, K.; DeFalco, T.A.; Moeder, W.; Yoshioka, K. The Arabidopsis cyclic nucleotide-gated ion channels AtCNGC2 and AtCNGC4 work in the same signaling pathway to regulate pathogen defense and floral transition. Plant Physiol. 2013, 163, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Dong, C.; Zhang, Y.; Zhu, J.; Dai, H.; Bai, S. An apple cyclic nucleotide-gated ion channel gene highly responsive to Botryosphaeria dothidea infection enhances the susceptibility of Nicotiana benthamiana to bacterial and fungal pathogens. Plant Sci. 2018, 269, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Yu, I.C.; Parker, J.; Bent, A.F. Gene-for-gene disease resistance without the hypersensitive response in Arabidopsis dnd1 mutant. Proc. Natl. Acad. Sci. USA 1998, 95, 7819–7824. [Google Scholar] [CrossRef]
- Jurkowski, G.I.; Smith, R.K., Jr.; Yu, I.C.; Ham, J.H.; Sharma, S.B.; Klessig, D.F.; Fengler, K.A.; Bent, A.F. Arabidopsis DND2, a second cyclic nucleotide-gated ion channel gene for which mutation causes the defense, no death phenotype. Mol. Plant Microbe Interact. 2004, 17, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.G.; da Cunha, L.; McFall, A.J.; Belkhadir, Y.; DebRoy, S.; Dangl, J.L.; Mackey, D. Two Pseudomonas syringae type III effectors inhibit RIN4-regulated basal defense in Arabidopsis. Cell 2005, 121, 749–759. [Google Scholar] [CrossRef]
- Liu, J.; Elmore, J.M.; Fuglsang, A.T.; Palmgren, M.G.; Staskwicz, B.J.; Coaker, G. RIN4 functions with plasma membrane H+-ATPases to regulate stomatal apertures during pathogen attack. PLoS Biol. 2009, 7, e1000139. [Google Scholar] [CrossRef]
- Yamada, K.; Yamashita-Yamada, M.; Hirase, T.; Fujiwara, T.; Tsuda, K.; Hiruma, K.; Saijo, Y. Danger peptide receptor signaling in plants ensures basal immunity upon pathogen-induced depletion of BAK1. EMBO J. 2016, 35, 46–61. [Google Scholar] [CrossRef]
- Yasuda, S.; Okada, K.; Saijo, Y. A look at plant immunity through the window of the multitasking coreceptor BAK1. Curr. Opin. Plant Biol. 2017, 38, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Azevedo, R.; Roque-Barreira, M.C.; Gay, N.J. Targeting and recognition of Toll-like receptors by plant and pathogen pectins. Front. Immunol. 2017, 8, 1820. [Google Scholar] [CrossRef]
- Salmeron, J.M.; Oldroyd, G.E.; Rommens, C.M.; Scofield, S.R.; Kim, H.S.; Lavelle, D.T.; Dahlbeck, D.; Staskawicz, B.J. Tomato Prf is a member of the leucine-rich repeat class of plant disease resistance genes and lies embedded within the Pto kinase gene cluster. Cell 1996, 86, 123–133. [Google Scholar] [CrossRef]
- Gao, Y.L.; Wang, B.W.; Xu, Z.L.; Li, M.Y.; Song, Z.B.; Li, W.Z.; Li, Y.P. Tobacco serine/threonine protein kinase gene NrSTK enhances black shank resistance. Genet. Mol. Res. 2015, 14, 16415–16424. [Google Scholar] [CrossRef]
- Yang, X.; Deng, F.; Ramonell, K.M. Receptor-like kinases and receptor-like proteins: Keys to pathogen recognition and defense signaling in plant innate immunity. Front. Biol. 2012, 7, 155–166. [Google Scholar] [CrossRef]
- Underwood, W.; Somerville, S.C. Phosphorylation is required for the pathogen defense function of the Arabidopsis PEN3 ABC transporter. Plant Signal. Behav. 2017, 12, e1379644. [Google Scholar] [CrossRef] [PubMed]
- Meng, D.; Dubin, M.; Zhang, P.; Osborne, E.J.; Stegle, O.; Clark, R.M.; Nordborg, M. Limited contribution of DNA methylation variation to expression regulation in Arabidopsis thaliana. PLoS Genet. 2016, 12, e1006141. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Zhang, J.; Liu, Y.; Liu, S.; Liu, Z.; Duan, Z.; Wang, Z.; Zhu, B.; Guo, Y.L.; Tian, Z. DNA methylation footprints during soybean domestication and improvement. Genome Biol. 2018, 19, 128. [Google Scholar] [CrossRef] [PubMed]
- Zilberman, D.; Gehring, M.; Tran, R.K.; Ballinger, T.; Henikoff, S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat. Genet. 2007, 39, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Gent, J.I.; Ellis, N.A.; Guo, L.; Harkess, A.E.; Yao, Y.; Zhang, X.; Dawe, R.K. CHH islands: De novo DNA methylation in near-gene chromatin regulation in maize. Genome Res. 2013, 23, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Prado, J.S.; Abulfaraj, A.A.; Rayapuram, N.; Benhamed, M.; Hirt, H. Plant immunity: From signaling to epigenetic control of defense. Trends Plant Sci. 2018, 23, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Pendon, J.A.; Li, F.; Li, W.X.; Ding, S.W. Suppression of antiviral silencing by cucumber mosaic virus 2b protein in Arabidopsis is associated with drastically reduced accumulation of three classes of viral small interfering RNAs. Plant Cell 2007, 19, 2053–2063. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Zhang, H.M.; Adams, M.J.; Yang, J.; Peng, J.J.; Antoniw, J.F.; Zhou, Y.J.; Chen, J.P. Characterization of siRNAs derived from Rice stripe virus in infected rice plants by deep sequencing. Arch. Virol. 2010, 155, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Dowen, R.H.; Pelizzola, M.; Schmitz, R.J.; Lister, R.; Dowen, J.M.; Nery, J.R.; Dixon, J.E.; Ecker, J.R. Widespread dynamic DNA methylation in response to biotic stress. Proc. Natl. Acad. Sci. USA 2012, 109, 2183–2191. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Terms | ck | 48 hpi | 25 dpi |
|---|---|---|---|
| Raw reads | 91,070,116 | 80,314,902 | 73,989,862 |
| Raw bases | 13,660,517,400 | 11,098,479,300 | 12,047,235,300 |
| Clean reads | 83,839,958 | 71,659,074 | 67,011,742 |
| Clean bases | 12,402,189,963 | 9,905,599,786 | 10,592,716,908 |
| Uniquely mapped reads | 75,798,928 | 6,402,9094 | 60,394,949 |
| Uniquely mapped rates (%) | 90.41 | 89.35 | 90.13 |
| Clean reads with unknown bases (Ns) | 104,845 | 160,432 | 150,563 |
| Quality score 20 (Q20) (%) | 97.14 | 97.11 | 97.09 |
| Quality score 30 (Q30) (%) | 90.84 | 90.82 | 90.80 |
| GC (%) | 19.87 | 19.79 | 20.29 |
| Coverage (%) | 82.05 | 74.54 | 72.33 |
| Strand depth | 12.06 | 10.49 | 11.62 |
| Depth >= 4 (%) | 60.73 | 51.57 | 54.15 |
| Conversion rate (%) | 99.61 | 99.60 | 99.64 |
| Comparison | DMRs Total | DMRs Hyper | DMRs Hypo | Intragenic DMRs | Intragenic Hyper | Intragenic Hypo | Intergenic DMRs | Intergenic Hyper | Intergenic Hypo |
|---|---|---|---|---|---|---|---|---|---|
| 48h_ck_CG | 205 | 23 | 182 | 39 | 11 | 28 | 166 | 12 | 154 |
| 48h_ck_CHG | 205 | 29 | 176 | 41 | 11 | 30 | 164 | 18 | 146 |
| 48h_ck_CHH | 16,073 | 10,631 | 5442 | 1293 | 860 | 433 | 14,790 | 9771 | 5009 |
| 48h_ck_Total | 16,483 | 10,683 | 5800 | 1373 | 882 | 491 | 15,120 | 9801 | 5309 |
| 25d_ck_CG | 61 | 19 | 42 | 11 | 3 | 8 | 50 | 16 | 34 |
| 25d_ck.CHG | 96 | 63 | 33 | 31 | 21 | 10 | 65 | 42 | 23 |
| 25d_ck_CHH | 24,193 | 23,004 | 1189 | 2112 | 2021 | 91 | 22,081 | 20,983 | 1098 |
| 25d_ck_Total | 24,350 | 23,086 | 1264 | 2154 | 2045 | 109 | 22,196 | 21,041 | 1155 |
| 25d_48h_CG | 181 | 28 | 153 | 28 | 6 | 22 | 153 | 22 | 131 |
| 25d_48h_CHG | 215 | 12 | 203 | 36 | 3 | 33 | 179 | 9 | 170 |
| 25d_48h_CHH | 30,127 | 3909 | 26,218 | 2502 | 292 | 2210 | 27,625 | 3617 | 24,008 |
| 25d_48h_Total | 30,523 | 3949 | 26,574 | 2566 | 301 | 2265 | 27,957 | 3648 | 24,309 |
| Genes | Annotation | FPKM_ck | FPKM_48h | FPKM_25d | log2(48h_ck) | log2(25d_ck) | log2(25d_48h) |
|---|---|---|---|---|---|---|---|
| ClCG08G009740 | DCL3 | 6.63 | 5.95 | 5.49 | −0.16 (down) | −0.27 (down) | −0.11 (down) |
| ClCG10G005250 | DCL3 | 4.08 | 5.28 | 2.96 | 0.37 (up) | −0.46 (down) | −0.84 (down) |
| ClCG02G002110 | DCL3 | 1.49 | 1.83 | 0.00 | 0.30 (up) | Inf (down) | Inf (down) |
| ClCG06G012100 | DCL4 | 2.37 | 2.63 | 1.78 | 0.15 (up) | −0.41 (down) | −0.56 (down) |
| ClCG03G010530 | DCL2 | 5.64 | 5.38 | 21.89 | −0.07 (down) | 1.96 (up) | 2.03 (up) |
| ClCG02G024220 | AGO4 | 44.49 | 38.07 | 20.30 | −0.23 (down) | −1.13 (down) | −0.91 (down) |
| ClCG09G011170 | AGO6 | 1.40 | 0.70 | 0.00 | −0.99 (down) | Inf (down) | Inf (down) |
| ClCG00G004530 | AGO10 | 1.71 | 0.86 | 0.80 | −1.00 (down) | −1.10 (down) | −0.10 (down) |
| ClCG04G012520 | AGO1B | 40.68 | 34.46 | 49.82 | −0.24 (down) | 0.29 (up) | 0.53 (up) |
| ClCG05G023990 | AGO | 9.93 | 6.31 | 6.98 | −0.65 (down) | −0.51 (down) | 0.15 (up) |
| ClCG05G004800 | AGO | 40.68 | 34.46 | 49.82 | −0.24 (down) | 0.29 (up) | 0.53 (up) |
| ClCG01G014010 | AGO | 5.19 | 6.18 | 6.52 | 0.25 (up) | 0.33 (up) | 0.08 (up) |
| ClCG03G013970 | CMT | 6.68 | 2.12 | 5.87 | −1.65 (down) | −0.19 (down) | 1.47 (up) |
| ClCG10G001140 | CMT | 15.47 | 17.87 | 14.80 | 0.21 (up) | −0.06 (down) | −0.27 (down) |
| ClCG11G000400 | CMT | 6.53 | 6.09 | 3.37 | −0.10 (down) | −0.96 (down) | −0.85 (down) |
| ClCG10G014050 | DRM2 | 20.84 | 19.90 | 14.12 | −0.07 (down) | −0.56 (down) | −0.49 (down) |
| ClCG06G004300 | DRM2 | 8.24 | 8.25 | 17.88 | 0.00 | 1.12 (up) | 1.12 (up) |
| ClCG01G006600 | RDR | 0.06 | 0.41 | 0.00 | 2.76 (up) | Inf (down) | Inf (down) |
| ClCG01G006450 | RDR | 0.28 | 0.02 | 1.04 | −4.04 (down) | 1.86 (up) | 5.90 (up) |
| ClCG08G013320 | RDR6 | 7.14 | 4.69 | 1.40 | −0.61 (down) | −2.35 (down) | −1.74 (down) |
| ClCG07G005340 | RDR1a | 0.86 | 1.12 | 1.18 | 0.38 (up) | 0.46 (up) | 0.07 (up) |
| ClCG06G016860 | RDR2 | 3.61 | 2.93 | 1.83 | −0.30 (down) | −0.98 (down) | −0.68 (down) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Y.; Fan, M.; He, Y. DNA Methylation Analysis of the Citrullus lanatus Response to Cucumber Green Mottle Mosaic Virus Infection by Whole-Genome Bisulfite Sequencing. Genes 2019, 10, 344. https://doi.org/10.3390/genes10050344
Sun Y, Fan M, He Y. DNA Methylation Analysis of the Citrullus lanatus Response to Cucumber Green Mottle Mosaic Virus Infection by Whole-Genome Bisulfite Sequencing. Genes. 2019; 10(5):344. https://doi.org/10.3390/genes10050344
Chicago/Turabian StyleSun, Yuyan, Min Fan, and Yanjun He. 2019. "DNA Methylation Analysis of the Citrullus lanatus Response to Cucumber Green Mottle Mosaic Virus Infection by Whole-Genome Bisulfite Sequencing" Genes 10, no. 5: 344. https://doi.org/10.3390/genes10050344
APA StyleSun, Y., Fan, M., & He, Y. (2019). DNA Methylation Analysis of the Citrullus lanatus Response to Cucumber Green Mottle Mosaic Virus Infection by Whole-Genome Bisulfite Sequencing. Genes, 10(5), 344. https://doi.org/10.3390/genes10050344