The Alternative Splicing Landscape of Brassica napus Infected with Leptosphaeria maculans

 ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Downloading of RNA-seq Data
2.2. Assembly of Putative Transcripts and Analysis of the AS Landscape
2.3. Weighted Gene Co-expression Network Analysis of Overlapping Differentially Spliced Genes
2.4. Functional Enrichment and Clustering
3. Results
3.1. Putative Transcript Assembly
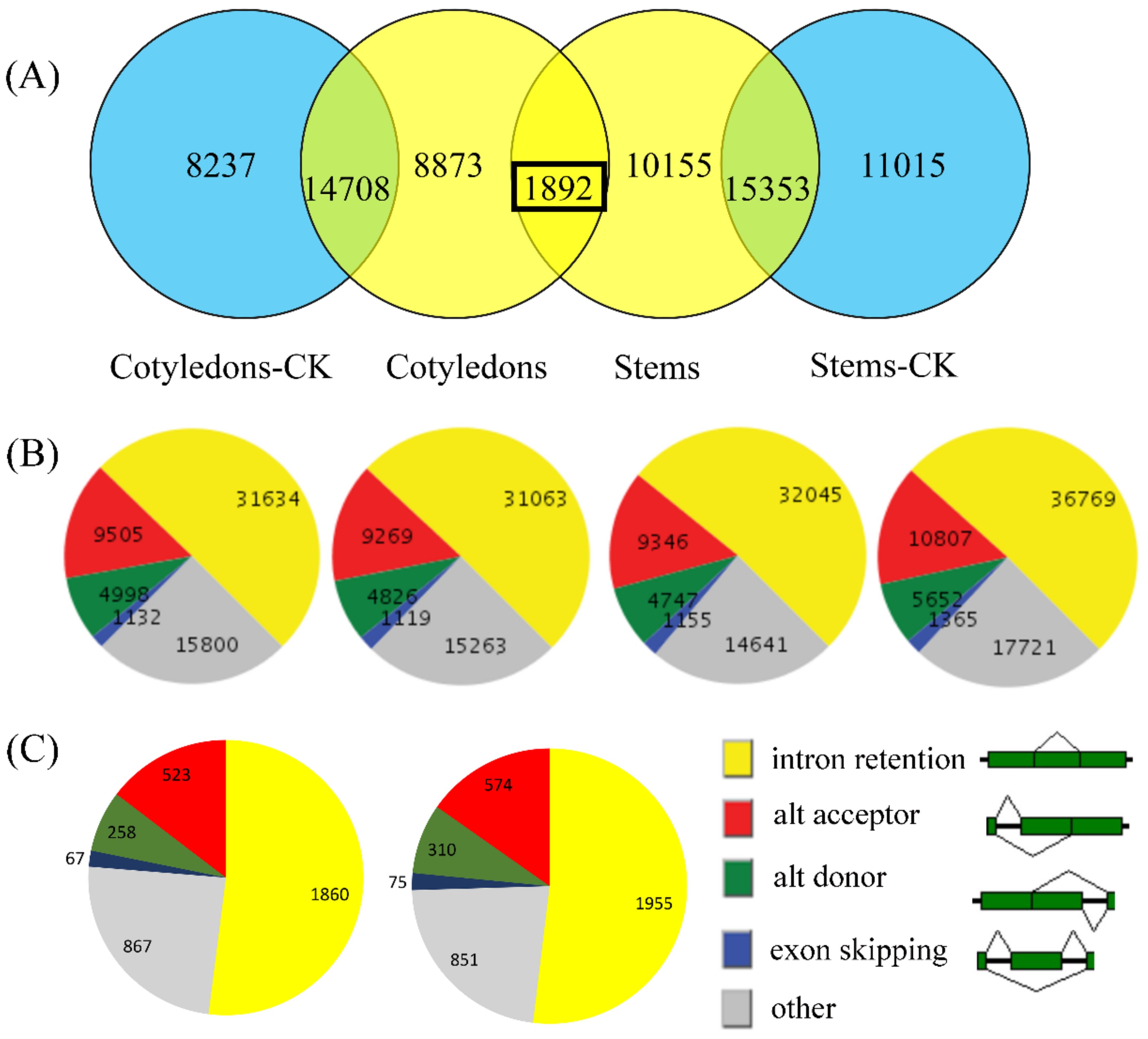
3.2. Analysis of the AS Landscape
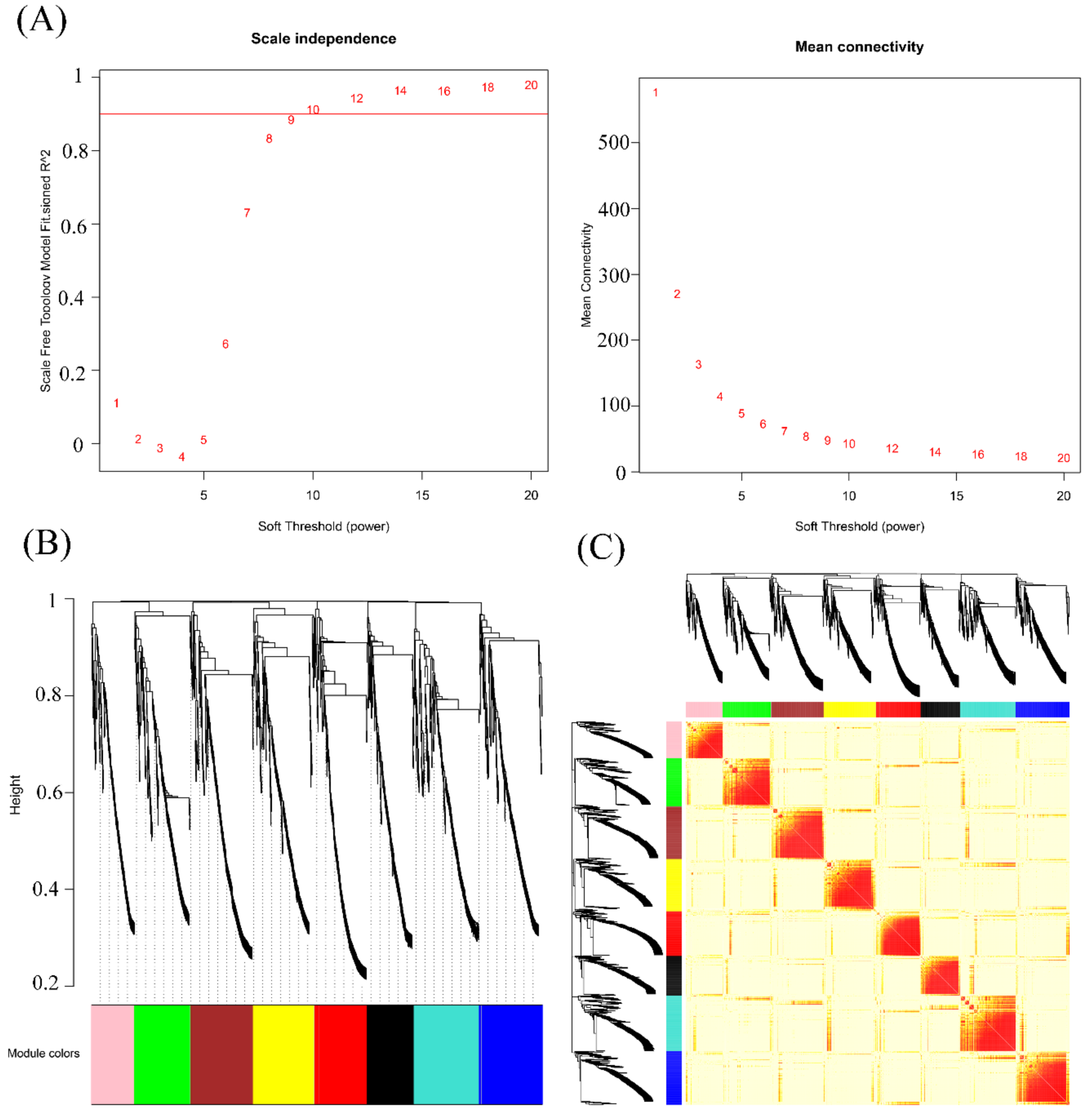
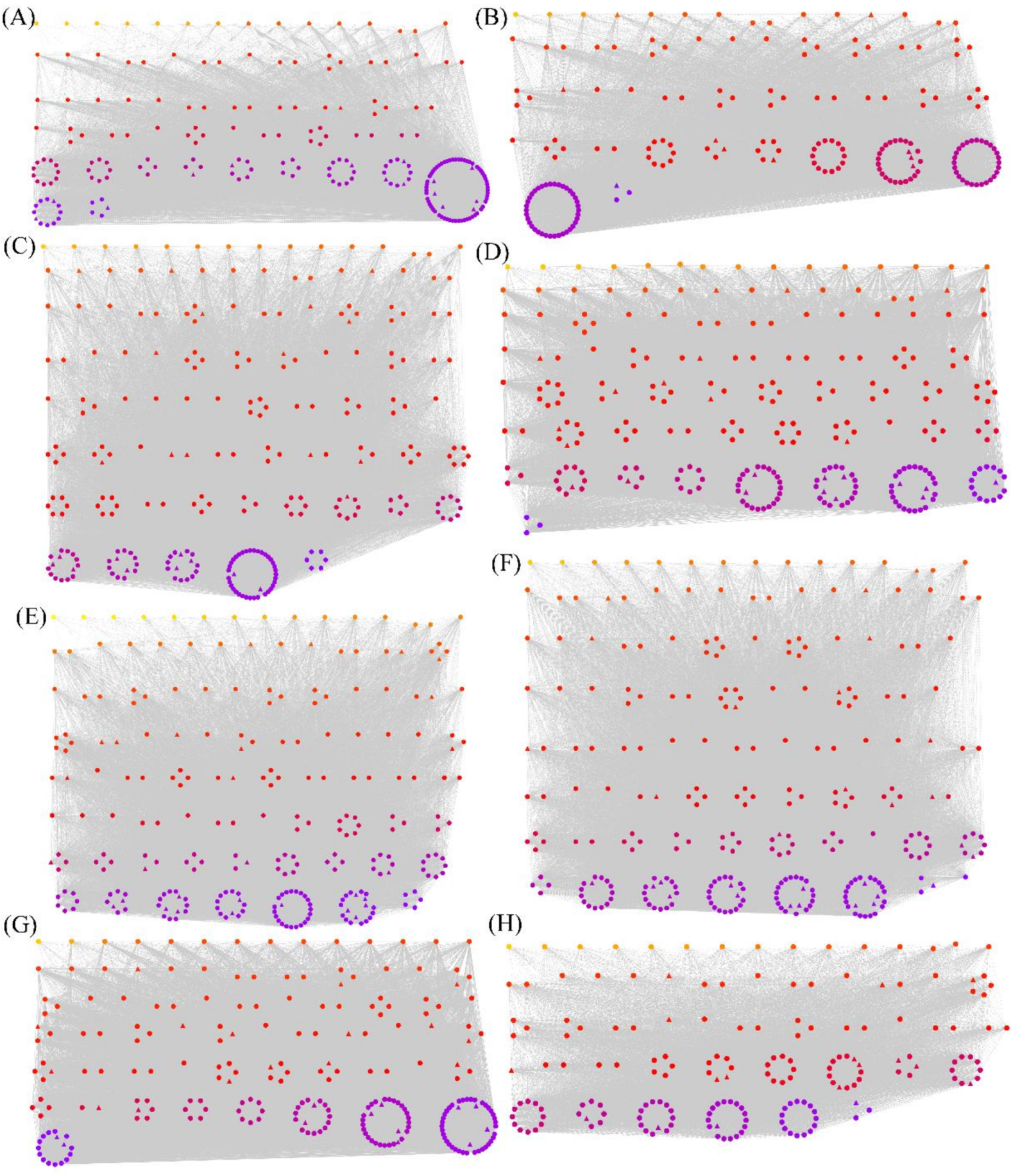
3.3. WGCNA of Overlapping Differentially Spliced Genes
3.4. The Hub Genes and TFs in the Modules Identified by WGCNA
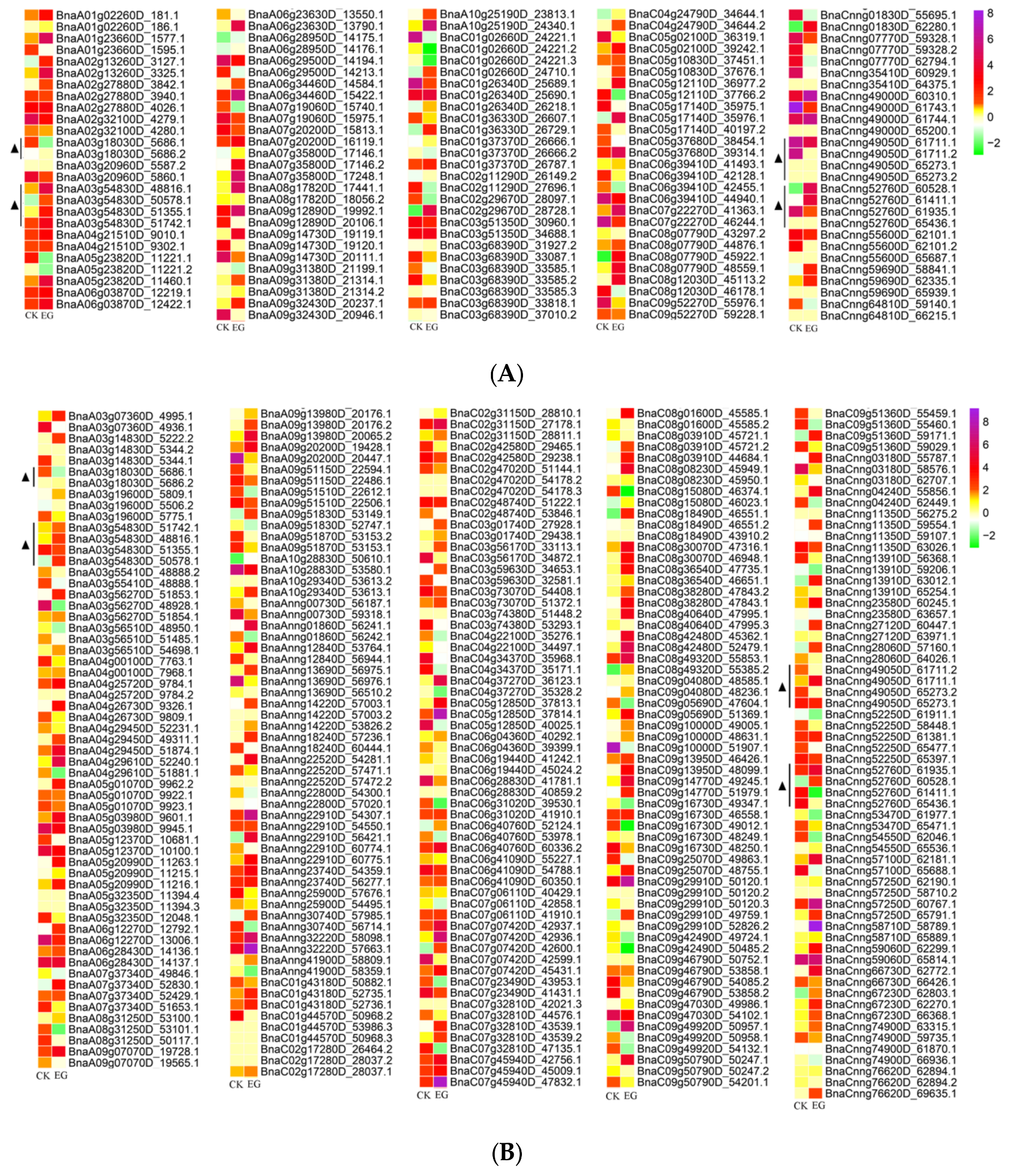
3.5. Identification of Differentially Expressed Genes in Mock-Infected and Infected Samples among Differentially Spliced Genes
4. Discussion
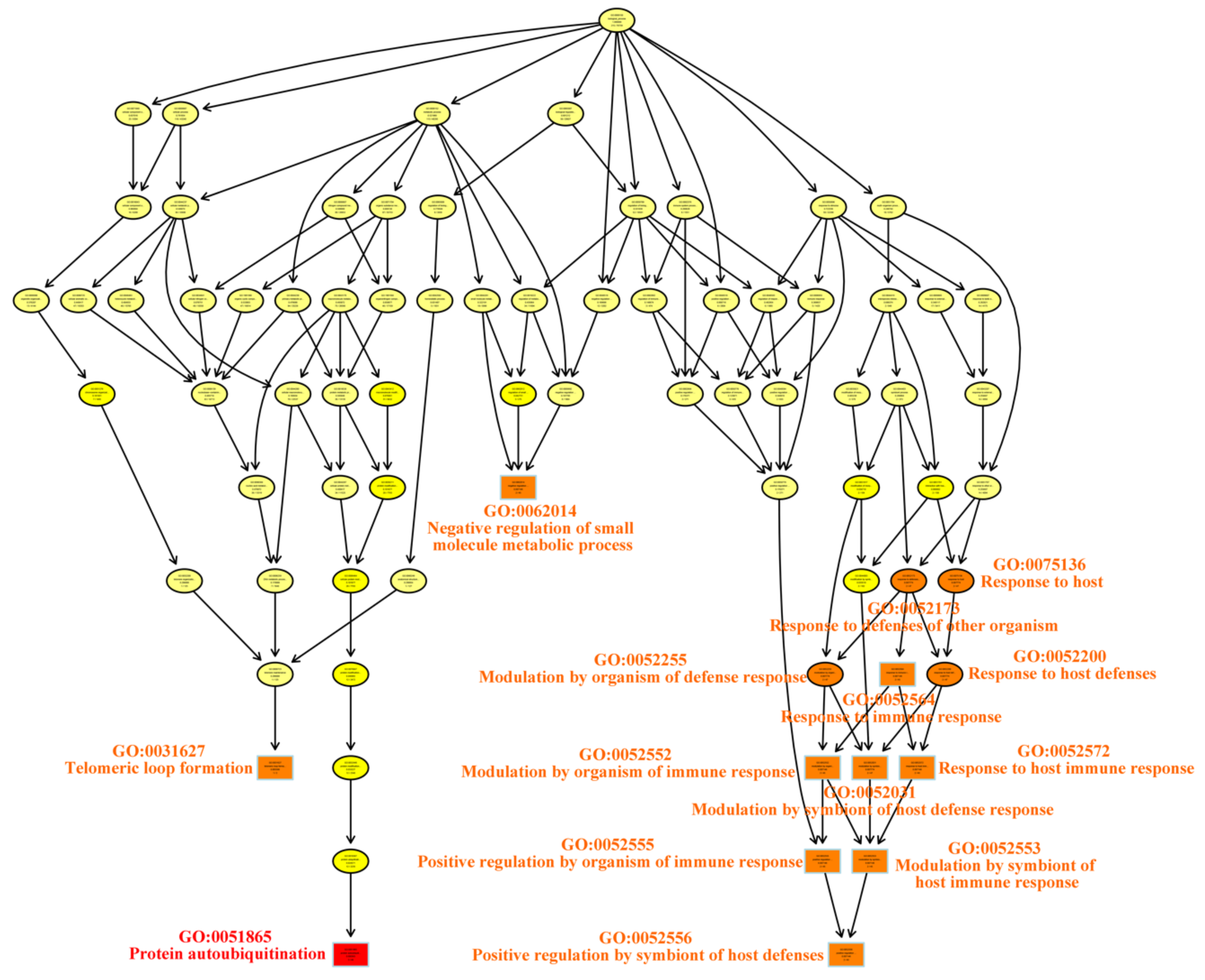
4.1. Enriched Pathways and Hub Genes Identified by WGCNA
4.2. The Roles of TFs in the Defense Response to Pathogens among the AS Genes
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gilbert, W. Why genes in pieces? Nature 1978, 271, 501. [Google Scholar] [CrossRef]
- Mandadi, K.K.; Scholthof, K.B. Genome-wide analysis of alternative splicing landscapes modulated during plant-virus interactions in Brachypodium distachyon. Plant Cell. 2015, 27, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Rickard, S.; Luo, S.; Irina, K.; Lu, Z.; Christine, M.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative Isoform Regulation in Human Tissue Transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Filichkin, S.A.; Priest, H.D.; Givan, S.A.; Shen, R.; Bryant, D.W.; Fox, S.E.; Wong, W.K.; Mockler, T.C. Genome-wide mapping of alternative splicing in Arabidopsis thaliana. Genome Res. 2010, 20, 45–58. [Google Scholar] [CrossRef]
- Shen, Y.; Tian, Z. Global dissection of alternative splicing in paleopolyploid soybean. Plant Cell 2014, 26, 996–1008. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Xiao, G.; Zhu, Y.-X. Single-Nucleotide Resolution Mapping of the Gossypium raimondii Transcriptome Reveals a New Mechanism for Alternative Splicing of Introns. Plant Cell 2014, 7, 829–840. [Google Scholar]
- Thatcher, S.R.; Li, B. Genome-Wide Analysis of Alternative Splicing in Zea mays: Landscape and Genetic Regulation. Plant Cell 2014, 26, 3472–3487. [Google Scholar] [CrossRef]
- Zhang, G.; Guo, G.; Hu, X.; Yong, Z.; Li, Q.; Li, R.; Zhuang, R.; Lu, Z.; He, Z.; Fang, X. Deep RNA sequencing at single base-pair resolution reveals high complexity of the rice transcriptome. Genome Res. 2010, 20, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Green, R.E.; Brenner, S.E. Evidence for the Widespread Coupling of Alternative Splicing and Nonsense-Mediated mRNA Decay in Humans. Proc. Natl. Acad. Sci. USA 2003, 100, 189–192. [Google Scholar]
- Modrek, B.; Lee, C. A genomic view of alternative splicing. Nat. Genet. 2002, 30, 13–19. [Google Scholar] [PubMed]
- Sultan, M.; Schulz, M.H.; Richard, H.; Magen, A.; Klingenhoff, A.; Scherf, M.; Seifert, M.; Borodina, T.; Soldatov, A.; Parkhomchuk, D. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science 2008, 321, 956–960. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Hou, H.; Song, H.; Lin, K.; Zhang, Z.; Hu, J.; Pang, E. The comparison of alternative splicing among the multiple tissues in cucumber. BMC Plant Biol. 2018, 18, 5. [Google Scholar] [CrossRef] [PubMed]
- Crosetto, P.; Deparis, S.; Fourestey, G.; Quarteroni, A. Alternative splicing at the intersection of biological timing, development, and stress responses. Plant Cell 2013, 25, 3640–3656. [Google Scholar]
- Drechsel, G.; Kahles, A.; Kesarwani, A.K.; Stauffer, E.; Behr, J.; Drewe, P.; Rätsch, G.; Wachter, A. Nonsense-Mediated Decay of Alternative Precursor mRNA Splicing Variants Is a Major Determinant of the Arabidopsis Steady State Transcriptome. Plant Cell 2013, 25, 3726–3742. [Google Scholar] [CrossRef] [PubMed]
- Lareau, L.F.; Brooks, A.N.; Soergel, D.A.; Meng, Q.; Brenner, S.E. The coupling of alternative splicing and nonsense-mediated mRNA decay. Adv. Exp. Med. Biol. 2007, 623, 190–211. [Google Scholar]
- Luo, Z.; Yang, Q.; Yang, L. RNA Structure Switches RBP Binding. Mol. Cell 2016, 64, 219–220. [Google Scholar] [CrossRef][Green Version]
- Breathnach, R.; Benoist, C.; O’Hare, K.; Gannon, F.; Chambon, P. Ovalbumin gene: Evidence for a leader sequence in mRNA and DNA sequences at the exon-intron boundaries. Proc. Natl. Acad. Sci. USA 1978, 75, 4853–4857. [Google Scholar] [CrossRef]
- Fu, X.D.; Ares, M.Jr. Context-dependent control of alternative splicing by RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 689–701. [Google Scholar] [CrossRef]
- Ast, G. How did alternative splicing evolve? Nat. Rev. Genet. 2004, 5, 773–782. [Google Scholar] [CrossRef]
- Vuong, C.K.; Black, D.L.; Zheng, S. The neurogenetics of alternative splicing. Nat. Rev. Neurosci. 2016, 17, 265–281. [Google Scholar] [CrossRef]
- Li, W.; Lin, W.D.; Ray, P.; Lan, P.; Schmidt, W. Genome-wide detection of condition-sensitive alternative splicing in Arabidopsis roots. Plant Physiol. 2013, 162, 1750–1763. [Google Scholar] [CrossRef] [PubMed]
- Tress, M.L.; Abascal, F.; Valencia, A. Alternative Splicing May Not Be the Key to Proteome Complexity. Trends Biochem. Sci. 2016, 42, 98–110. [Google Scholar] [CrossRef]
- Chang, C.Y.; Tu, S.L. Genome-Wide Analysis of Heat-Sensitive Alternative Splicing in Physcomitrella patens. Plant Physiol. 2014, 165, 826–840. [Google Scholar] [CrossRef] [PubMed]
- Kalyna, M.; Simpson, C.G.; Syed, N.H.; Lewandowska, D.; Marquez, Y.; Kusenda, B.; Marshall, J.; Fuller, J.; Cardle, L.; Mcnicol, J. Alternative splicing and nonsense-mediated decay modulate expression of important regulatory genes in Arabidopsis. Nucleic Acids Res. 2012, 40, 2454–2469. [Google Scholar] [CrossRef]
- Jeong, B.; Lin, Y.; Joe, A.; Guo, M.; Korneli, C.; Yang, H.; Wang, P.; Yu, M.; Cerny, R.L.; Staiger, D. Structure Function Analysis of an ADP-ribosyltransferase Type III Effector and Its RNA-binding Target in Plant Immunity. J. Biol. Chem. 2011, 286, 43272–43282. [Google Scholar] [CrossRef]
- Liu, J.; Chen, X.; Liang, X.; Zhou, X.; Yang, F.; Liu, J.; He, S.Y.; Guo, Z. Alternative splicing of rice WRKY62 and WRKY76 transcription factor genes in pathogen defense. Plant Physiol. 2016, 171, 1427–1442. [Google Scholar] [CrossRef]
- Yang, S.; Tang, F.; Zhu, H. Alternative splicing in plant immunity. Int. J. Mol. Sci. 2014, 15, 10424–10445. [Google Scholar] [CrossRef]
- Fitt, B.D.L.; Brun, H.; Barbetti, M.J.; Rimmer, S.R. World-Wide Importance of Phoma Stem Canker (Leptosphaeria maculans and L. biglobosa) on Oilseed Rape (Brassica napus); Springer: Dordrecht, The Netherlands, 2006; pp. 3–15. [Google Scholar]
- Chalhoub, B.; Denoeud, F.; Liu, S.; Parkin, I.A.P.; Tang, H.; Wang, X.; Chiquet, J.; Belcram, H.; Tong, C.; Samans, B. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 2014, 345, 950–953. [Google Scholar] [CrossRef]
- Wang, X.; Wang, H.; Wang, J.; Sun, R.; Wu, J.; Liu, S.; Bai, Y.; Mun, J.H.; Bancroft, I.; Cheng, F.; et al. The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 2011, 43, 1035–1039. [Google Scholar] [CrossRef]
- Liu, S.; Liu, Y.; Yang, X.; Tong, C.; Edwards, D.; Parkin, I.A.P.; Zhao, M.; Ma, J.; Yu, J.; Huang, S.; et al. The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat. Commun. 2014, 5, 3930. [Google Scholar] [CrossRef]
- Dineshkumar, S.P.; Baker, B.J. Alternatively spliced N resistance gene transcripts: Their possible role in tobacco mosaic virus resistance. Proc. Natl. Acad. Sci. USA 2000, 97, 1908–1913. [Google Scholar] [CrossRef]
- Piron, F.; Nicolaï, M.; Minoïa, S.; Piednoir, E.; Moretti, A.; Salgues, A.; Zamir, D.; Caranta, C.; Bendahmane, A. An induced mutation in tomato eIF4E leads to immunity to two potyviruses. Plos ONE 2010, 5, e11313. [Google Scholar] [CrossRef]
- Fan, X.; Munson, M. Petabytes in Motion: Ultra High Speed Transport of Media Files: A Theoretical Study and its Engineering Practice of Aspera fasp. In Proceedings of the SMPTE Tech Conference & Expo, Hollywood, CA, USA, 26–28 October 2010; pp. 2–13. [Google Scholar]
- Wajid, B.; Serpedin, E. Do it yourself guide to genome assembly. Brief. Funct. Genom. 2016, 15, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Marc, L.; Bjoern, U. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.; Davis, C.A.; Felix, S.; Jorg, D.; Chris, Z.; Sonali, J.; Philippe, B.; Mark, C.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar]
- Sreya, G.; Chon-Kit Kenneth, C. Analysis of RNA-Seq Data Using TopHat and Cufflinks. Methods Mol. Biol. 2016, 1374, 339–361. [Google Scholar]
- Sylvain, F.; Michael, S. ASTALAVISTA: Dynamic and flexible analysis of alternative splicing events in custom gene datasets. Nucleic Acids Res. 2007, 35, W297–W299. [Google Scholar]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Kohl, M.; Wiese, S.; Warscheid, B. Cytoscape: Software for visualization and analysis of biological networks. Methods Mol. Biol. 2011, 696, 291–303. [Google Scholar] [PubMed]
- O’Connor, B.P. SPSS and SAS programs for determining the number of components using parallel analysis and Velicer’s MAP test. Behav. Res. Methods Instrum. Comput. 2000, 32, 396–402. [Google Scholar] [CrossRef]
- Li, D.; Dye, T.D. Power and stability properties of resampling-based multiple testing procedures with applications to gene oncology studies. Comput. Math. Methods Med. 2013, 2013, 610297. [Google Scholar] [CrossRef]
- Lalitah, S. Primer Premier 5. Biotech Softw. Internet Rep. 2000, 1, 270–272. [Google Scholar] [CrossRef]
- Yin, L.; Cai, Z.; Zhu, B.; Xu, C. Identification of Key Pathways and Genes in the Dynamic Progression of HCC Based on WGCNA. Genes 2018, 9, 92. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; He, F.; Berkowitz, O.; Liu, J.; Cao, P.; Tang, M.; Shi, H.; Wang, W.; Li, Q.; Shen, Z.; et al. Alternative Splicing Plays a Critical Role in Maintaining Mineral Nutrient Homeostasis in Rice (Oryza sativa). Plant Cell 2018, 30, 2267–2285. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.D.; Šašek, V.; Burketová, L.; Čopíková, J.; Synytsya, A.; Jindřichová, B.; Valentová, O. Cell Wall Components of Leptosphaeria maculans Enhance Resistance of Brassica napus. J. Agric. Food Chem. 2013, 61, 5207–5214. [Google Scholar] [CrossRef]
- Rouxel, T.; Grandaubert, J.; Hane, J.K.; Hoede, C.; Wouw, A.P.V.D.; Couloux, A.; Dominguez, V.; Anthouard, V.; Bally, P.; Bourras, S. Effector diversification within compartments of the Leptosphaeria maculans genome affected by Repeat-Induced Point mutations. Nat. Commun. 2011, 2, 202. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Peng, G.; Kutcher, H.R.; Balesdent, M.H.; Delourme, R.; Fernando, W.G.D. Breakdown of Rlm3 resistance in the Brassica napus–Leptosphaeria maculans pathosystem in western Canada. Eur. J. Plant Pathol. 2016, 145, 659–674. [Google Scholar] [CrossRef]
- Kei, I.; Motoaki, S.; Tetsuya, S.; Masakazu, S.; Kenji, A.; Tetsuro, T.; Akihiko, K.; Kazuo, S. Genome-wide analysis of alternative pre-mRNA splicing in Arabidopsis thaliana based on full-length cDNA sequences. Nucleic Acids Res. 2004, 32, 5096–5103. [Google Scholar]
- Li, Y.; Yin, H.; Wang, Q.; Zhao, X.M.; Du, Y.G.; Li, F.L. Oligochitosan induced Brassica napus L. production of NO and H2O2 and their physiological function. Carbohydr. Polym. 2009, 75, 612–617. [Google Scholar] [CrossRef]
- Soanes, D.M.; Alam, I.; Cornell, M.; Wong, H.M.; Hedeler, C.; Paton, N.W.; Rattray, M.; Hubbard, S.J.; Oliver, S.G.; Talbot, N.J. Comparative genome analysis of filamentous fungi reveals gene family expansions associated with fungal pathogenesis. Plos ONE 2008, 3, e2300. [Google Scholar] [CrossRef]
- Remy, E.; Meyer, M.; Blaise, F.; Simon, U.K.; Kuhn, D.; Balesdent, M.H.; Rouxel, T. A key enzyme of the Leloir pathway is involved in pathogenicity of Leptosphaeria maculans toward oilseed rape. Mol. Plant Microbe Interact. 2009, 22, 725–736. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Annis, S.L.; Goodwin, P.H. Comparison of cell wall-degrading enzymes produced by highly and weakly virulent isolates of Leptosphaeria maculans, in culture. Microbiol. Res. 1996, 151, 401–406. [Google Scholar] [CrossRef]
- Estavillo, G.M.; Crisp, P.A.; Wannarat, P.; Markus, W.; Derek, C.; Chris, C.; Estelle, G.; James, W.; Pascale, D.; Hélène, J. Evidence for a SAL1-PAP chloroplast retrograde pathway that functions in drought and high light signaling in Arabidopsis. Plant Cell 2011, 23, 3992–4012. [Google Scholar] [CrossRef]
- Xin, X.F.; Nomura, K.; Ding, X.; Chen, X.; Wang, K.; Aung, K.; Uribe, F.; Rosa, B.; Yao, J.; Chen, J. Pseudomonas syringae Effector Avirulence Protein E Localizes to the Host Plasma Membrane and Down-Regulates the Expression of the Nonrace-Specific Disease Resistance1/Harpin-Induced1-Like13 Gene Required for Antibacterial Immunity in Arabidopsis. Plant Physiol. 2015, 169, 793–802. [Google Scholar]
- Sun, Y.; Fan, X.Y.; Cao, D.M.; Tang, W.; He, K.; Zhu, J.Y.; He, J.X.; Bai, M.Y.; Zhu, S.; Oh, E. Integration of Brassinosteroid Signal Transduction with the Transcription Network for Plant Growth Regulation in Arabidopsis. Dev. Cell 2010, 19, 765–777. [Google Scholar] [CrossRef]
- Ma, S.; Gong, Q.; Bohnert, H.J. Dissecting salt stress pathways. J. Exp. Bot. 2006, 57, 1097–1107. [Google Scholar] [CrossRef]
- Jin, J.; Zhang, H.; Kong, L.; Gao, G.; Luo, J. PlantTFDB 3.0: A portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res. 2014, 42, D1182–D1187. [Google Scholar] [CrossRef]
- Daurelio, L.D.; Romero, M.S.; Petrocelli, S.; Merelo, P.; Cortadi, A.A.; Talón, M.; Tadeo, F.R.; Orellano, E.G. Characterization of Citrus sinensis transcription factors closely associated with the non-host response to Xanthomonas campestris pv. vesicatoria. J. Plant Physiol. 2013, 170, 934–942. [Google Scholar] [CrossRef]
- Hyung-Sae, K.; Sung Jin, K.; Nazia, A.; Bressan, R.A.; Dae-Jin, Y.; Sang-Dong, Y.; Suk-Yun, K.; Sang-Bong, C. The DOF transcription factor Dof5.1 influences leaf axial patterning by promoting Revoluta transcription in Arabidopsis. Plant J. 2010, 64, 524–535. [Google Scholar]
- Wang, Q.J.; Chen, L.G.; Di-Qiu, Y.U. Overexpression of AtWRKY71 Affects Plant’s Defense Response to Pseudomonas syringae. Plant Divers. Resour. 2015, 37, 577–585. [Google Scholar]
- Sun, T.J.; Lu, Y.; Narusaka, M.; Shi, C.; Yang, Y.B.; Wu, J.X.; Zeng, H.Y.; Narusaka, Y.; Yao, N. A novel pyrimidin-like plant activator stimulates plant disease resistance and promotes growth. Plos ONE 2015, 10, e0123227. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Term | Annotation | Significant | Expected | Classic Fisher | |
|---|---|---|---|---|---|
| GO:0051865 | Protein Autoubiquitination | 49 | 3 | 0.14 | 0.00036 |
| GO:0031627 | Telomeric Loop Formation | 2 | 1 | 0.01 | 0.00560 |
| GO:0052552 | Modulation by Organism of Immune Response | 45 | 2 | 0.13 | 0.00715 |
| GO:0052553 | Modulation by Symbiont of Host Immune Response | 45 | 2 | 0.13 | 0.00715 |
| GO:0052555 | Positive Regulation by Organism of Immune Response | 45 | 2 | 0.13 | 0.00715 |
| GO:0052556 | Positive Regulation by Symbiont of Host | 45 | 2 | 0.13 | 0.00715 |
| GO:0052564 | Response to Immune Response | 45 | 2 | 0.13 | 0.00715 |
| GO:0052572 | Response to Host Immune Response | 45 | 2 | 0.13 | 0.00715 |
| GO:0062014 | Negative Regulation of Small Molecule Members | 45 | 2 | 0.13 | 0.00715 |
| GO:0052031 | Modulation by Symbiont of Host Defense Response | 47 | 2 | 0.13 | 0.00777 |
| GO:0052173 | Response to Defenses of Other Organism Immune Response | 47 | 2 | 0.13 | 0.00777 |
| GO:0052200 | Response to Host Defenses | 47 | 2 | 0.13 | 0.00777 |
| GO:0052255 | Modulation by Organism of Defense Response | 47 | 2 | 0.13 | 0.00777 |
| GO:0075136 | Response to Host | 47 | 2 | 0.13 | 0.00777 |
| GO:0018107 | Peptidyl-threonine Phosphorylation | 3 | 1 | 0.01 | 0.00838 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, J.-Q.; Wei, L.-J.; Lin, A.; Zhang, C.; Sun, W.; Yang, B.; Lu, K.; Li, J.-N. The Alternative Splicing Landscape of Brassica napus Infected with Leptosphaeria maculans. Genes 2019, 10, 296. https://doi.org/10.3390/genes10040296
Ma J-Q, Wei L-J, Lin A, Zhang C, Sun W, Yang B, Lu K, Li J-N. The Alternative Splicing Landscape of Brassica napus Infected with Leptosphaeria maculans. Genes. 2019; 10(4):296. https://doi.org/10.3390/genes10040296
Chicago/Turabian StyleMa, Jin-Qi, Li-Juan Wei, Ai Lin, Chao Zhang, Wei Sun, Bo Yang, Kun Lu, and Jia-Na Li. 2019. "The Alternative Splicing Landscape of Brassica napus Infected with Leptosphaeria maculans" Genes 10, no. 4: 296. https://doi.org/10.3390/genes10040296
APA StyleMa, J.-Q., Wei, L.-J., Lin, A., Zhang, C., Sun, W., Yang, B., Lu, K., & Li, J.-N. (2019). The Alternative Splicing Landscape of Brassica napus Infected with Leptosphaeria maculans. Genes, 10(4), 296. https://doi.org/10.3390/genes10040296