Insights into the Link between the Organization of DNA Replication and the Mutational Landscape


{kind=link}
Abstract
:1. Introduction
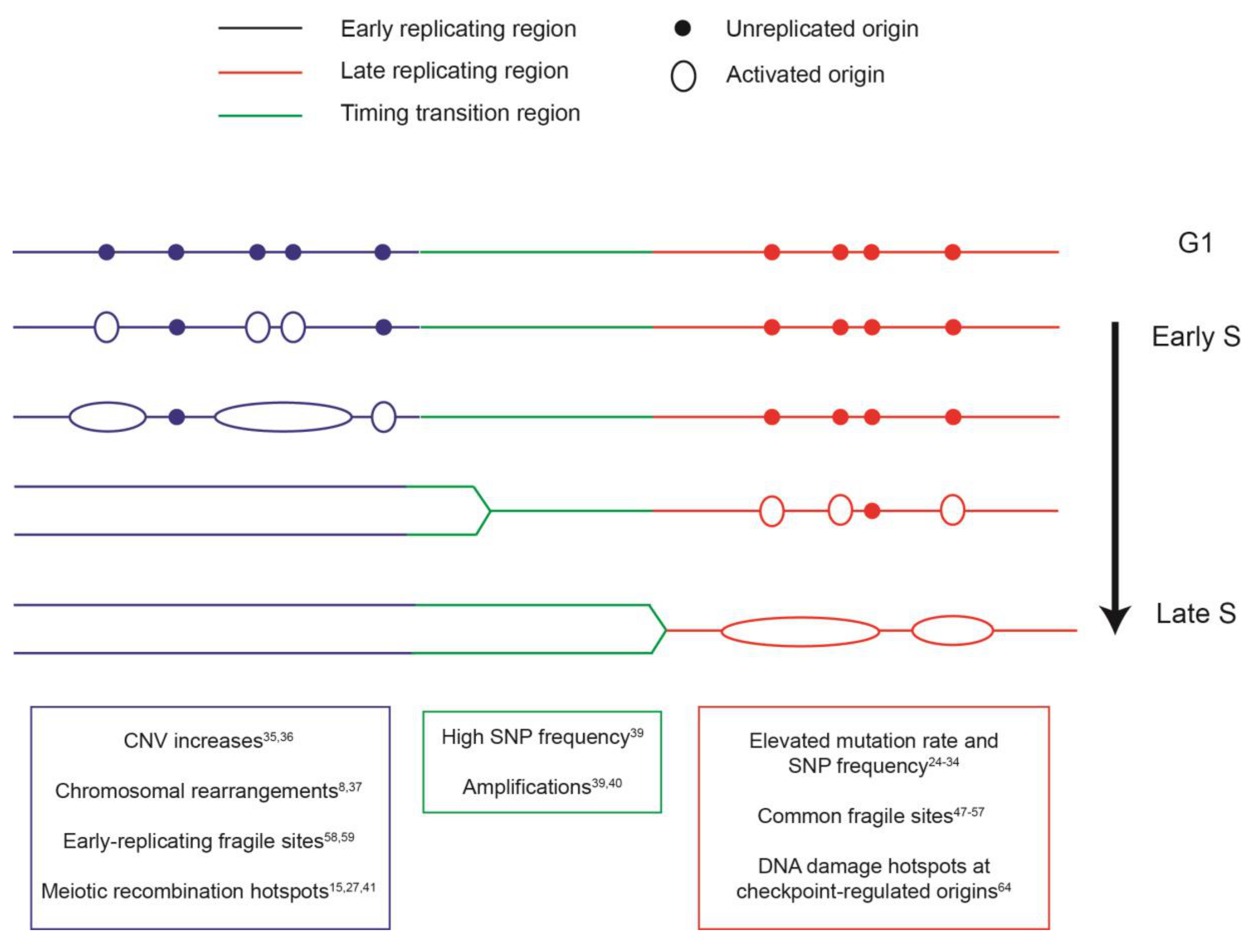
2. The Spatial and Temporal Organization of DNA Replication
3. Coupling between the Replication Program and Mutational Landscape
4. The Replication Program and Genome Instability Hotspots
5. Mechanisms Underlying the Profile of Genetic Variation
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mott, M.L.; Berger, J.M. DNA replication initiation: Mechanisms and regulation in bacteria. Nat. Rev. Microbiol. 2007, 5, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Méchali, M. Eukaryotic DNA replication origins: Many choices for appropriate answers. Nat. Rev. Mol. Cell Biol. 2010, 11, 728–738. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.K.; Arcangioli, B.; Baker, S.P.; Bensimon, A.; Rhind, N. DNA replication origins fire stochastically in fission yeast. Mol. Biol. Cell 2006, 17, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Taljanidisz, J.; Popowski, J.; Sarkar, N. Temporal order of gene replication in Chinese hamster ovary cells. Mol. Cell. Biol. 1989, 9, 2881–2889. [Google Scholar] [CrossRef]
- Desprat, R.; Thierry-Mieg, D.; Lailler, N.; Lajugie, J.; Schildkraut, C.; Thierry-Mieg, J.; Bouhassira, E.E. Predictable dynamic program of timing of DNA replication in human cells. Genome Res. 2009, 19, 2288–2299. [Google Scholar] [CrossRef]
- Heichinger, C.; Penkett, C.J.; Bähler, J.; Nurse, P. Genome-wide characterization of fission yeast DNA replication origins. EMBO J. 2006, 25, 5171–5179. [Google Scholar] [CrossRef] [PubMed]
- Ryba, T.; Hiratani, I.; Lu, J.; Itoh, M.; Kulik, M.; Zhang, J.; Schulz, T.C.; Robins, A.J.; Dalton, S.; Gilbert, D.M. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Res. 2010, 20, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Yaffe, E.; Farkash-Amar, S.; Polten, A.; Yakhini, Z.; Tanay, A.; Simon, I. Comparative analysis of DNA replication timing reveals conserved large-scale chromosomal architecture. PLoS Genet. 2010, 6, e1001011. [Google Scholar] [CrossRef] [PubMed]
- Pope, B.D.; Ryba, T.; Dileep, V.; Yue, F.; Wu, W.; Denas, O.; Vera, D.L.; Wang, Y.; Hansen, R.S.; Canfield, T.K.; et al. Topologically associating domains are stable units of replication-timing regulation. Nature 2014, 515, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Malyavantham, K.S.; Bhattacharya, S.; Alonso, W.D.; Acharya, R.; Berezney, R. Spatio-temporal dynamics of replication and transcription sites in the mammalian cell nucleus. Chromosoma 2008, 117, 553–567. [Google Scholar] [CrossRef]
- Berezney, R.; Dubey, D.D.; Huberman, J.A. Heterogeneity of eukaryotic replicons, replicon clusters, and replication foci. Chromosoma 2000, 108, 471–484. [Google Scholar] [CrossRef]
- Heun, P.; Laroche, T.; Shimada, K.; Furrer, P.; Gasser, S.M. Chromosome dynamics in the yeast interphase nucleus. Science 2001, 294, 2181–2186. [Google Scholar] [CrossRef]
- Muller, C.A.; Nieduszynski, C.A. Conservation of replication timing reveals global and local regulation of replication origin activity. Genome Res. 2012, 22, 1953–1962. [Google Scholar] [CrossRef]
- Perrot, A.; Millington, C.L.; Gómez-Escoda, B.; Schausi-Tiffoche, D.; Wu, P.-Y.J. CDK activity provides temporal and quantitative cues for organizing genome duplication. PLoS Genet. 2018, 14, e1007214. [Google Scholar] [CrossRef]
- Wu, P.-Y.J.; Nurse, P. Replication origin selection regulates the distribution of meiotic recombination. Mol. Cell 2014, 53, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Pope, B.D.; Hiratani, I.; Gilbert, D.M. Domain-wide regulation of DNA replication timing during mammalian development. Chromosome Res. 2010, 18, 127–136. [Google Scholar] [CrossRef]
- Hiratani, I.; Ryba, T.; Itoh, M.; Yokochi, T.; Schwaiger, M.; Chang, C.-W.; Lyou, Y.; Townes, T.M.; Schübeler, D.; Gilbert, D.M. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol. 2008, 6, e245. [Google Scholar] [CrossRef] [PubMed]
- Stambrook, P.J.; Flickinger, R.A. Changes in chromosomal DNA replication patterns in developing frog embryos. J. Exp. Zool. 1970, 174, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Martínez, M.; Pinzón, N.; Ghommidh, C.; Beyne, E.; Seitz, H.; Cayrou, C.; Méchali, M. The gastrula transition reorganizes replication-origin selection in Caenorhabditis elegans. Nat. Struct. Mol. Biol. 2017, 24, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Siefert, J.C.; Georgescu, C.; Wren, J.D.; Koren, A.; Sansam, C.L. DNA replication timing during development anticipates transcriptional programs and parallels enhancer activation. Genome Res. 2017, 27, 1406–1416. [Google Scholar] [CrossRef]
- MacAlpine, D.M.; Rodríguez, H.K.; Bell, S.P. Coordination of replication and transcription along a Drosophila chromosome. Genes Dev. 2004, 18, 3094–3105. [Google Scholar] [CrossRef]
- Pourkarimi, E.; Bellush, J.M.; Whitehouse, I. Spatiotemporal coupling and decoupling of gene transcription with DNA replication origins during embryogenesis in C. elegans. eLife 2016, 5, e21728. [Google Scholar] [CrossRef]
- Müller, C.A.; Nieduszynski, C.A. DNA replication timing influences gene expression level. J. Cell Biol. 2017, 216, 1907–1914. [Google Scholar] [CrossRef] [PubMed]
- Stamatoyannopoulos, J.A.; Adzhubei, I.; Thurman, R.E.; Kryukov, G.V.; Mirkin, S.M.; Sunyaev, S.R. Human mutation rate associated with DNA replication timing. Nat. Genet. 2009, 41, 393–395. [Google Scholar] [CrossRef]
- Lang, G.I.; Murray, A.W. Mutation rates across budding yeast chromosome VI are correlated with replication timing. Genome Biol. Evol. 2011, 3, 799–811. [Google Scholar] [CrossRef]
- Koren, A.; Polak, P.; Nemesh, J.; Michaelson, J.J.; Sebat, J.; Sunyaev, S.R.; McCarroll, S.A. Differential relationship of DNA replication timing to different forms of human mutation and variation. Am. J. Hum. Genet. 2012, 91, 1033–1040. [Google Scholar] [CrossRef]
- Yehuda, Y.; Blumenfeld, B.; Mayorek, N.; Makedonski, K.; Vardi, O.; Cohen-Daniel, L.; Mansour, Y.; Baror-Sebban, S.; Masika, H.; Farago, M.; et al. Germline DNA replication timing shapes mammalian genome composition. Nucleic Acids Res. 2018, 46, 8299–8310. [Google Scholar] [CrossRef] [PubMed]
- Agier, N.; Fischer, G. The mutational profile of the yeast genome is shaped by replication. Mol. Biol. Evol. 2012, 29, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.C.; Pink, C.J.; Hurst, L.D. Late-replicating domains have higher divergence and diversity in Drosophila melanogaster. Mol. Biol. Evol. 2012, 29, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-L.; Rappailles, A.; Duquenne, L.; Huvet, M.; Guilbaud, G.; Farinelli, L.; Audit, B.; d’Aubenton-Carafa, Y.; Arneodo, A.; Hyrien, O.; et al. Impact of replication timing on non-CpG and CpG substitution rates in mammalian genomes. Genome Res. 2010, 20, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Woo, Y.H.; Li, W.-H. DNA replication timing and selection shape the landscape of nucleotide variation in cancer genomes. Nat. Commun. 2012, 3, 1004. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; De, S.; Michor, F. DNA replication timing and higher-order nuclear organization determine single-nucleotide substitution patterns in cancer genomes. Nat. Commun. 2013, 4, 1502. [Google Scholar] [CrossRef] [PubMed]
- Polak, P.; Karlić, R.; Koren, A.; Thurman, R.; Sandstrom, R.; Lawrence, M.; Reynolds, A.; Rynes, E.; Vlahoviček, K.; Stamatoyannopoulos, J.A.; et al. Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature 2015, 518, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- De, S.; Michor, F. DNA replication timing and long-range DNA interactions predict mutational landscapes of cancer genomes. Nat. Biotechnol. 2011, 29, 1103–1108. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Li, H.; Hu, M.; Sasaki, T.; Baccei, A.; Gilbert, D.M.; Liu, J.S.; Collins, J.J.; Lerou, P.H. The distribution of genomic variations in human iPSCs is related to replication-timing reorganization during reprogramming. Cell Rep. 2014, 7, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Shugay, M.; Ortiz de Mendíbil, I.; Vizmanos, J.L.; Novo, F.J. Genomic hallmarks of genes involved in chromosomal translocations in hematological cancer. PLoS Comput. Biol. 2012, 8, e1002797. [Google Scholar] [CrossRef] [PubMed]
- Guan, Z.; Hughes, C.M.; Kosiyatrakul, S.; Norio, P.; Sen, R.; Fiering, S.; Allis, C.D.; Bouhassira, E.E.; Schildkraut, C.L. Decreased replication origin activity in temporal transition regions. J. Cell Biol. 2009, 187, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Fujiyama, A.; Ichiba, Y.; Hattori, M.; Yada, T.; Sakaki, Y.; Ikemura, T. Chromosome-wide assessment of replication timing for human chromosomes 11q and 21q: Disease-related genes in timing-switch regions. Hum. Mol. Genet. 2002, 11, 13–21. [Google Scholar] [CrossRef]
- Watanabe, Y.; Ikemura, T.; Sugimura, H. Amplicons on human chromosome 11q are located in the early/late-switch regions of replication timing. Genomics 2004, 84, 796–805. [Google Scholar] [CrossRef] [PubMed]
- Halldorsson, B.V.; Palsson, G.; Stefansson, O.A.; Jonsson, H.; Hardarson, M.T.; Eggertsson, H.P.; Gunnarsson, B.; Oddsson, A.; Halldorsson, G.H.; Zink, F.; et al. Characterizing mutagenic effects of recombination through a sequence-level genetic map. Science 2019, 363, eaau1043. [Google Scholar] [CrossRef] [PubMed]
- McCarroll, R.M.; Fangman, W.L. Time of replication of yeast centromeres and telomeres. Cell 1988, 54, 505–513. [Google Scholar] [CrossRef]
- Ahmad, K.; Henikoff, S. Centromeres are specialized replication domains in heterochromatin. J. Cell Biol. 2001, 153, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-M.; Dubey, D.D.; Huberman, J.A. Early-replicating heterochromatin. Genes Dev. 2003, 17, 330–335. [Google Scholar] [CrossRef]
- Koren, A.; Tsai, H.-J.; Tirosh, I.; Burrack, L.S.; Barkai, N.; Berman, J. Epigenetically-inherited centromere and neocentromere DNA replicates earliest in S-phase. PLoS Genet 2010, 6, e1001068. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Bachant, J.; Collingwood, D.; Raghuraman, M.K.; Brewer, B.J. Centromere replication timing determines different forms of genomic instability in Saccharomyces cerevisiae checkpoint mutants during replication stress. Genetics 2009, 183, 1249–1260. [Google Scholar] [CrossRef] [PubMed]
- Glover, T.W.; Berger, C.; Coyle, J.; Echo, B. DNA polymerase α inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Hum. Genet. 1984, 67, 136–142. [Google Scholar] [CrossRef]
- Admire, A.; Shanks, L.; Danzl, N.; Wang, M.; Weier, U.; Stevens, W.; Hunt, E.; Weinert, T. Cycles of chromosome instability are associated with a fragile site and are increased by defects in DNA replication and checkpoint controls in yeast. Genes Dev. 2006, 20, 159–173. [Google Scholar] [CrossRef]
- Glover, T.W.; Wilson, T.E.; Arlt, M.F. Fragile sites in cancer: More than meets the eye. Nat. Rev. Cancer 2017, 17, 489–501. [Google Scholar] [CrossRef]
- Di Rienzi, S.C.; Collingwood, D.; Raghuraman, M.K.; Brewer, B.J. Fragile genomic sites are associated with origins of replication. Genome Biol. Evol. 2009, 1, 350–363. [Google Scholar] [CrossRef] [PubMed]
- Debatisse, M.; Le Tallec, B.; Letessier, A.; Dutrillaux, B.; Brison, O. Common fragile sites: Mechanisms of instability revisited. Trends Genet. 2012, 28, 22–32. [Google Scholar] [CrossRef]
- Arlt, M.F.; Durkin, S.G.; Ragland, R.L.; Glover, T.W. Common fragile sites as targets for chromosome rearrangements. DNA Repair 2006, 5, 1126–1135. [Google Scholar] [CrossRef] [PubMed]
- Arlt, M.F.; Miller, D.E.; Beer, D.G.; Glover, T.W. Molecular characterization of FRAXB and comparative common fragile site instability in cancer cells. Genes Chromosomes Cancer 2002, 33, 82–92. [Google Scholar] [CrossRef]
- Burrow, A.A.; Williams, L.E.; Pierce, L.C.T.; Wang, Y.-H. Over half of breakpoints in gene pairs involved in cancer-specific recurrent translocations are mapped to human chromosomal fragile sites. Bmc Genom. 2009, 10, 59. [Google Scholar] [CrossRef] [PubMed]
- Le Beau, M.M.; Rassool, F.V.; Neilly, M.E.; Espinosa, R.; Glover, T.W.; Smith, D.I.; McKeithan, T.W. Replication of a common fragile site, FRA3B, occurs late in S phase and is delayed further upon induction: Implications for the mechanism of fragile site induction. Hum. Mol. Genet. 1998, 7, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Letessier, A.; Millot, G.A.; Koundrioukoff, S.; Lachagès, A.-M.; Vogt, N.; Hansen, R.S.; Malfoy, B.; Brison, O.; Debatisse, M. Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature 2011, 470, 120–123. [Google Scholar] [CrossRef]
- Blin, M.; Le Tallec, B.; Nähse, V.; Schmidt, M.; Brossas, C.; Millot, G.A.; Prioleau, M.-N.; Debatisse, M. Transcription-dependent regulation of replication dynamics modulates genome stability. Nat. Struct. Mol. Biol. 2019, 26, 58–66. [Google Scholar] [CrossRef]
- Barlow, J.H.; Faryabi, R.B.; Callén, E.; Wong, N.; Malhowski, A.; Chen, H.T.; Gutierrez-Cruz, G.; Sun, H.-W.; McKinnon, P.; Wright, G.; et al. Identification of early replicating fragile sites that contribute to genome instability. Cell 2013, 152, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Macheret, M.; Halazonetis, T.D. Intragenic origins due to short G1 phases underlie oncogene-induced DNA replication stress. Nature 2018, 555, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.-Y.; Smith, S.; Ceschia, A.; Torres-Rosell, J.; Aragón, L.; Myung, K. Smc5-Smc6 complex suppresses gross chromosomal rearrangements mediated by break-induced replications. DNA Repair 2008, 7, 1426–1436. [Google Scholar] [CrossRef] [PubMed]
- Dunn, B.; Sherlock, G. Reconstruction of the genome origins and evolution of the hybrid lager yeast Saccharomyces pastorianus. Genome Res. 2008, 18, 1610–1623. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.L.; Byrne, K.P.; Wolfe, K.H. Additions, losses, and rearrangements on the evolutionary route from a reconstructed ancestor to the modern Saccharomyces cerevisiae genome. PLoS Genet. 2009, 5, e1000485. [Google Scholar] [CrossRef] [PubMed]
- Lujan, S.A.; Clausen, A.R.; Clark, A.B.; MacAlpine, H.K.; MacAlpine, D.M.; Malc, E.P.; Mieczkowski, P.A.; Burkholder, A.B.; Fargo, D.C.; Gordenin, D.A.; et al. Heterogeneous polymerase fidelity and mismatch repair bias genome variation and composition. Genome Res. 2014, 24, 1751–1764. [Google Scholar] [CrossRef]
- Gómez-Escoda, B.; Wu, P.-Y.J. The organization of genome duplication is a critical determinant of the landscape of genome maintenance. Genome Res. 2018, 28, 1179–1192. [Google Scholar] [CrossRef]
- Feng, W.; Di Rienzi, S.C.; Raghuraman, M.K.; Brewer, B.J. Replication stress-induced chromosome breakage is correlated with replication fork progression and is preceded by single-stranded DNA formation. G3 2011, 1, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.A.; Sterling, J.; Thompson, C.; Harris, S.; Mav, D.; Shah, R.; Klimczak, L.J.; Kryukov, G.V.; Malc, E.; Mieczkowski, P.A.; et al. Clustered mutations in yeast and in human cancers can arise from damaged long single-strand DNA regions. Mol. Cell 2012, 46, 424–435. [Google Scholar] [CrossRef]
- Chan, K.; Sterling, J.F.; Roberts, S.A.; Bhagwat, A.S.; Resnick, M.A.; Gordenin, D.A. Base damage within single-strand DNA underlies in vivo hypermutability induced by a ubiquitous environmental agent. PLoS Genet. 2012, 8, e1003149. [Google Scholar] [CrossRef] [PubMed]
- Chabes, A.; Georgieva, B.; Domkin, V.; Zhao, X.; Rothstein, R.; Thelander, L. Survival of DNA damage in yeast directly depends on increased dNTP levels allowed by relaxed feedback inhibition of ribonucleotide reductase. Cell 2003, 112, 391–401. [Google Scholar] [CrossRef]
- Poli, J.; Tsaponina, O.; Crabbé, L.; Keszthelyi, A.; Pantesco, V.; Chabes, A.; Lengronne, A.; Pasero, P. dNTP pools determine fork progression and origin usage under replication stress. EMBO J. 2012, 31, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, T.; Ragu, S.; Magdalou, I.; Machon, C.; Dardillac, E.; Técher, H.; Guitton, J.; Debatisse, M.; Lopez, B.S. Slow replication fork velocity of Homologous recombination-defective cells results from Endogenous oxidative stress. PLoS Genet. 2016, 12, e1006007. [Google Scholar] [CrossRef]
- Kumar, D.; Viberg, J.; Nilsson, A.K.; Chabes, A. Highly mutagenic and severely imbalanced dNTP pools can escape detection by the S-phase checkpoint. Nucleic Acids Res. 2010, 38, 3975–3983. [Google Scholar] [CrossRef] [PubMed]
- Watt, D.L.; Buckland, R.J.; Lujan, S.A.; Kunkel, T.A.; Chabes, A. Genome-wide analysis of the specificity and mechanisms of replication infidelity driven by imbalanced dNTP pools. Nucleic Acids Res. 2016, 44, 1669–1680. [Google Scholar] [CrossRef] [PubMed]
- Toledo, L.I.; Altmeyer, M.; Rask, M.-B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 2013, 155, 1088–1103. [Google Scholar] [CrossRef]
- Hereford, L.M.; Osley, M.A.; Ludwig, T.R.; McLaughlin, C.S. Cell-cycle regulation of yeast histone mRNA. Cell 1981, 24, 367–375. [Google Scholar] [CrossRef]
- Osley, M.A. The regulation of histone synthesis in the cell cycle. Annu. Rev. Biochem. 1991, 60, 827–861. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; McKillop-Smith, S.; Müller, B. The human histone gene expression regulator HBP/SLBP is required for histone and DNA synthesis, cell cycle progression and cell proliferation in mitotic cells. J. Cell Sci. 2004, 117, 6043–6051. [Google Scholar] [CrossRef] [PubMed]
- Mejlvang, J.; Feng, Y.; Alabert, C.; Neelsen, K.J.; Jasencakova, Z.; Zhao, X.; Lees, M.; Sandelin, A.; Pasero, P.; Lopes, M.; et al. New histone supply regulates replication fork speed and PCNA unloading. J. Cell Biol. 2014, 204, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Prado, F.; Aguilera, A. Partial depletion of histone H4 increases homologous recombination-mediated genetic instability. Mol. Cell. Biol. 2005, 25, 1526–1536. [Google Scholar] [CrossRef] [PubMed]
- Collins, N.; Poot, R.A.; Kukimoto, I.; García-Jiménez, C.; Dellaire, G.; Varga-Weisz, P.D. An ACF1-ISWI chromatin-remodeling complex is required for DNA replication through heterochromatin. Nat. Genet. 2002, 32, 627–632. [Google Scholar] [CrossRef]
- Schuster-Böckler, B.; Lehner, B. Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature 2012, 488, 504–507. [Google Scholar] [CrossRef]
- French, S. Consequences of replication fork movement through transcription units in vivo. Science 1992, 258, 1362–1365. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, A.M.; Newlon, C.S. DNA replication fork pause sites dependent on transcription. Science 1996, 272, 1030–1033. [Google Scholar] [CrossRef] [PubMed]
- Prado, F.; Aguilera, A. Impairment of replication fork progression mediates RNA polII transcription-associated recombination. EMBO J. 2005, 24, 1267–1276. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Million-Weaver, S.; Chattopadhyay, S.; Sokurenko, E.; Merrikh, H. Accelerated gene evolution through replication-transcription conflicts. Nature 2013, 495, 512–515. [Google Scholar] [CrossRef] [PubMed]
- Azvolinsky, A.; Giresi, P.G.; Lieb, J.D.; Zakian, V.A. Highly transcribed RNA polymerase II genes are impediments to replication fork progression in Saccharomyces cerevisiae. Mol. Cell 2009, 34, 722–734. [Google Scholar] [CrossRef]
- Helmrich, A.; Ballarino, M.; Tora, L. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell 2011, 44, 966–977. [Google Scholar] [CrossRef]
- Wansink, D.G.; Manders, E.E.; van der Kraan, I.; Aten, J.A.; van Driel, R.; de Jong, L. RNA polymerase II transcription is concentrated outside replication domains throughout S-phase. J. Cell Sci. 1994, 107 Pt 6, 1449–1456. [Google Scholar]
- Wang, J.D.; Berkmen, M.B.; Grossman, A.D. Genome-wide coorientation of replication and transcription reduces adverse effects on replication in Bacillus subtilis. Proc. Natl. Acad. Sci. USA 2007, 104, 5608–5613. [Google Scholar] [CrossRef] [PubMed]
- Huvet, M.; Nicolay, S.; Touchon, M.; Audit, B.; d’Aubenton-Carafa, Y.; Arneodo, A.; Thermes, C. Human gene organization driven by the coordination of replication and transcription. Genome Res. 2007, 17, 1278–1285. [Google Scholar] [CrossRef]
- Marsolier-Kergoat, M.-C.; Goldar, A. DNA replication induces compositional biases in yeast. Mol. Biol. Evol. 2012, 29, 893–904. [Google Scholar] [CrossRef] [PubMed]
- Sale, J.E. Translesion DNA synthesis and mutagenesis in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012708. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, E.C. Suffering in silence: The tolerance of DNA damage. Nat. Rev. Mol. Cell Biol. 2005, 6, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Plachta, M.; Halas, A.; McIntyre, J.; Sledziewska-Gojska, E. The steady-state level and stability of TLS polymerase eta are cell cycle dependent in the yeast S. cerevisiae. DNA Repair 2015, 29, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Seplyarskiy, V.B.; Bazykin, G.A.; Soldatov, R.A. Polymerase ζ activity is linked to replication timing in Humans: evidence from mutational signatures. Mol. Biol. Evol. 2015, 32, 3158–3172. [Google Scholar] [PubMed]
- Supek, F.; Lehner, B. Differential DNA mismatch repair underlies mutation rate variation across the human genome. Nature 2015, 521, 81–84. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaboriaud, J.; Wu, P.-Y.J. Insights into the Link between the Organization of DNA Replication and the Mutational Landscape. Genes 2019, 10, 252. https://doi.org/10.3390/genes10040252
Gaboriaud J, Wu P-YJ. Insights into the Link between the Organization of DNA Replication and the Mutational Landscape. Genes. 2019; 10(4):252. https://doi.org/10.3390/genes10040252
Chicago/Turabian StyleGaboriaud, Julia, and Pei-Yun Jenny Wu. 2019. "Insights into the Link between the Organization of DNA Replication and the Mutational Landscape" Genes 10, no. 4: 252. https://doi.org/10.3390/genes10040252
APA StyleGaboriaud, J., & Wu, P.-Y. J. (2019). Insights into the Link between the Organization of DNA Replication and the Mutational Landscape. Genes, 10(4), 252. https://doi.org/10.3390/genes10040252