Post-Translational Modifications of the Mini-Chromosome Maintenance Proteins in DNA Replication

Abstract
:1. Introduction
2. Overview of the MCM Proteins
3. Involvement of the MCM Proteins in Human Disease
4. Involvement of the MCM Proteins in Human Cancer
5. MCM Protein PTMs Modulate DNA Replication and the Replication Stress Response
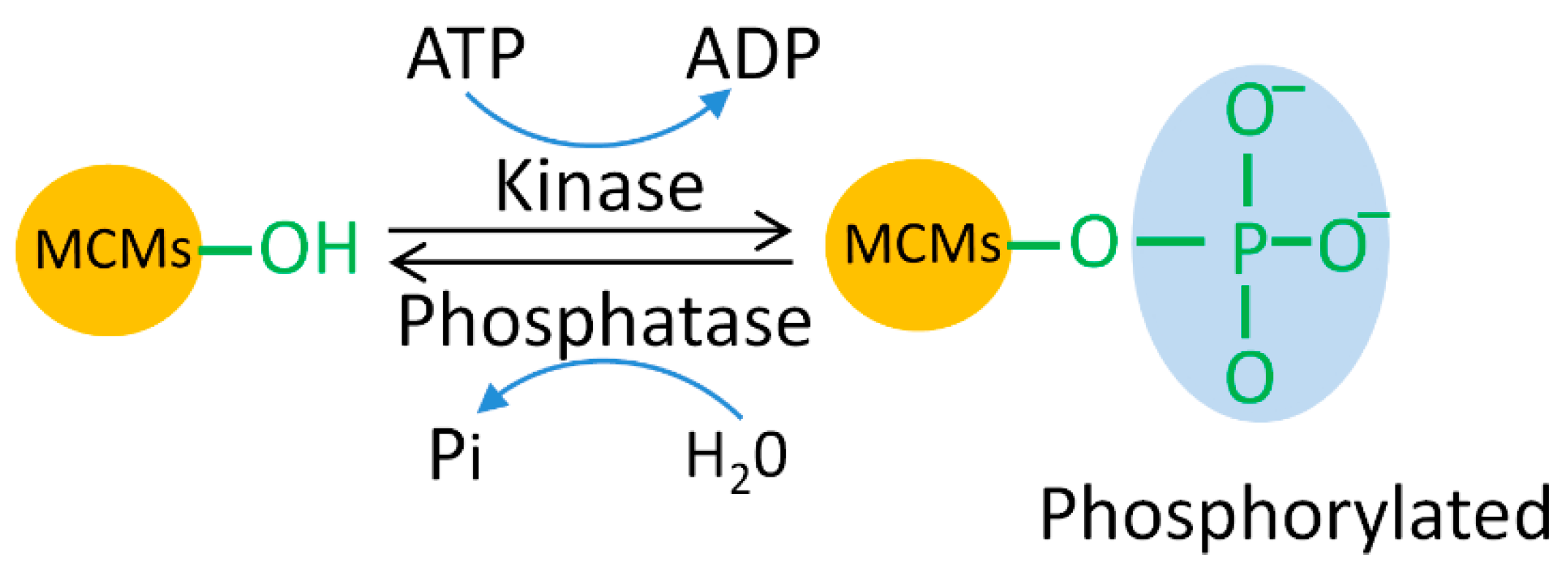
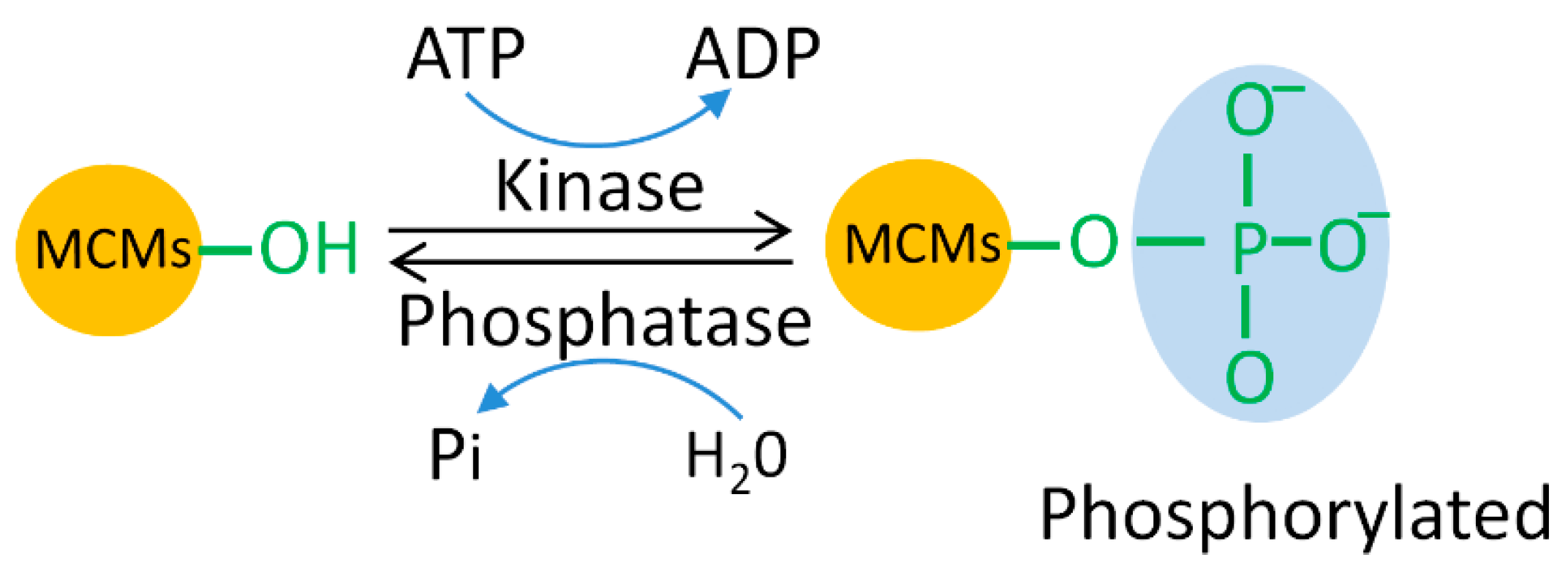
5.1. MCM Phosphorylation
5.1.1. CDK-Dependent Phosphorylation of MCMs
5.1.2. CDC7-Dependent Phosphorylation of MCMs
5.1.3. ATM/ATR-Dependent Phosphorylation of MCMs
5.1.4. Other Kinase-Dependent Phosphorylation of MCMs
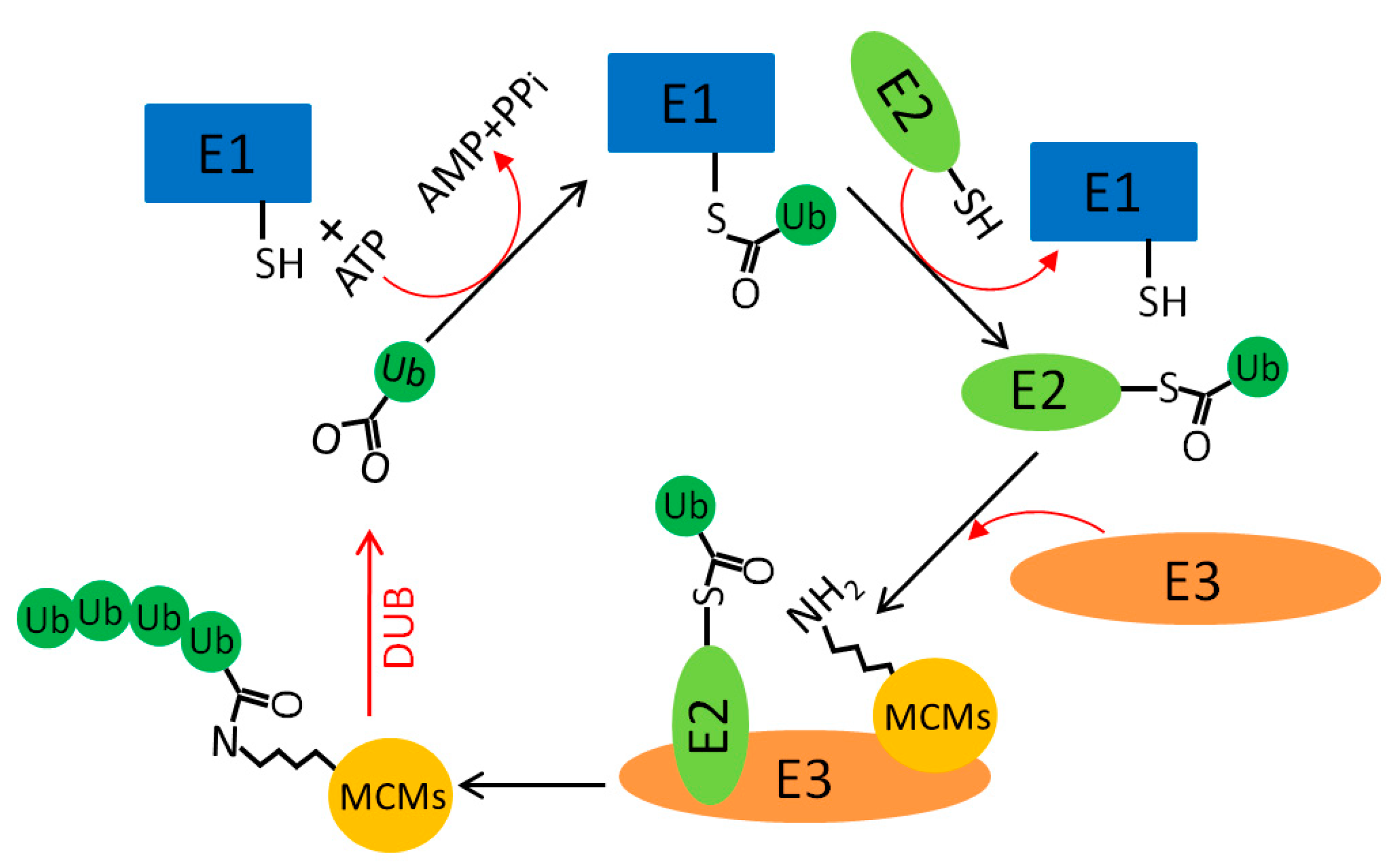
5.2. MCM Ubiquitination
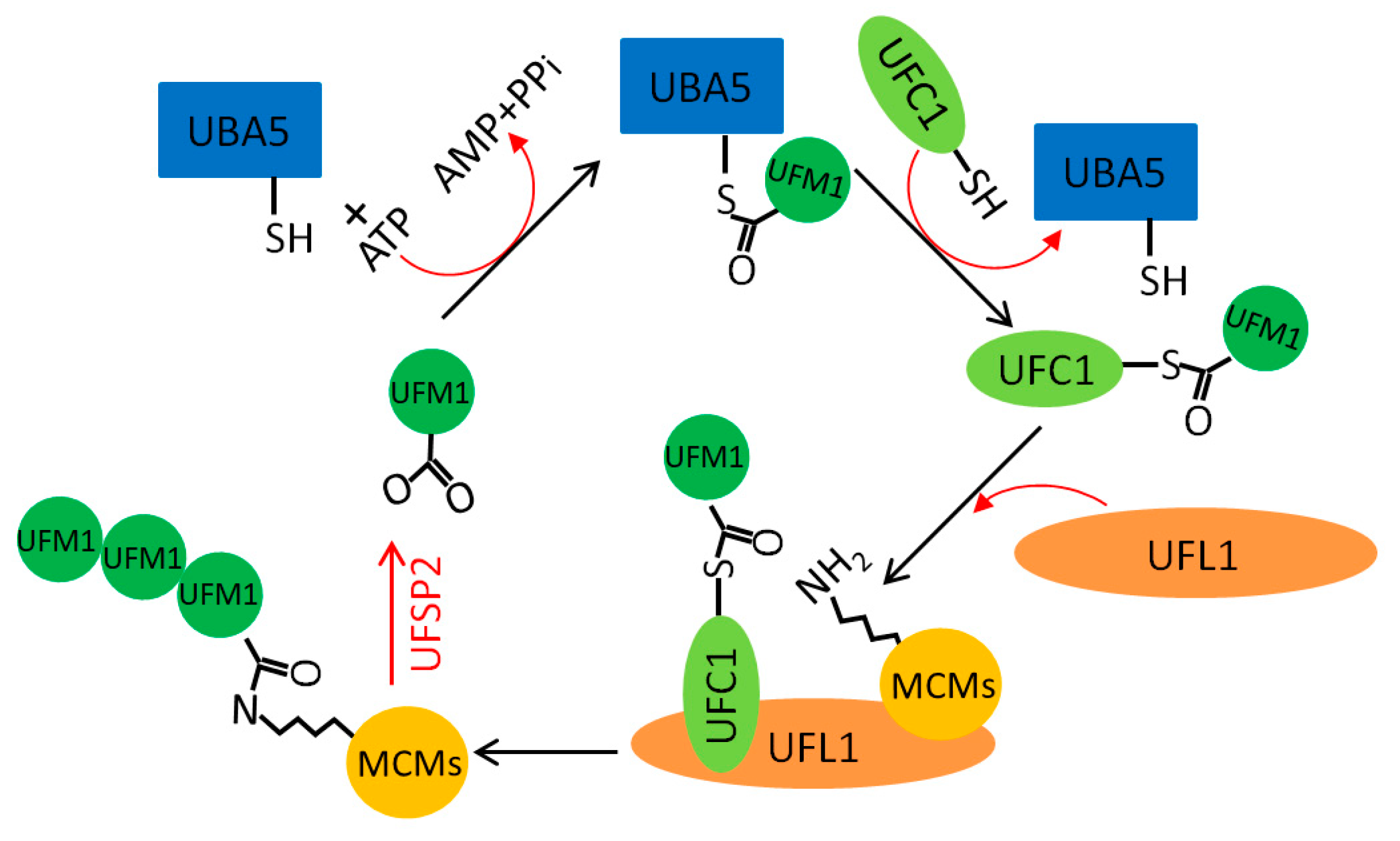
5.3. MCM UFMylation
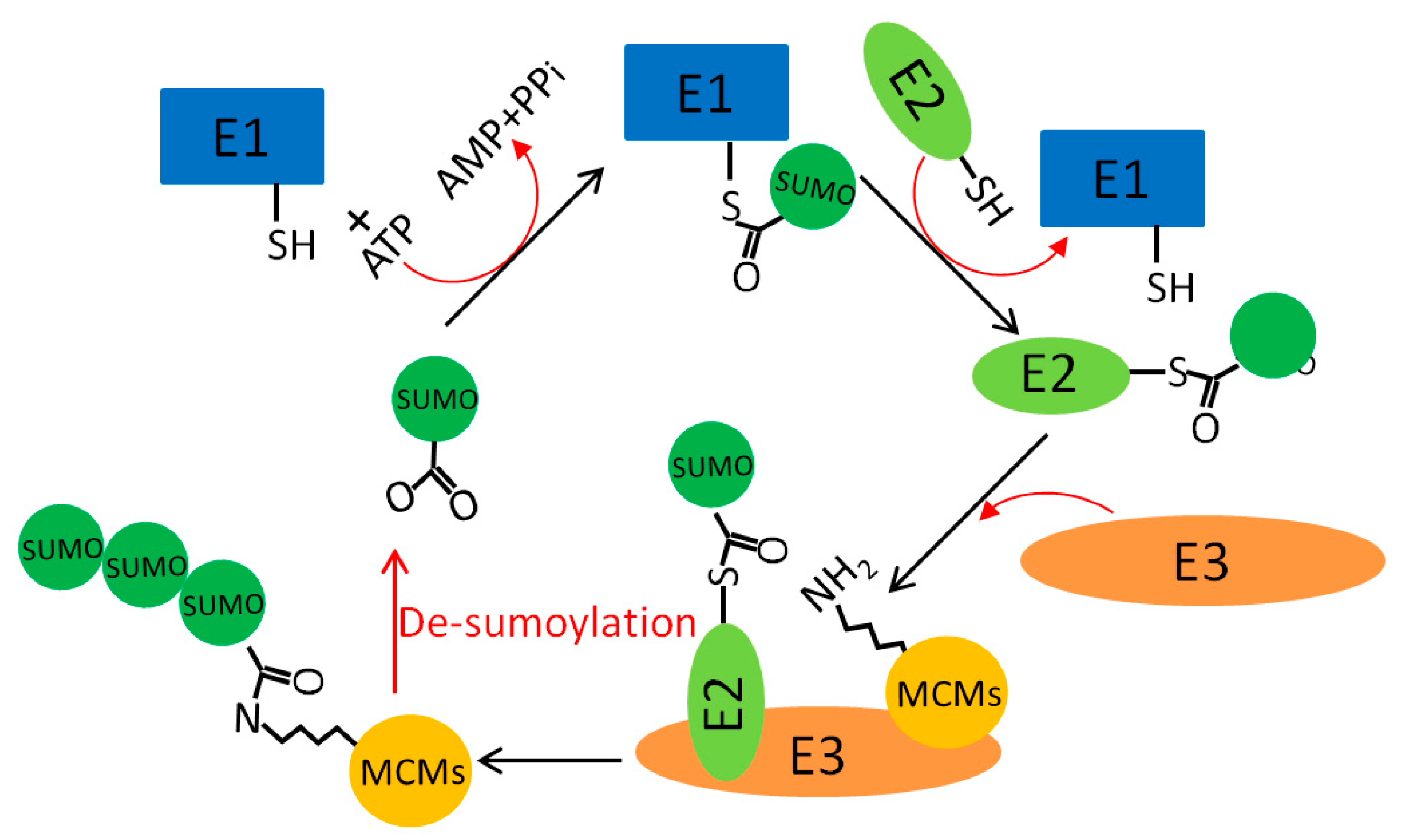
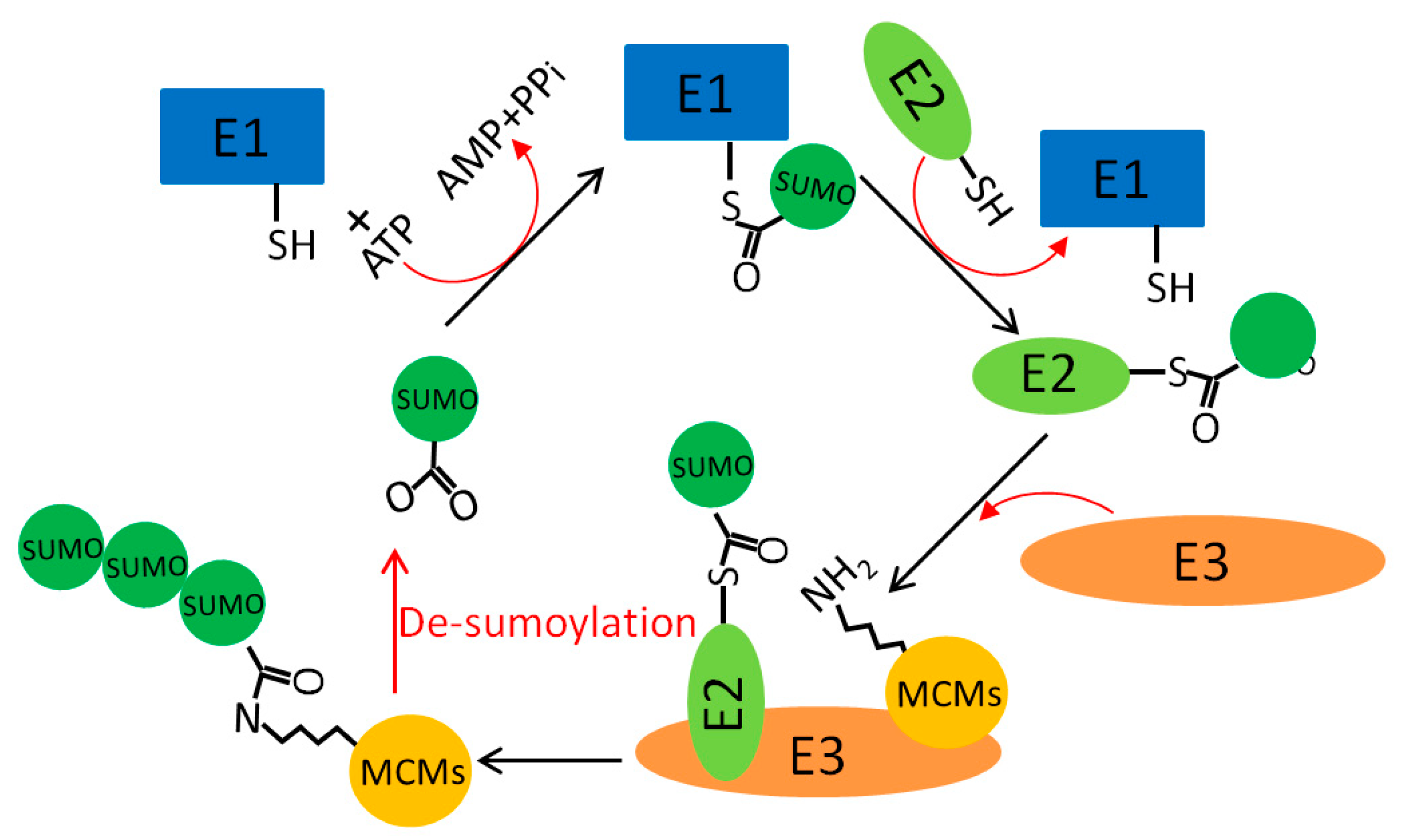
5.4. MCM Sumoylation
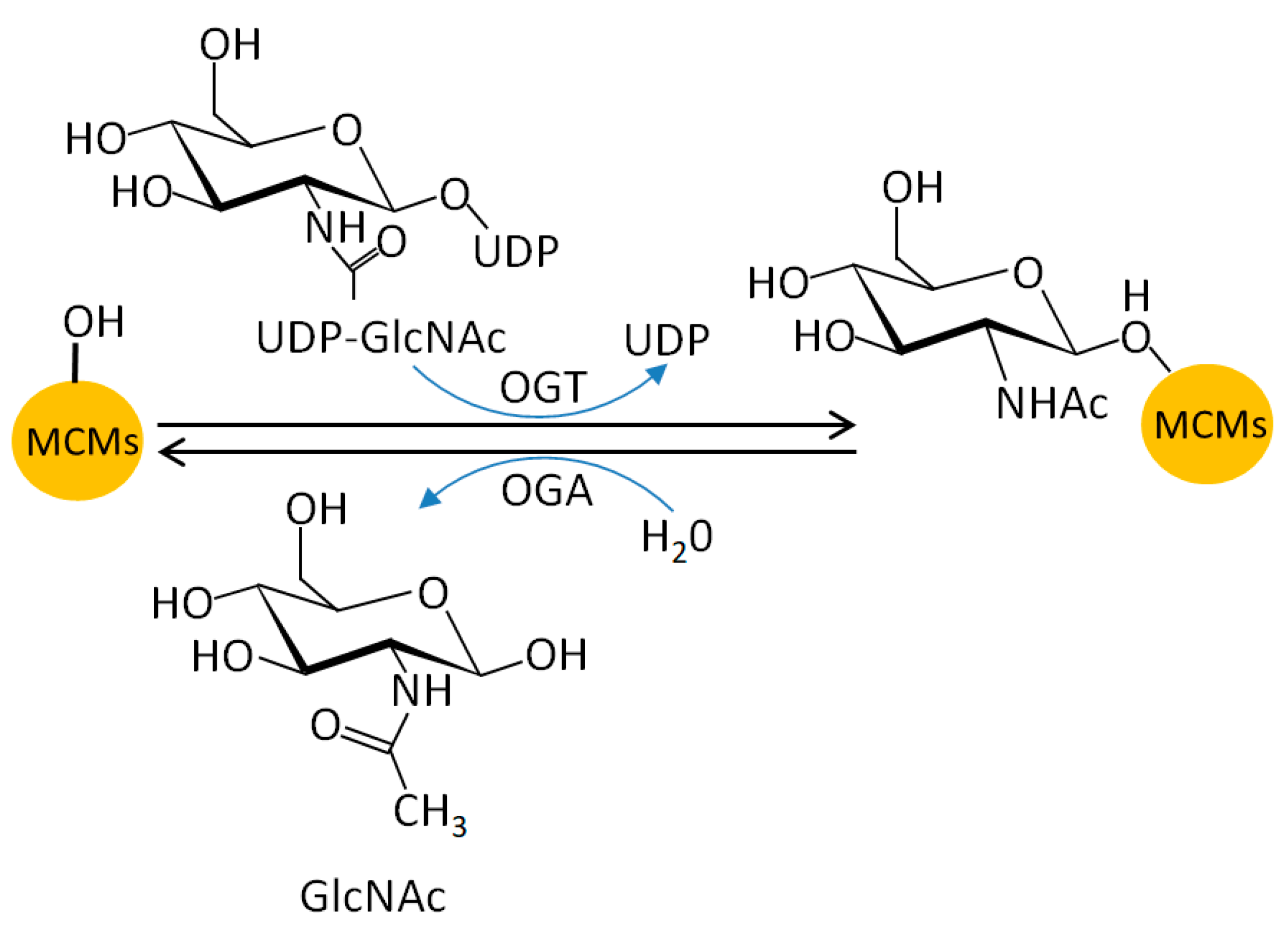
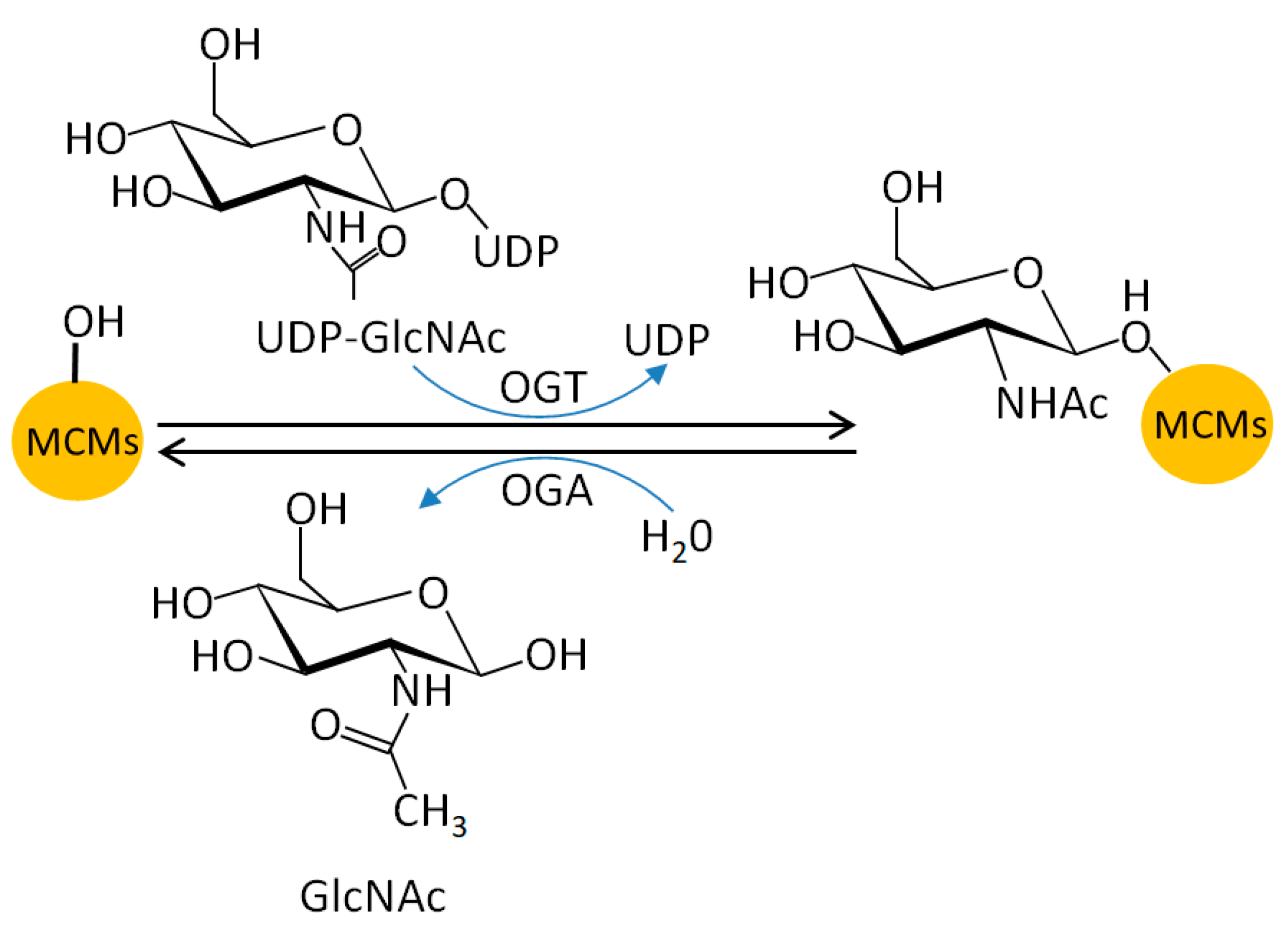
5.5. MCM O-N-acetyl-D-glucosamine (GlcNAc)ylation
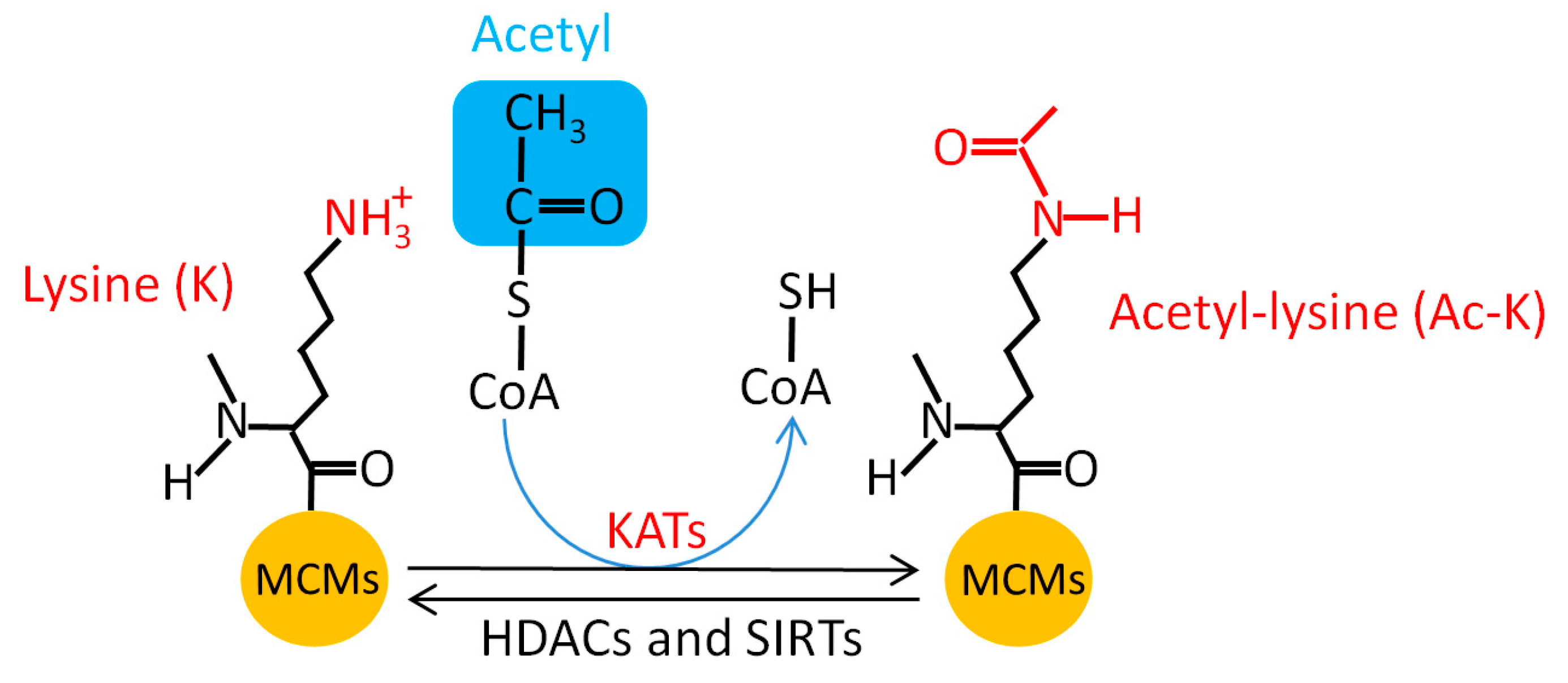
5.6. MCM Acetylation
5.7. Other PTMs of the MCM Proteins
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gaillard, H.; Garcia-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef]
- Masai, H.; You, Z.; Arai, K. Control of DNA replication: Regulation and activation of eukaryotic replicative helicase, MCM. IUBMB Life 2005, 57, 323–335. [Google Scholar] [CrossRef]
- Lujan, S.A.; Williams, J.S.; Kunkel, T.A. DNA Polymerases Divide the Labor of Genome Replication. Trends Cell Biol. 2016, 26, 640–654. [Google Scholar] [CrossRef]
- Forment, J.V.; O’Connor, M.J. Targeting the replication stress response in cancer. Pharmacol. Ther. 2018, 188, 155–167. [Google Scholar] [CrossRef] [PubMed]
- D’Angiolella, V.; Donato, V.; Forrester, F.M.; Jeong, Y.T.; Pellacani, C.; Kudo, Y.; Saraf, A.; Florens, L.; Washburn, M.P.; Pagano, M. Cyclin F-mediated degradation of ribonucleotide reductase M2 controls genome integrity and DNA repair. Cell 2012, 149, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Dehe, P.M.; Gaillard, P.H. Control of structure-specific endonucleases to maintain genome stability. Nat. Rev. Mol. Cell Biol. 2017, 18, 315–330. [Google Scholar] [CrossRef]
- Bailis, J.M.; Forsburg, S.L. MCM proteins: DNA damage, mutagenesis and repair. Curr. Opin. Genet. Dev. 2004, 14, 17–21. [Google Scholar] [CrossRef]
- Seo, Y.S.; Kang, Y.H. The Human Replicative Helicase, the CMG Complex, as a Target for Anti-cancer Therapy. Front. Mol. Biosci. 2018, 5, 26. [Google Scholar] [CrossRef] [PubMed]
- Forsburg, S.L. Eukaryotic MCM proteins: Beyond replication initiation. Microbiol. Mol. Biol. Rev. 2004, 68, 109–131. [Google Scholar] [CrossRef] [PubMed]
- Shore, P.; Sharrocks, A.D. The MADS-box family of transcription factors. Eur. J. Biochem. 1995, 229, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Das, S.P.; Rhind, N. How and why multiple MCMs are loaded at origins of DNA replication. Bioessays 2016, 38, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.D.; Botchan, M.R. The minichromosome maintenance replicative helicase. Cold Spring Harb. Perspect. Biol. 2013, 5, a012807. [Google Scholar] [CrossRef] [PubMed]
- Douglas, M.E.; Ali, F.A.; Costa, A.; Diffley, J.F.X. The mechanism of eukaryotic CMG helicase activation. Nature 2018, 555, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Musialek, M.W.; Rybaczek, D. Behavior of replication origins in Eukaryota—Spatio-temporal dynamics of licensing and firing. Cell Cycle 2015, 14, 2251–2264. [Google Scholar] [CrossRef]
- Ibarra, A.; Schwob, E.; Mendez, J. Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. Proc. Natl. Acad. Sci. USA 2008, 105, 8956–8961. [Google Scholar] [CrossRef]
- Ge, X.Q.; Jackson, D.A.; Blow, J.J. Dormant origins licensed by excess Mcm2-7 are required for human cells to survive replicative stress. Genes Dev. 2007, 21, 3331–3341. [Google Scholar] [CrossRef]
- Volkening, M.; Hoffmann, I. Involvement of human MCM8 in prereplication complex assembly by recruiting hcdc6 to chromatin. Mol. Cell. Biol. 2005, 25, 1560–1568. [Google Scholar] [CrossRef] [PubMed]
- Gambus, A.; Blow, J.J. Mcm8 and Mcm9 form a dimeric complex in Xenopus laevis egg extract that is not essential for DNA replication initiation. Cell Cycle 2013, 12, 1225–1232. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Im, J.S.; Shibata, E.; Park, J.; Handa, N.; Kowalczykowski, S.C.; Dutta, A. MCM8-9 complex promotes resection of double-strand break ends by MRE11-RAD50-NBS1 complex. Nat. Commun. 2015, 6, 7744. [Google Scholar] [CrossRef]
- Nishimura, K.; Ishiai, M.; Horikawa, K.; Fukagawa, T.; Takata, M.; Takisawa, H.; Kanemaki, M.T. Mcm8 and Mcm9 form a complex that functions in homologous recombination repair induced by DNA interstrand crosslinks. Mol. Cell 2012, 47, 511–522. [Google Scholar] [CrossRef]
- Looke, M.; Maloney, M.F.; Bell, S.P. Mcm10 regulates DNA replication elongation by stimulating the CMG replicative helicase. Genes Dev. 2017, 31, 291–305. [Google Scholar] [CrossRef]
- Langston, L.D.; Mayle, R.; Schauer, G.D.; Yurieva, O.; Zhang, D.; Yao, N.Y.; Georgescu, R.E.; O’Donnell, M.E. Mcm10 promotes rapid isomerization of CMG-DNA for replisome bypass of lagging strand DNA blocks. Elife 2017, 6. [Google Scholar] [CrossRef]
- Chattopadhyay, S.; Bielinsky, A.K. Human Mcm10 regulates the catalytic subunit of DNA polymerase-α and prevents DNA damage during replication. Mol. Biol. Cell 2007, 18, 4085–4095. [Google Scholar] [CrossRef]
- Mayle, R.; Langston, L.; Molloy, K.R.; Zhang, D.; Chait, B.T.; O’Donnell, M.E. Mcm10 has potent strand-annealing activity and limits translocase-mediated fork regression. Proc. Natl. Acad. Sci. USA 2019, 116, 798–803. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Weng, C.; Zhang, H.; Sun, J.; Yuan, Y. A Direct Interaction between P53-Binding Protein 1 and Minichromosome Maintenance Complex in Hepg2 Cells. Cell. Physiol. Biochem. 2018, 47, 2350–2359. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Luo, H.L.; Pan, H.; Qiu, C.; Hao, T.F.; Zhu, Z.M. Interaction between RAD51 and MCM Complex Is Essential for RAD51 Foci Forming in Colon Cancer HCT116 Cells. Biochemistry 2018, 83, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wang, Q.; Dong, C.; Chen, S.; Qi, Y.; Liu, Y. Whole Exome Sequencing Identified MCM2 as a Novel Causative Gene for Autosomal Dominant Nonsyndromic Deafness in a Chinese Family. PLoS ONE 2015, 10, e0133522. [Google Scholar] [CrossRef]
- Bayazit, Y.A.; Yilmaz, M. An overview of hereditary hearing loss. ORL J. Otorhinolaryngol. Relat. Spec. 2006, 68, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Casey, J.P.; Nobbs, M.; McGettigan, P.; Lynch, S.; Ennis, S. Recessive mutations in MCM4/PRKDC cause a novel syndrome involving a primary immunodeficiency and a disorder of DNA repair. J. Med. Genet. 2012, 49, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.R.; Guasti, L.; Meimaridou, E.; Chuang, C.H.; Schimenti, J.C.; King, P.J.; Costigan, C.; Clark, A.J.; Metherell, L.A. MCM4 mutation causes adrenal failure, short stature, and natural killer cell deficiency in humans. J. Clin. Investig. 2012, 122, 814–820. [Google Scholar] [CrossRef]
- Gineau, L.; Cognet, C.; Kara, N.; Lach, F.P.; Dunne, J.; Veturi, U.; Picard, C.; Trouillet, C.; Eidenschenk, C.; Aoufouchi, S.; et al. Partial MCM4 deficiency in patients with growth retardation, adrenal insufficiency, and natural killer cell deficiency. J. Clin. Investig. 2012, 122, 821–832. [Google Scholar] [CrossRef]
- Eidenschenk, C.; Dunne, J.; Jouanguy, E.; Fourlinnie, C.; Gineau, L.; Bacq, D.; McMahon, C.; Smith, O.; Casanova, J.L.; Abel, L.; et al. A novel primary immunodeficiency with specific natural-killer cell deficiency maps to the centromeric region of chromosome 8. Am. J. Hum. Genet. 2006, 78, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Shima, N.; Alcaraz, A.; Liachko, I.; Buske, T.R.; Andrews, C.A.; Munroe, R.J.; Hartford, S.A.; Tye, B.K.; Schimenti, J.C. A viable allele of Mcm4 causes chromosome instability and mammary adenocarcinomas in mice. Nat. Genet. 2007, 39, 93–98. [Google Scholar] [CrossRef]
- Shalev, S.A.; Hall, J.G. Another adult with Meier-Gorlin syndrome--insights into the natural history. Clin. Dysmorphol. 2003, 12, 167–169. [Google Scholar] [CrossRef]
- Vetro, A.; Savasta, S.; Russo Raucci, A.; Cerqua, C.; Sartori, G.; Limongelli, I.; Forlino, A.; Maruelli, S.; Perucca, P.; Vergani, D.; et al. MCM5: A new actor in the link between DNA replication and Meier-Gorlin syndrome. Eur. J. Hum. Genet. 2017, 25, 646–650. [Google Scholar] [CrossRef]
- Ryu, S.; Holzschuh, J.; Erhardt, S.; Ettl, A.K.; Driever, W. Depletion of minichromosome maintenance protein 5 in the zebrafish retina causes cell-cycle defect and apoptosis. Proc. Natl. Acad. Sci. USA 2005, 102, 18467–18472. [Google Scholar] [CrossRef]
- Fauchereau, F.; Shalev, S.; Chervinsky, E.; Beck-Fruchter, R.; Legois, B.; Fellous, M.; Caburet, S.; Veitia, R.A. A non-sense MCM9 mutation in a familial case of primary ovarian insufficiency. Clin. Genet. 2016, 89, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Tenenbaum-Rakover, Y.; Weinberg-Shukron, A.; Renbaum, P.; Lobel, O.; Eideh, H.; Gulsuner, S.; Dahary, D.; Abu-Rayyan, A.; Kanaan, M.; Levy-Lahad, E.; et al. Minichromosome maintenance complex component 8 (MCM8) gene mutations result in primary gonadal failure. J. Med. Genet. 2015, 52, 391–399. [Google Scholar] [CrossRef]
- Wood-Trageser, M.A.; Gurbuz, F.; Yatsenko, S.A.; Jeffries, E.P.; Kotan, L.D.; Surti, U.; Ketterer, D.M.; Matic, J.; Chipkin, J.; Jiang, H.; et al. MCM9 mutations are associated with ovarian failure, short stature, and chromosomal instability. Am. J. Hum. Genet. 2014, 95, 754–762. [Google Scholar] [CrossRef] [PubMed]
- AlAsiri, S.; Basit, S.; Wood-Trageser, M.A.; Yatsenko, S.A.; Jeffries, E.P.; Surti, U.; Ketterer, D.M.; Afzal, S.; Ramzan, K.; Faiyaz-Ul Haque, M.; et al. Exome sequencing reveals MCM8 mutation underlies ovarian failure and chromosomal instability. J. Clin. Investig. 2015, 125, 258–262. [Google Scholar] [CrossRef]
- Stolk, L.; Zhai, G.; van Meurs, J.B.; Verbiest, M.M.; Visser, J.A.; Estrada, K.; Rivadeneira, F.; Williams, F.M.; Cherkas, L.; Deloukas, P.; et al. Loci at chromosomes 13, 19 and 20 influence age at natural menopause. Nat. Genet. 2009, 41, 645–647. [Google Scholar] [CrossRef]
- Lutzmann, M.; Grey, C.; Traver, S.; Ganier, O.; Maya-Mendoza, A.; Ranisavljevic, N.; Bernex, F.; Nishiyama, A.; Montel, N.; Gavois, E.; et al. MCM8- and MCM9-deficient mice reveal gametogenesis defects and genome instability due to impaired homologous recombination. Mol. Cell 2012, 47, 523–534. [Google Scholar] [CrossRef]
- Wojnar, A.; Kobierzycki, C.; Krolicka, A.; Pula, B.; Podhorska-Okolow, M.; Dziegiel, P. Correlation of Ki-67 and MCM-2 proliferative marker expression with grade of histological malignancy (G) in ductal breast cancers. Folia Histochem. Cytobiol. 2010, 48, 442–446. [Google Scholar] [CrossRef]
- Burger, M. MCM2 and MCM5 as prognostic markers in colon cancer: A worthwhile approach. Dig. Dis. Sci. 2009, 54, 197–198. [Google Scholar] [CrossRef]
- Ren, B.; Yu, G.; Tseng, G.C.; Cieply, K.; Gavel, T.; Nelson, J.; Michalopoulos, G.; Yu, Y.P.; Luo, J.H. MCM7 amplification and overexpression are associated with prostate cancer progression. Oncogene 2006, 25, 1090–1098. [Google Scholar] [CrossRef]
- Neves, H.; Kwok, H.F. In sickness and in health: The many roles of the minichromosome maintenance proteins. Biochim. Biophys. Acta 2017, 1868, 295–308. [Google Scholar] [CrossRef]
- Simon, N.; Bochman, M.L.; Seguin, S.; Brodsky, J.L.; Seibel, W.L.; Schwacha, A. Ciprofloxacin is an inhibitor of the Mcm2-7 replicative helicase. Biosci. Rep. 2013, 33. [Google Scholar] [CrossRef]
- Toyokawa, G.; Masuda, K.; Daigo, Y.; Cho, H.S.; Yoshimatsu, M.; Takawa, M.; Hayami, S.; Maejima, K.; Chino, M.; Field, H.I.; et al. Minichromosome Maintenance Protein 7 is a potential therapeutic target in human cancer and a novel prognostic marker of non-small cell lung cancer. Mol. Cancer 2011, 10, 65. [Google Scholar] [CrossRef]
- Ishimi, Y.; Sugiyama, T.; Nakaya, R.; Kanamori, M.; Kohno, T.; Enomoto, T.; Chino, M. Effect of heliquinomycin on the activity of human minichromosome maintenance 4/6/7 helicase. FEBS J. 2009, 276, 3382–3391. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, S.C.; Ku, J.L. Metformin increases chemo-sensitivity via gene downregulation encoding DNA replication proteins in 5-Fu resistant colorectal cancer cells. Oncotarget 2017, 8, 56546–56557. [Google Scholar] [CrossRef]
- Guan, Y.B.; Yang, D.R.; Nong, S.J.; Ni, J.; Hu, C.H.; Li, J.; Zhu, J.; Shan, Y.X. Breviscapine (BVP) inhibits prostate cancer progression through damaging DNA by minichromosome maintenance protein-7 (MCM-7) modulation. Biomed. Pharmacother. 2017, 93, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Mio, C.; Lavarone, E.; Conzatti, K.; Baldan, F.; Toffoletto, B.; Puppin, C.; Filetti, S.; Durante, C.; Russo, D.; Orlacchio, A.; et al. MCM5 as a target of BET inhibitors in thyroid cancer cells. Endocr. Relat. Cancer 2016, 23, 335–347. [Google Scholar] [CrossRef]
- Zhang, X.; Teng, Y.; Yang, F.; Wang, M.; Hong, X.; Ye, L.G.; Gao, Y.N.; Chen, G.Y. MCM2 is a therapeutic target of lovastatin in human non-small cell lung carcinomas. Oncol. Rep. 2015, 33, 2599–2605. [Google Scholar] [CrossRef]
- Majid, S.; Dar, A.A.; Saini, S.; Chen, Y.; Shahryari, V.; Liu, J.; Zaman, M.S.; Hirata, H.; Yamamura, S.; Ueno, K.; et al. Regulation of minichromosome maintenance gene family by microRNA-1296 and genistein in prostate cancer. Cancer Res. 2010, 70, 2809–2818. [Google Scholar] [CrossRef]
- Kwon, H.J.; Hong, Y.K.; Park, C.; Choi, Y.H.; Yun, H.J.; Lee, E.W.; Kim, B.W. Widdrol induces cell cycle arrest, associated with MCM down-regulation, in human colon adenocarcinoma cells. Cancer Lett 2010, 290, 96–103. [Google Scholar] [CrossRef]
- Simon, N.E.; Schwacha, A. The Mcm2-7 replicative helicase: A promising chemotherapeutic target. Biomed. Res. Int. 2014, 2014, 549719. [Google Scholar] [CrossRef]
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef] [PubMed]
- Chi, Y.; Welcker, M.; Hizli, A.A.; Posakony, J.J.; Aebersold, R.; Clurman, B.E. Identification of CDK2 substrates in human cell lysates. Genome Biol. 2008, 9, R149. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.I.; Aggarwal, P.; Diehl, J.A. Phosphorylation of MCM3 on Ser-112 regulates its incorporation into the MCM2-7 complex. Proc. Natl. Acad. Sci. USA 2008, 105, 8079–8084. [Google Scholar] [CrossRef]
- Li, J.; Deng, M.; Wei, Q.; Liu, T.; Tong, X.; Ye, X. Phosphorylation of MCM3 protein by cyclin E/cyclin-dependent kinase 2 (Cdk2) regulates its function in cell cycle. J. Biol. Chem. 2011, 286, 39776–39785. [Google Scholar] [CrossRef]
- Schumann, M.; Malesevic, M.; Hinze, E.; Mathea, S.; Meleshin, M.; Schutkowski, M.; Haehnel, W.; Schiene-Fischer, C. Regulation of the Minichromosome Maintenance Protein 3 (MCM3) Chromatin Binding by the Prolyl Isomerase Pin1. J. Mol. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Li, J.; Liu, T.; Tong, X.; Ye, X. Phosphorylation of minichromosome maintenance protein 7 (MCM7) by cyclin/cyclin-dependent kinase affects its function in cell cycle regulation. J. Biol. Chem. 2013, 288, 19715–19725. [Google Scholar] [CrossRef] [PubMed]
- Chuang, L.C.; Teixeira, L.K.; Wohlschlegel, J.A.; Henze, M.; Yates, J.R.; Mendez, J.; Reed, S.I. Phosphorylation of Mcm2 by Cdc7 promotes pre-replication complex assembly during cell-cycle re-entry. Mol. Cell 2009, 35, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Komamura-Kohno, Y.; Karasawa-Shimizu, K.; Saitoh, T.; Sato, M.; Hanaoka, F.; Tanaka, S.; Ishimi, Y. Site-specific phosphorylation of MCM4 during the cell cycle in mammalian cells. FEBS J. 2006, 273, 1224–1239. [Google Scholar] [CrossRef]
- Moritani, M.; Ishimi, Y. Inhibition of DNA binding of MCM2-7 complex by phosphorylation with cyclin-dependent kinases. J. Biochem. 2013, 154, 363–372. [Google Scholar] [CrossRef]
- Masai, H.; Arai, K. Cdc7 kinase complex: A key regulator in the initiation of DNA replication. J. Cell. Physiol. 2002, 190, 287–296. [Google Scholar] [CrossRef]
- Jares, P.; Donaldson, A.; Blow, J.J. The Cdc7/Dbf4 protein kinase: Target of the S phase checkpoint? EMBO Rep. 2000, 1, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, T.; Ficarro, S.B.; Jiang, W. Essential role of phosphorylation of MCM2 by Cdc7/Dbf4 in the initiation of DNA replication in mammalian cells. Mol. Biol. Cell 2006, 17, 4459–4472. [Google Scholar] [CrossRef]
- Montagnoli, A.; Valsasina, B.; Brotherton, D.; Troiani, S.; Rainoldi, S.; Tenca, P.; Molinari, A.; Santocanale, C. Identification of Mcm2 phosphorylation sites by S-phase-regulating kinases. J. Biol. Chem. 2006, 281, 10281–10290. [Google Scholar] [CrossRef]
- Charych, D.H.; Coyne, M.; Yabannavar, A.; Narberes, J.; Chow, S.; Wallroth, M.; Shafer, C.; Walter, A.O. Inhibition of Cdc7/Dbf4 kinase activity affects specific phosphorylation sites on MCM2 in cancer cells. J. Cell. Biochem. 2008, 104, 1075–1086. [Google Scholar] [CrossRef] [PubMed]
- Cortez, D.; Glick, G.; Elledge, S.J. Minichromosome maintenance proteins are direct targets of the ATM and ATR checkpoint kinases. Proc. Natl. Acad. Sci. USA 2004, 101, 10078–10083. [Google Scholar] [CrossRef]
- Cho, W.H.; Lee, Y.J.; Kong, S.I.; Hurwitz, J.; Lee, J.K. CDC7 kinase phosphorylates serine residues adjacent to acidic amino acids in the minichromosome maintenance 2 protein. Proc. Natl. Acad. Sci. USA 2006, 103, 11521–11526. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef]
- Han, X.; Mayca Pozo, F.; Wisotsky, J.N.; Wang, B.; Jacobberger, J.W.; Zhang, Y. Phosphorylation of Minichromosome Maintenance 3 (MCM3) by Checkpoint Kinase 1 (Chk1) Negatively Regulates DNA Replication and Checkpoint Activation. J. Biol. Chem. 2015, 290, 12370–12378. [Google Scholar] [CrossRef] [PubMed]
- Blasius, M.; Forment, J.V.; Thakkar, N.; Wagner, S.A.; Choudhary, C.; Jackson, S.P. A phospho-proteomic screen identifies substrates of the checkpoint kinase Chk1. Genome Biol. 2011, 12, R78. [Google Scholar] [CrossRef] [PubMed]
- Litchfield, D.W. Protein kinase CK2: Structure, regulation and role in cellular decisions of life and death. Biochem. J. 2003, 369, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Bialik, S.; Berissi, H.; Kimchi, A. A high throughput proteomics screen identifies novel substrates of death-associated protein kinase. Mol. Cell. Proteom. 2008, 7, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Drissi, R.; Dubois, M.L.; Douziech, M.; Boisvert, F.M. Quantitative Proteomics Reveals Dynamic Interactions of the Minichromosome Maintenance Complex (MCM) in the Cellular Response to Etoposide Induced DNA Damage. Mol. Cell. Proteom. 2015, 14, 2002–2013. [Google Scholar] [CrossRef]
- FitzGerald, J.; Murillo, L.S.; O’Brien, G.; O’Connell, E.; O’Connor, A.; Wu, K.; Wang, G.N.; Rainey, M.D.; Natoni, A.; Healy, S.; et al. A high through-put screen for small molecules modulating MCM2 phosphorylation identifies Ryuvidine as an inducer of the DNA damage response. PLoS ONE 2014, 9, e98891. [Google Scholar] [CrossRef]
- Perrera, C.; Colombo, R.; Valsasina, B.; Carpinelli, P.; Troiani, S.; Modugno, M.; Gianellini, L.; Cappella, P.; Isacchi, A.; Moll, J.; et al. Identification of Myb-binding protein 1A (MYBBP1A) as a novel substrate for aurora B kinase. J. Biol. Chem. 2010, 285, 11775–11785. [Google Scholar] [CrossRef]
- Zhou, H.; Di Palma, S.; Preisinger, C.; Peng, M.; Polat, A.N.; Heck, A.J.; Mohammed, S. Toward a comprehensive characterization of a human cancer cell phosphoproteome. J. Proteome Res. 2013, 12, 260–271. [Google Scholar] [CrossRef]
- Olsen, J.V.; Vermeulen, M.; Santamaria, A.; Kumar, C.; Miller, M.L.; Jensen, L.J.; Gnad, F.; Cox, J.; Jensen, T.S.; Nigg, E.A.; et al. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 2010, 3, ra3. [Google Scholar] [CrossRef]
- Mayya, V.; Lundgren, D.H.; Hwang, S.I.; Rezaul, K.; Wu, L.; Eng, J.K.; Rodionov, V.; Han, D.K. Quantitative phosphoproteomic analysis of T cell receptor signaling reveals system-wide modulation of protein-protein interactions. Sci. Signal. 2009, 2, ra46. [Google Scholar] [CrossRef]
- Daub, H.; Olsen, J.V.; Bairlein, M.; Gnad, F.; Oppermann, F.S.; Korner, R.; Greff, Z.; Keri, G.; Stemmann, O.; Mann, M. Kinase-selective enrichment enables quantitative phosphoproteomics of the kinome across the cell cycle. Mol. Cell 2008, 31, 438–448. [Google Scholar] [CrossRef]
- Dephoure, N.; Zhou, C.; Villen, J.; Beausoleil, S.A.; Bakalarski, C.E.; Elledge, S.J.; Gygi, S.P. A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. USA 2008, 105, 10762–10767. [Google Scholar] [CrossRef]
- Shi, Y.; Dodson, G.E.; Mukhopadhyay, P.S.; Shanware, N.P.; Trinh, A.T.; Tibbetts, R.S. Identification of carboxyl-terminal MCM3 phosphorylation sites using polyreactive phosphospecific antibodies. J. Biol. Chem. 2007, 282, 9236–9243. [Google Scholar] [CrossRef]
- Rigbolt, K.T.; Prokhorova, T.A.; Akimov, V.; Henningsen, J.; Johansen, P.T.; Kratchmarova, I.; Kassem, M.; Mann, M.; Olsen, J.V.; Blagoev, B. System-wide temporal characterization of the proteome and phosphoproteome of human embryonic stem cell differentiation. Sci. Signal. 2011, 4, rs3. [Google Scholar] [CrossRef]
- Olsen, J.V.; Blagoev, B.; Gnad, F.; Macek, B.; Kumar, C.; Mortensen, P.; Mann, M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 2006, 127, 635–648. [Google Scholar] [CrossRef]
- Beausoleil, S.A.; Villen, J.; Gerber, S.A.; Rush, J.; Gygi, S.P. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 2006, 24, 1285–1292. [Google Scholar] [CrossRef]
- Yu, L.R.; Zhu, Z.; Chan, K.C.; Issaq, H.J.; Dimitrov, D.S.; Veenstra, T.D. Improved titanium dioxide enrichment of phosphopeptides from HeLa cells and high confident phosphopeptide identification by cross-validation of MS/MS and MS/MS/MS spectra. J. Proteome Res. 2007, 6, 4150–4162. [Google Scholar] [CrossRef]
- Kudoh, A.; Daikoku, T.; Ishimi, Y.; Kawaguchi, Y.; Shirata, N.; Iwahori, S.; Isomura, H.; Tsurumi, T. Phosphorylation of MCM4 at sites inactivating DNA helicase activity of the MCM4-MCM6-MCM7 complex during Epstein-Barr virus productive replication. J. Virol. 2006, 80, 10064–10072. [Google Scholar] [CrossRef] [PubMed]
- Ishimi, Y.; Komamura-Kohno, Y.; Kwon, H.J.; Yamada, K.; Nakanishi, M. Identification of MCM4 as a target of the DNA replication block checkpoint system. J. Biol. Chem. 2003, 278, 24644–24650. [Google Scholar] [CrossRef] [PubMed]
- Ishimi, Y.; Komamura-Kohno, Y. Phosphorylation of Mcm4 at specific sites by cyclin-dependent kinase leads to loss of Mcm4,6,7 helicase activity. J. Biol. Chem. 2001, 276, 34428–34433. [Google Scholar] [CrossRef]
- Pan, C.; Olsen, J.V.; Daub, H.; Mann, M. Global effects of kinase inhibitors on signaling networks revealed by quantitative phosphoproteomics. Mol. Cell. Proteom. 2009, 8, 2796–2808. [Google Scholar] [CrossRef]
- Raijmakers, R.; Kraiczek, K.; de Jong, A.P.; Mohammed, S.; Heck, A.J. Exploring the human leukocyte phosphoproteome using a microfluidic reversed-phase-TiO2-reversed-phase high-performance liquid chromatography phosphochip coupled to a quadrupole time-of-flight mass spectrometer. Anal. Chem. 2010, 82, 824–832. [Google Scholar] [CrossRef] [PubMed]
- Gauci, S.; Helbig, A.O.; Slijper, M.; Krijgsveld, J.; Heck, A.J.; Mohammed, S. Lys-N and trypsin cover complementary parts of the phosphoproteome in a refined SCX-based approach. Anal. Chem. 2009, 81, 4493–4501. [Google Scholar] [CrossRef]
- Ruse, C.I.; McClatchy, D.B.; Lu, B.; Cociorva, D.; Motoyama, A.; Park, S.K.; Yates, J.R., 3rd. Motif-specific sampling of phosphoproteomes. J. Proteome Res. 2008, 7, 2140–2150. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, N.; Masuda, T.; Shinoda, K.; Nakamura, A.; Tomita, M.; Ishihama, Y. Phosphopeptide enrichment by aliphatic hydroxy acid-modified metal oxide chromatography for nano-LC-MS/MS in proteomics applications. Mol. Cell. Proteom. 2007, 6, 1103–1109. [Google Scholar] [CrossRef]
- Huang, T.H.; Huo, L.; Wang, Y.N.; Xia, W.; Wei, Y.; Chang, S.S.; Chang, W.C.; Fang, Y.F.; Chen, C.T.; Lang, J.Y.; et al. Epidermal growth factor receptor potentiates MCM7-mediated DNA replication through tyrosine phosphorylation of Lyn kinase in human cancers. Cancer Cell 2013, 23, 796–810. [Google Scholar] [CrossRef]
- Campbell, S.J.; Edwards, R.A.; Leung, C.C.; Neculai, D.; Hodge, C.D.; Dhe-Paganon, S.; Glover, J.N. Molecular insights into the function of RING finger (RNF)-containing proteins hRNF8 and hRNF168 in Ubc13/Mms2-dependent ubiquitylation. J. Biol. Chem. 2012, 287, 23900–23910. [Google Scholar] [CrossRef]
- Yau, R.G.; Doerner, K.; Castellanos, E.R.; Haakonsen, D.L.; Werner, A.; Wang, N.; Yang, X.W.; Martinez-Martin, N.; Matsumoto, M.L.; Dixit, V.M.; et al. Assembly and Function of Heterotypic Ubiquitin Chains in Cell-Cycle and Protein Quality Control. Cell 2017, 171, 918–933 e920. [Google Scholar] [CrossRef]
- Guervilly, J.H.; Takedachi, A.; Naim, V.; Scaglione, S.; Chawhan, C.; Lovera, Y.; Despras, E.; Kuraoka, I.; Kannouche, P.; Rosselli, F.; et al. The SLX4 complex is a SUMO E3 ligase that impacts on replication stress outcome and genome stability. Mol. Cell 2015, 57, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Pickart, C.M.; Eddins, M.J. Ubiquitin: Structures, functions, mechanisms. Biochim. Biophys. Acta 2004, 1695, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Udeshi, N.D.; Svinkina, T.; Mertins, P.; Kuhn, E.; Mani, D.R.; Qiao, J.W.; Carr, S.A. Refined preparation and use of anti-diglycine remnant (K-epsilon-GG) antibody enables routine quantification of 10,000s of ubiquitination sites in single proteomics experiments. Mol. Cell. Proteom. 2013, 12, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Bennett, E.J.; Huttlin, E.L.; Guo, A.; Li, J.; Possemato, A.; Sowa, M.E.; Rad, R.; Rush, J.; Comb, M.J.; et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 2011, 44, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.A.; Beli, P.; Weinert, B.T.; Nielsen, M.L.; Cox, J.; Mann, M.; Choudhary, C. A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol. Cell. Proteom. 2011, 10, M111 013284. [Google Scholar] [CrossRef] [PubMed]
- Mulvaney, K.M.; Matson, J.P.; Siesser, P.F.; Tamir, T.Y.; Goldfarb, D.; Jacobs, T.M.; Cloer, E.W.; Harrison, J.S.; Vaziri, C.; Cook, J.G.; et al. Identification and Characterization of MCM3 as a Kelch-like ECH-associated Protein 1 (KEAP1) Substrate. J. Biol. Chem. 2016, 291, 23719–23733. [Google Scholar] [CrossRef] [PubMed]
- Moreno, S.P.; Bailey, R.; Campion, N.; Herron, S.; Gambus, A. Polyubiquitylation drives replisome disassembly at the termination of DNA replication. Science 2014, 346, 477–481. [Google Scholar] [CrossRef]
- Maric, M.; Maculins, T.; De Piccoli, G.; Labib, K. Cdc48 and a ubiquitin ligase drive disassembly of the CMG helicase at the end of DNA replication. Science 2014, 346, 1253596. [Google Scholar] [CrossRef]
- Bailey, R.; Priego Moreno, S.; Gambus, A. Termination of DNA replication forks: “Breaking up is hard to do”. Nucleus 2015, 6, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Maculins, T.; Nkosi, P.J.; Nishikawa, H.; Labib, K. Tethering of SCF(Dia2) to the Replisome Promotes Efficient Ubiquitylation and Disassembly of the CMG Helicase. Curr. Biol. 2015, 25, 2254–2259. [Google Scholar] [CrossRef]
- Franz, A.; Orth, M.; Pirson, P.A.; Sonneville, R.; Blow, J.J.; Gartner, A.; Stemmann, O.; Hoppe, T. CDC-48/p97 coordinates CDT-1 degradation with GINS chromatin dissociation to ensure faithful DNA replication. Mol. Cell 2011, 44, 85–96. [Google Scholar] [CrossRef]
- Dewar, J.M.; Low, E.; Mann, M.; Raschle, M.; Walter, J.C. CRL2(Lrr1) promotes unloading of the vertebrate replisome from chromatin during replication termination. Genes Dev. 2017, 31, 275–290. [Google Scholar] [CrossRef]
- Wu, R.A.; Semlow, D.R.; Kamimae-Lanning, A.N.; Kochenova, O.V.; Chistol, G.; Hodskinson, M.R.; Amunugama, R.; Sparks, J.L.; Wang, M.; Deng, L.; et al. TRAIP is a master regulator of DNA interstrand crosslink repair. Nature 2019, 567, 267–272. [Google Scholar] [CrossRef]
- Kaur, M.; Khan, M.M.; Kar, A.; Sharma, A.; Saxena, S. CRL4-DDB1-VPRBP ubiquitin ligase mediates the stress triggered proteolysis of Mcm10. Nucleic Acids Res. 2012, 40, 7332–7346. [Google Scholar] [CrossRef]
- Sharma, A.; Kaur, M.; Kar, A.; Ranade, S.M.; Saxena, S. Ultraviolet radiation stress triggers the down-regulation of essential replication factor Mcm10. J. Biol. Chem. 2010, 285, 8352–8362. [Google Scholar] [CrossRef]
- Lien, W.C.; Chen, T.Y.; Sheu, S.Y.; Lin, T.C.; Kang, F.C.; Yu, C.H.; Kuan, T.S.; Huang, B.M.; Wang, C.Y. 7-hydroxy-staurosporine, UCN-01, induces DNA damage response, and autophagy in human osteosarcoma U2-OS cells. J. Cell. Biochem. 2018, 119, 4729–4741. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Chiba, T.; Tatsumi, K.; Iemura, S.; Tanida, I.; Okazaki, N.; Ueno, T.; Kominami, E.; Natsume, T.; Tanaka, K. A novel protein-conjugating system for Ufm1, a ubiquitin-fold modifier. EMBO J. 2004, 23, 1977–1986. [Google Scholar] [CrossRef]
- Wang, Z.; Zhu, W.G.; Xu, X. Ubiquitin-like modifications in the DNA damage response. Mutat. Res. 2017, 803-805, 56–75. [Google Scholar] [CrossRef]
- Wei, Y.; Xu, X. UFMylation: A Unique & Fashionable Modification for Life. Genom. Proteom. Bioinform. 2016, 14, 140–146. [Google Scholar] [CrossRef]
- Li, J.; Yue, G.; Ma, W.; Zhang, A.; Zou, J.; Cai, Y.; Tang, X.; Wang, J.; Liu, J.; Li, H.; et al. Ufm1-Specific Ligase Ufl1 Regulates Endoplasmic Reticulum Homeostasis and Protects Against Heart Failure. Circ. Heart Fail. 2018, 11, e004917. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, Y.; Song, L.; Zeng, L.; Yi, W.; Liu, T.; Chen, H.; Wang, M.; Ju, Z.; Cong, Y.S. A critical role of DDRGK1 in endoplasmic reticulum homoeostasis via regulation of IRE1alpha stability. Nat. Commun. 2017, 8, 14186. [Google Scholar] [CrossRef]
- Zhang, M.; Zhu, X.; Zhang, Y.; Cai, Y.; Chen, J.; Sivaprakasam, S.; Gurav, A.; Pi, W.; Makala, L.; Wu, J.; et al. RCAD/Ufl1, a Ufm1 E3 ligase, is essential for hematopoietic stem cell function and murine hematopoiesis. Cell Death Differ. 2015, 22, 1922–1934. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, M.; Wu, J.; Lei, G.; Li, H. Transcriptional regulation of the Ufm1 conjugation system in response to disturbance of the endoplasmic reticulum homeostasis and inhibition of vesicle trafficking. PLoS ONE 2012, 7, e48587. [Google Scholar] [CrossRef]
- Yang, R.; Wang, H.; Kang, B.; Chen, B.; Shi, Y.; Yang, S.; Sun, L.; Liu, Y.; Xiao, W.; Zhang, T.; et al. CDK5RAP3, a UFL1 substrate adaptor, is crucial for liver development. Development 2019, 146. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Itakura, E.; Weber, K.P.; Hegde, R.S.; de Bono, M. An ER complex of ODR-4 and ODR-8/Ufm1 specific protease 2 promotes GPCR maturation by a Ufm1-independent mechanism. PLoS Genet. 2014, 10, e1004082. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Pi, W.; Sivaprakasam, S.; Zhu, X.; Zhang, M.; Chen, J.; Makala, L.; Lu, C.; Wu, J.; Teng, Y.; et al. UFBP1, a Key Component of the Ufm1 Conjugation System, Is Essential for Ufmylation-Mediated Regulation of Erythroid Development. PLoS Genet. 2015, 11, e1005643. [Google Scholar] [CrossRef]
- Yoo, H.M.; Kang, S.H.; Kim, J.Y.; Lee, J.E.; Seong, M.W.; Lee, S.W.; Ka, S.H.; Sou, Y.S.; Komatsu, M.; Tanaka, K.; et al. Modification of ASC1 by UFM1 is crucial for ERalpha transactivation and breast cancer development. Mol. Cell 2014, 56, 261–274. [Google Scholar] [CrossRef]
- Merbl, Y.; Refour, P.; Patel, H.; Springer, M.; Kirschner, M.W. Profiling of ubiquitin-like modifications reveals features of mitotic control. Cell 2013, 152, 1160–1172. [Google Scholar] [CrossRef] [PubMed]
- DeJesus, R.; Moretti, F.; McAllister, G.; Wang, Z.; Bergman, P.; Liu, S.; Frias, E.; Alford, J.; Reece-Hoyes, J.S.; Lindeman, A.; et al. Functional CRISPR screening identifies the ufmylation pathway as a regulator of SQSTM1/p62. Elife 2016, 5. [Google Scholar] [CrossRef]
- Wang, Z.; Gong, Y.; Peng, B.; Shi, R.; Fan, D.; Zhao, H.; Zhu, M.; Zhang, H.; Lou, Z.; Zhou, J.; et al. MRE11 UFMylation promotes ATM activation. Nucleic Acids Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Qin, B.; Yu, J.; Nowsheen, S.; Wang, M.; Tu, X.; Liu, T.; Li, H.; Wang, L.; Lou, Z. UFL1 promotes histone H4 ufmylation and ATM activation. Nat. Commun. 2019, 10, 1242. [Google Scholar] [CrossRef] [PubMed]
- Pirone, L.; Xolalpa, W.; Sigurethsson, J.O.; Ramirez, J.; Perez, C.; Gonzalez, M.; de Sabando, A.R.; Elortza, F.; Rodriguez, M.S.; Mayor, U.; et al. A comprehensive platform for the analysis of ubiquitin-like protein modifications using in vivo biotinylation. Sci. Rep. 2017, 7, 40756. [Google Scholar] [CrossRef] [PubMed]
- Hay, R.T. SUMO: A history of modification. Mol. Cell 2005, 18, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Seeler, J.S.; Dejean, A. SUMO and the robustness of cancer. Nat. Rev. Cancer 2017, 17, 184–197. [Google Scholar] [CrossRef]
- Cremona, C.A.; Sarangi, P.; Yang, Y.; Hang, L.E.; Rahman, S.; Zhao, X. Extensive DNA damage-induced sumoylation contributes to replication and repair and acts in addition to the Mec1 checkpoint. Mol. Cell 2012, 45, 422–432. [Google Scholar] [CrossRef]
- Elrouby, N.; Coupland, G. Proteome-wide screens for small ubiquitin-like modifier (SUMO) substrates identify Arabidopsis proteins implicated in diverse biological processes. Proc. Natl. Acad. Sci. USA 2010, 107, 17415–17420. [Google Scholar] [CrossRef]
- Golebiowski, F.; Matic, I.; Tatham, M.H.; Cole, C.; Yin, Y.; Nakamura, A.; Cox, J.; Barton, G.J.; Mann, M.; Hay, R.T. System-wide changes to SUMO modifications in response to heat shock. Sci. Signal. 2009, 2, ra24. [Google Scholar] [CrossRef]
- de Albuquerque, C.P.; Liang, J.; Gaut, N.J.; Zhou, H. Molecular Circuitry of the SUMO (Small Ubiquitin-like Modifier) Pathway in Controlling Sumoylation Homeostasis and Suppressing Genome Rearrangements. J. Biol. Chem. 2016, 291, 8825–8835. [Google Scholar] [CrossRef]
- Wei, L.; Zhao, X. A new MCM modification cycle regulates DNA replication initiation. Nat. Struct. Mol. Biol. 2016, 23, 209–216. [Google Scholar] [CrossRef]
- Hendriks, I.A.; D’Souza, R.C.; Yang, B.; Verlaan-de Vries, M.; Mann, M.; Vertegaal, A.C. Uncovering global SUMOylation signaling networks in a site-specific manner. Nat. Struct. Mol. Biol. 2014, 21, 927–936. [Google Scholar] [CrossRef]
- Wang, Z.; Udeshi, N.D.; Slawson, C.; Compton, P.D.; Sakabe, K.; Cheung, W.D.; Shabanowitz, J.; Hunt, D.F.; Hart, G.W. Extensive crosstalk between O-GlcNAcylation and phosphorylation regulates cytokinesis. Sci. Signal. 2010, 3, ra2. [Google Scholar] [CrossRef]
- Caldwell, S.A.; Jackson, S.R.; Shahriari, K.S.; Lynch, T.P.; Sethi, G.; Walker, S.; Vosseller, K.; Reginato, M.J. Nutrient sensor O-GlcNAc transferase regulates breast cancer tumorigenesis through targeting of the oncogenic transcription factor FoxM1. Oncogene 2010, 29, 2831–2842. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.R.; Song, M.; Lee, H.; Jeon, Y.; Choi, E.J.; Jang, H.J.; Moon, H.Y.; Byun, H.Y.; Kim, E.K.; Kim, D.H.; et al. O-GlcNAcase is essential for embryonic development and maintenance of genomic stability. Aging Cell 2012, 11, 439–448. [Google Scholar] [CrossRef]
- Slawson, C.; Zachara, N.E.; Vosseller, K.; Cheung, W.D.; Lane, M.D.; Hart, G.W. Perturbations in O-linked β-N-acetylglucosamine protein modification cause severe defects in mitotic progression and cytokinesis. J. Biol. Chem. 2005, 280, 32944–32956. [Google Scholar] [CrossRef]
- Nandi, A.; Sprung, R.; Barma, D.K.; Zhao, Y.; Kim, S.C.; Falck, J.R.; Zhao, Y. Global identification of O-GlcNAc-modified proteins. Anal. Chem. 2006, 78, 452–458. [Google Scholar] [CrossRef]
- Drougat, L.; Olivier-Van Stichelen, S.; Mortuaire, M.; Foulquier, F.; Lacoste, A.S.; Michalski, J.C.; Lefebvre, T.; Vercoutter-Edouart, A.S. Characterization of O-GlcNAc cycling and proteomic identification of differentially O-GlcNAcylated proteins during G1/S transition. Biochim. Biophys. Acta 2012, 1820, 1839–1848. [Google Scholar] [CrossRef] [PubMed]
- Leturcq, M.; Mortuaire, M.; Hardiville, S.; Schulz, C.; Lefebvre, T.; Vercoutter-Edouart, A.S. O-GlcNAc transferase associates with the MCM2-7 complex and its silencing destabilizes MCM-MCM interactions. Cell. Mol. Life Sci. 2018, 75, 4321–4339. [Google Scholar] [CrossRef]
- Gupta, R.; Brunak, S. Prediction of glycosylation across the human proteome and the correlation to protein function. Pac. Symp. Biocomput. 2002, 310–322. [Google Scholar]
- Chen, Q.; Yu, X. OGT restrains the expansion of DNA damage signaling. Nucleic Acids Res. 2016, 44, 9266–9278. [Google Scholar] [CrossRef]
- Narita, T.; Weinert, B.T.; Choudhary, C. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 156–174. [Google Scholar] [CrossRef]
- Bienvenut, W.V.; Sumpton, D.; Martinez, A.; Lilla, S.; Espagne, C.; Meinnel, T.; Giglione, C. Comparative large scale characterization of plant versus mammal proteins reveals similar and idiosyncratic N-α-acetylation features. Mol. Cell. Proteom. 2012, 11, M111 015131. [Google Scholar] [CrossRef]
- Van Damme, P.; Lasa, M.; Polevoda, B.; Gazquez, C.; Elosegui-Artola, A.; Kim, D.S.; De Juan-Pardo, E.; Demeyer, K.; Hole, K.; Larrea, E.; et al. N-terminal acetylome analyses and functional insights of the N-terminal acetyltransferase NatB. Proc. Natl. Acad. Sci. USA 2012, 109, 12449–12454. [Google Scholar] [CrossRef]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef]
- Takei, Y.; Swietlik, M.; Tanoue, A.; Tsujimoto, G.; Kouzarides, T.; Laskey, R. MCM3AP, a novel acetyltransferase that acetylates replication protein MCM3. EMBO Rep. 2001, 2, 119–123. [Google Scholar] [CrossRef]
- Fatoba, S.T.; Tognetti, S.; Berto, M.; Leo, E.; Mulvey, C.M.; Godovac-Zimmermann, J.; Pommier, Y.; Okorokov, A.L. Human SIRT1 regulates DNA binding and stability of the Mcm10 DNA replication factor via deacetylation. Nucleic Acids Res. 2013, 41, 4065–4079. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MCMs | Disease | Pathogenic Mutation | Reference |
|---|---|---|---|
| MCM2 | Non-syndromic sensorineural hearing loss | Arg44 to Cys (R44C) | [27] |
| MCM4 | Natural killer cell and glucocorticoid deficiency with DNA repair defect | Loss of the N-terminal sequence | [31,32] |
| MCM5 | Meier–Gorlin Syndrome 8 | 2-bp deletion in exon 7; Thr466 to Ile (T466I) | [35] |
| MCM8 | Premature ovarian failure 10 | Pro149 to Arg (P149R); c.1954-1G–A splice site mutation in intron 14; c.1469–1470insTA | [38,40] |
| MCM9 | Ovarian dysgenesis 4 | c.1732 + 2T–C transition in intron 9; Arg132 to Ter (R132X); Glu495 to Ter (E495X) | [37,39] |
| Cancer Type | No. of Cases | MCM2 | MCM3 | MCM4 | MCM6 | MCM7 | MCM8 | MCM9 | MCM10 |
|---|---|---|---|---|---|---|---|---|---|
| Stomach adenocarcinoma | 1131 | 3.45% (M) | - | 3.71% (M) | - | 5.13% (A) | 4.42% (M) | - | - |
| Serous ovarian cancer | 1191 | 3.78% (A) | - | 3.61% (A) | - | 4.11% (A) | 5.04% (A) | - | 4.79% (A) |
| Head and neck squamous cell carcinoma | 1332 | 3.38% (A) | - | 5.11% (A) | - | 5.03% (A) | - | - | - |
| Cutaneous melanoma | 892 | 3.03% (M) | - | - | - | - | 3.03% (M) | - | 3.14% (M) |
| Uterine endometrioid carcinoma | 1242 | 3.22% (M) | - | 3.7% (M) | - | - | - | - | - |
| Esophageal adenocarcinoma | 255 | - | 6.67% (A) | - | - | 12.55% (A) | 3.92% (A) | 3.53% (M) 3.92% (A) | - |
| Hepatocellular carcinoma | 798 | - | - | 5.26% (A) | - | - | - | - | - |
| Invasive breast carcinoma | 954 | - | - | 4.61% (A) | - | - | - | - | - |
| Pancreatic adenocarcinoma | 360 | - | - | 3.33% (A) | - | 3.33% (A) | - | - | - |
| Prostate adenocarcinoma | 1803 | - | - | 3.61% (D) | - | - | 6.16% (D) | - |
| Protein | Kinase | Phosphorylation Sites | Biological Significance | References |
|---|---|---|---|---|
| MCM2 | CDC7 | S4, S5, S7, S13, S26, S27, S31, S40, S41, S53, S108, S139, S220 | [63,69,70,72,79] | |
| S4, S5, S7 | Promotes MCM2 chromatin loading | [63] | ||
| CDK7 | S4, S5, S7, S13, S27, S40, S41, S53, T59, S108, S139 | [68,69] | ||
| CDK2 | S13, S27, S40, S41, S53, S108, S139 | [58,69] | ||
| ATR | S108 | Response to DNA damage | [71,73] | |
| CSNK2A1 | S13, S27, S40, S41, S53, S108, S139 | [69] | ||
| AURKA | S220 | [70,80] | ||
| Unknown | T25, S26, T35, T39, Y137, S381, S484, S566, S754 | [73,81,82,83,84,85] | ||
| S26, S27, S40, S41, S108, S139, S170 | Response to DNA damage | [78] | ||
| MCM3 | CDK1 | T722 | Promotes MCM3 chromatin loading | [60] |
| S112 | Promotes MCM2–7 complex formation and chromatin loading | [59] | ||
| S611, S719 | [59] | |||
| CDK2 | S711 | [58] | ||
| ATM | S535, S728 | Response to DNA damage | [71,86] | |
| ATR | S535 | Response to DNA damage | [71] | |
| CHK1 | S205 | Negatively regulates DNA replication | [74] | |
| DAPk1 | S160 | [77] | ||
| Unknown | S672, S711, T713, S728 | Response to DNA damage | [78] | |
| S681, S728, S734 | Response to DNA damage | [73] | ||
| T674, T725 | [73,82,83,85,87,88,89] | |||
| MCM4 | CDK1 | T7, T19, S32, S88, T110 | [64,81,82,85,90,91,92,93] | |
| CDK2 | T7, T19, S32, T110 | Release MCM complex from chromatin | [65] | |
| S3, T53, S54 | [64,81,82,85,90,91,92,93] | |||
| Unknown | S31, S120, S131, S326 | [81,82,84,87,88,94] | ||
| MCM5 | CHK1 | T633 | [75] | |
| Unknown | S2, S315, S605 | [81,82] | ||
| MCM6 | Unknown | S13, S762 | Response to DNA damage | [78] |
| T791 | Response to DNA damage | [73] | ||
| S219, T259, S271, T278, T380, S385, S413, S758 | [81,82,83,85,89,94,95,96,97,98] | |||
| MCM7 | CDK1 | S121 | [62,81,82,84,85] | |
| CDK2 | S121 | Promotes MCM7 chromatin loading and proper mitotic exit in M phase | [62] | |
| S365 | [62,81,82,84,85] | |||
| LYN | Y600 | [99] | ||
| Unknown | S549 | Response to DNA damage | [73] | |
| S314, S500, S678 | [73,99] | |||
| MCM8 | Unknown | S630 | [81] | |
| MCM9 | Unknown | S762, S802, S1109, S1069 | [81,82,85] | |
| MCM10 | Unknown | T85, S93, | [73,83,85] |
| Proteins | Potential Ubiquitination Sites |
|---|---|
| MCM2 | K178, K216, K224, K384, K462 #, K469, K476, K492, K505 #, K529 #, K538, K591, K613 ※,K722, K752, K837 #, K863 #, K868 #, K896 |
| MCM3 | K35, K152, K207, K230, K248, K266, K270 *, K283 *, K293, K301, K315 #, K351*, K413, K435*, K463, K579, K655 #, K656 #, K732, K748 * |
| MCM4 | K179, K216 #, K220 ※, K381, K413, K439, K450, K455, K47, K478 #, K536, K549, K578, K600, K627, K628, K746, K752, K762, K770, K814, K819※, K837, K858 |
| MCM5 | K141 ※, K220, K228, K256, K337, K351, K387, K392, K396, K407, K471, K499, K581, K583, K627, K696 ※ |
| MCM6 | K28, K102, K108 ※, K173, K197, K205 ※, K313, K326, K332, K365, K402, K407 ※, K422 ※, K486, K490, K517, K588, K599, K611, K643, K646, K654※, K733, K744, K754, K769, K775, K796 |
| MCM7 | K4, K10, K15, K28 ※, K29 ※, K32, K75 ※, K89, K96, K145, K159 #, K174, K231 ※, K236, K305, K308, K335, K351, K352, K387, K471, K557 ※, K569, K596 ※, K627, K641, K648 |
| MCM8 | K651 |
| MCM9 | K296, K446 |
| MCM10 | K134, K139, K313, K493, K520, K627, K665 |
| Proteins | Potential Sumoylation Sites |
|---|---|
| MCM3 | K248, K266, K732, K736 |
| MCM4 | K439, K477 |
| MCM7 | K4, K10, K15, K32, K159, K174, K231, K236, K308 |
| MCM10 | K134, K139, K313, K362, K482, K511, K534, K627, K665, K669, K675, K682, K695, K746, K762, K769, K854, K869 |
| Proteins | Potential O-GlcNAcylation Sites |
|---|---|
| MCM2 | S5, T25 **, S26, S27, S31, T35, T308, T313, S540, T546, S566, S608, T625, T845 |
| MCM3 | S118, T154, S170, S171, S302, S348, T368, T369 *, T383, S595 *, T610 *, S781 |
| MCM4 | S2 **, S3 *, S6, T7 *, S9, T19, S26 **, T53, S54 *, S70, S71, S77, S87 *, S97 *, S105, T369, T391, S406 *, T533, T611, S703, T778 |
| MCM5 | T111, S135 *, S136, T476, S600 *, S647, S654 |
| MCM6 | T266, S399, T419, S420 *, S507, S607 |
| MCM7 | S143, S401, T404, T405, T601, T654, S670 **, T717 |
| MCM8 | S338, S361, T484, S485, S555, S574, S621, T635, T839 |
| MCM9 | S91, S170, S171, T376, S468, T564, S659, S725 **, S727, T767 *, S768, S777, T864, T871 *, S874, S878, T879, S898, T946, S952, T990 *, S1067 **, S1069 *, S1088, T1092 **, T1093 *, S1099 *, S1143 |
| MCM10 | T96, T137, S143, S150, S171, T186, T192, S196, S202 **, S203 **, S205, T208 **, T217 *, S237, S261 *, S465 *, S555, S593, S598 *, S599 **, S600, T610 * |
| Protein | Potential Sites |
|---|---|
| MCM2 | K216, K469, K742, K896 |
| MCM3 | K152, K556, K559 |
| MCM4 | K123, K220, K439, K450, K627, K819, K858 |
| MCM5 | K220, K387, K392, K396, K581, K696 |
| MCM6 | K313, K344, K517, K599, K643, K646, K775 |
| MCM7 | K28, K29 |
| MCM10 | K267 *, K312 *, #, K318 *, K390 *, #, K657 *, K664 *, K681 *, K683 *, #, K745 *, #, K761 *, #, K768 *, #, K777, K783 *, K853 * |
| Protein | Modification | Modified Sites | Biological Significance | References |
|---|---|---|---|---|
| MCM2 | Phosphorylation | S4, S5, S7 | Promotes chromatin loading | [63] |
| S108 | Phosphorylated by ATR upon DNA damage | [71] | ||
| S27, S41, S139 | Promotes replication; response to DNA damage | [63,68,78] | ||
| Ubiquitination | K462, K505, K837, K863, K868 | Response to DNA damage | [81] | |
| MCM3 | Phosphorylation | S112 | Promotes MCM2–7 complex formation and chromatin loading | [59] |
| S205 | Phosphorylated by CHK1, negatively regulates DNA replication | [60] | ||
| S535 | Phosphorylated by ATM upon DNA damage | [71] | ||
| T722 | Promotes chromatin loading | [60] | ||
| S672, S711, T713, S728 | Response to DNA damage | [78] | ||
| Ubiquitination | K315, K655, K656 | Increased upon DNA damage | [81] | |
| MCM4 | Phosphorylation | T19, S32, T110 | Promotes MCM complex unloading from chromatin | [65] |
| Ubiquitination | K216, K478 | Response to DNA damage | [81] | |
| MCM6 | Phosphorylation | S13, S762 | Response to DNA damage | [78] |
| MCM7 | Phosphorylation | S121 | Promotes chromatin loading and proper mitotic exit | [62] |
| Ubiquitination | K159 | Response to DNA damage | [78] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Xu, X. Post-Translational Modifications of the Mini-Chromosome Maintenance Proteins in DNA Replication. Genes 2019, 10, 331. https://doi.org/10.3390/genes10050331
Li Z, Xu X. Post-Translational Modifications of the Mini-Chromosome Maintenance Proteins in DNA Replication. Genes. 2019; 10(5):331. https://doi.org/10.3390/genes10050331
Chicago/Turabian StyleLi, Zheng, and Xingzhi Xu. 2019. "Post-Translational Modifications of the Mini-Chromosome Maintenance Proteins in DNA Replication" Genes 10, no. 5: 331. https://doi.org/10.3390/genes10050331
APA StyleLi, Z., & Xu, X. (2019). Post-Translational Modifications of the Mini-Chromosome Maintenance Proteins in DNA Replication. Genes, 10(5), 331. https://doi.org/10.3390/genes10050331