H3K27me3 Depletion during Differentiation Promotes Myogenic Transcription in Porcine Satellite Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Isolation, Culture, and Differentiation of Porcine Satellite Cells (PSCs)
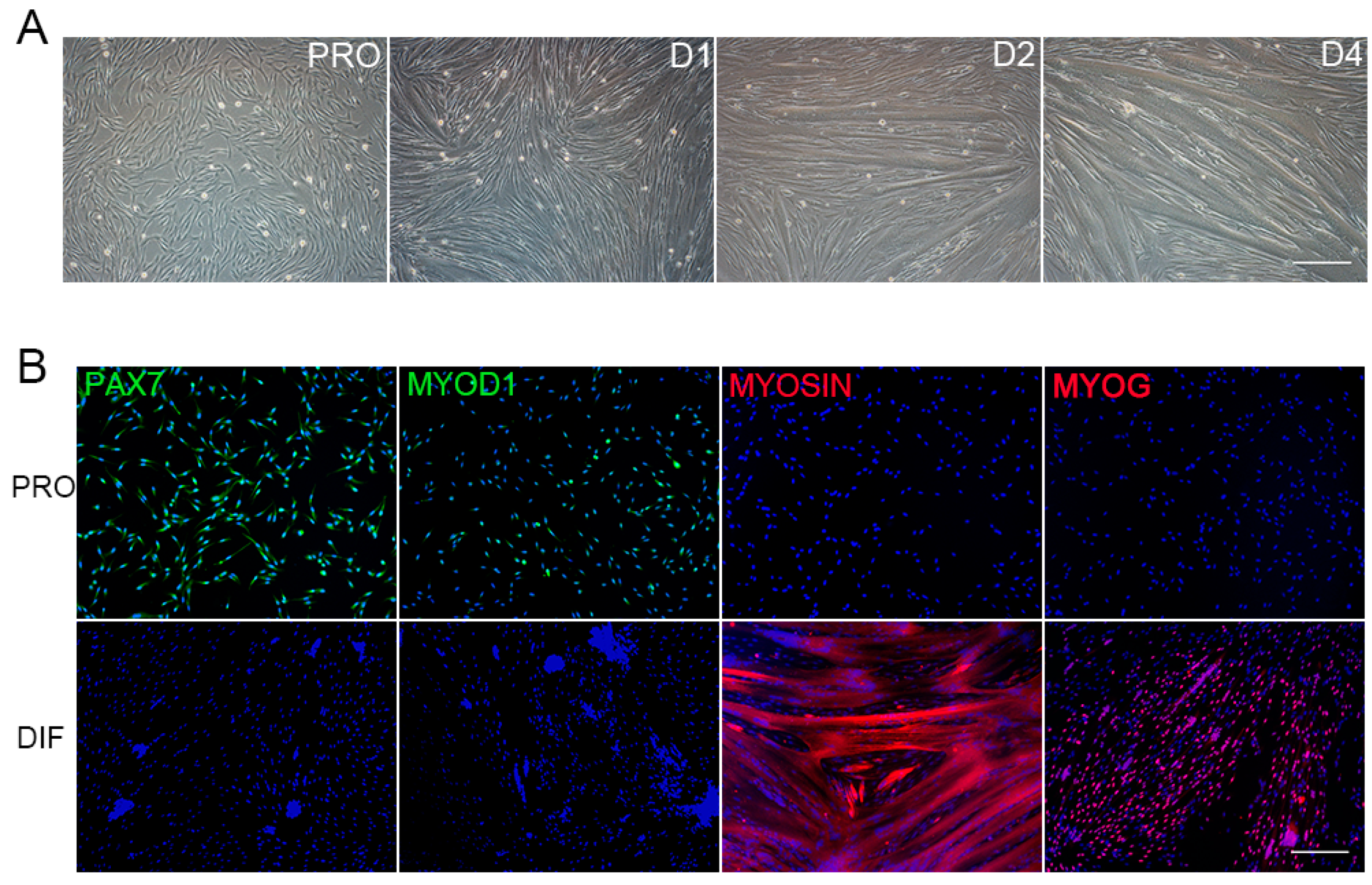
2.2. Immunofluorescence Assay of PSCs
2.3. Validation of Differentially Expressed Genes (DEGs) by Quantitative PCR (qPCR)
2.4. Chromatin Immunoprecipitation (ChIP) and ChIP-Seq Library Preparation
2.5. Library Construction and Sequencing
2.6. Data Preprocessing and Alignment
2.7. RNA-Seq Data Analysis
2.8. ChIP-Seq Data Analysis
2.9. Statistical Analysis
2.10. Data Availability
3. Results
3.1. The Morphology Characterization of PSCs
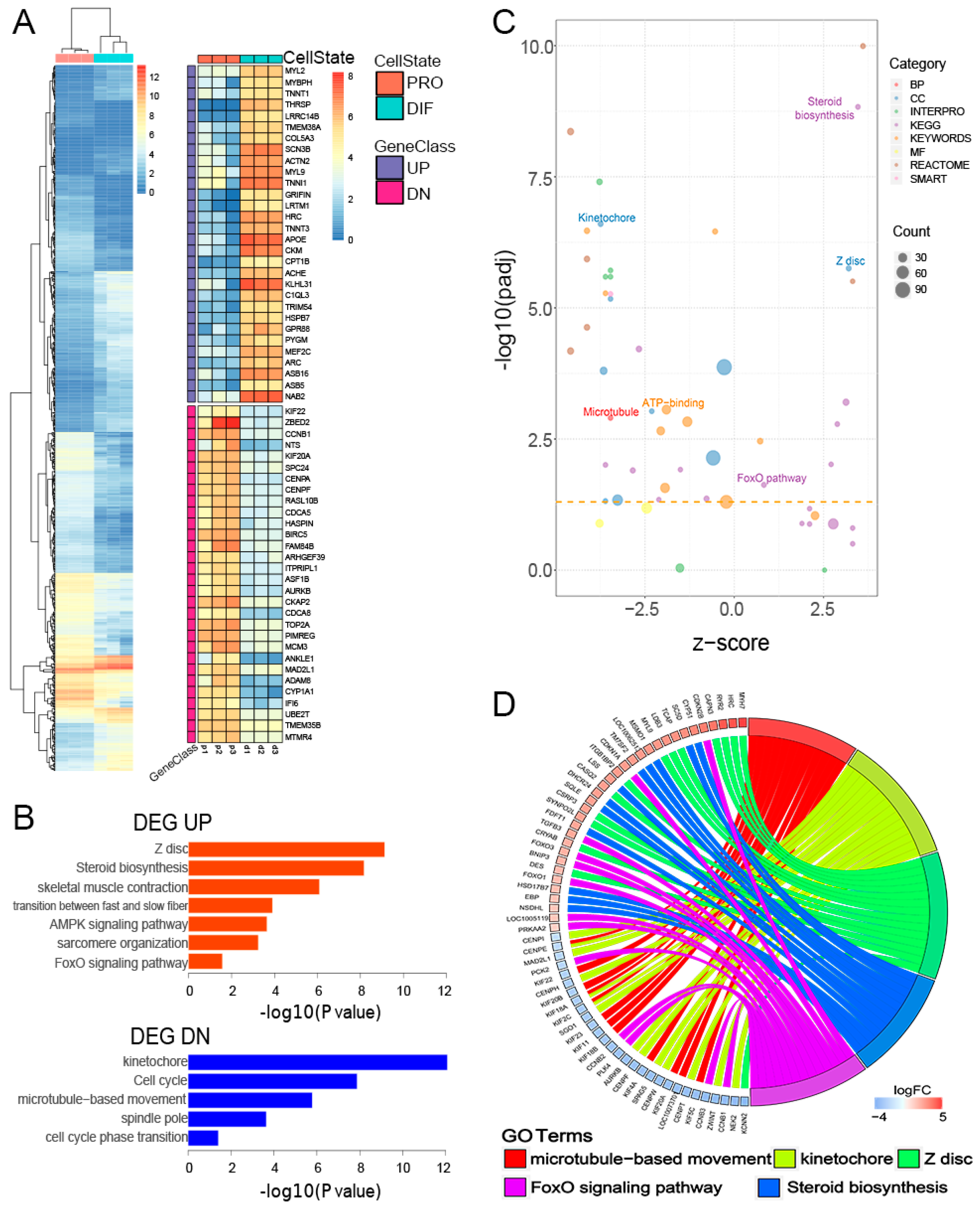
3.2. Characterization of Gene Transcription
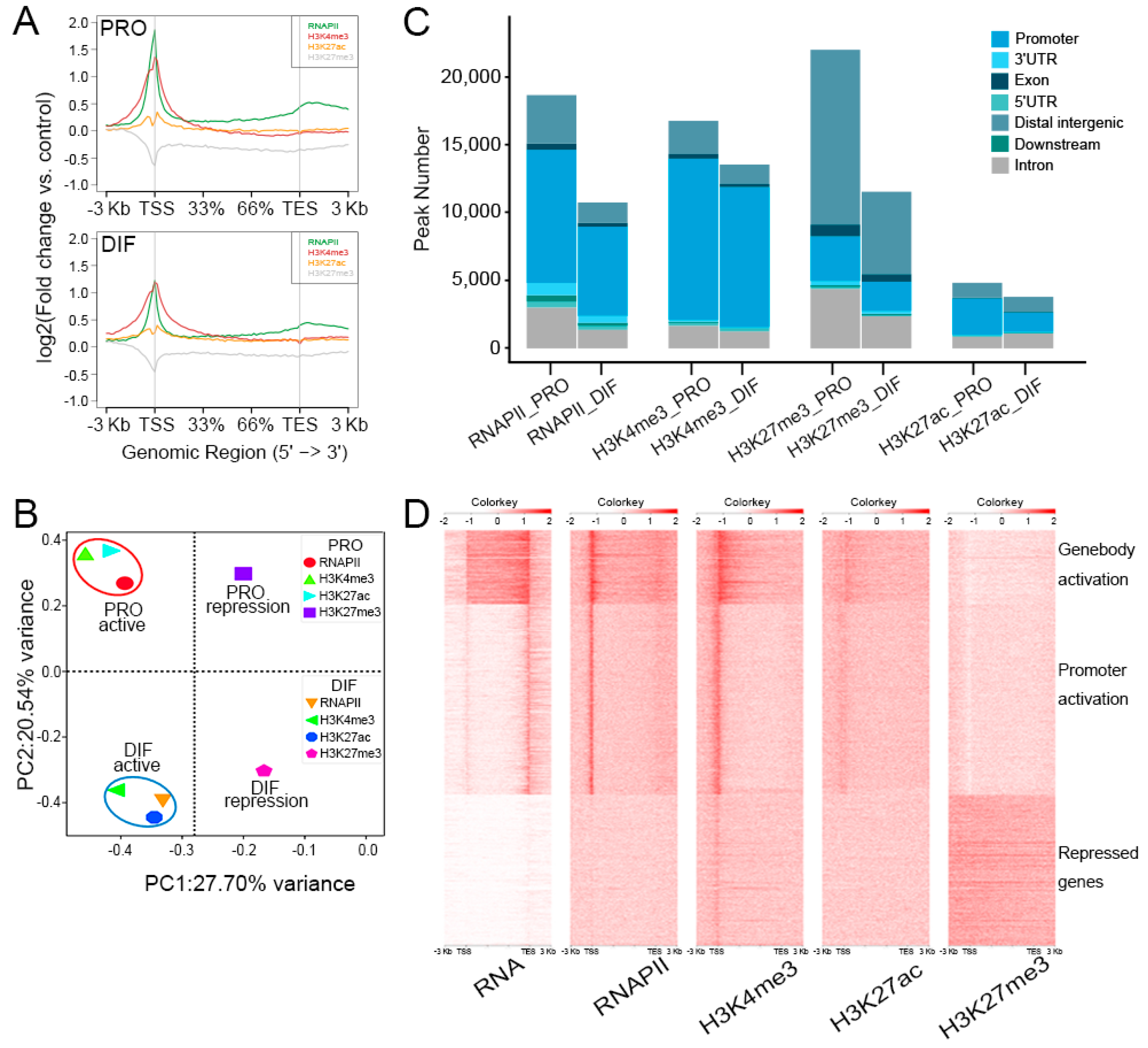
3.3. Epigenome Alteration in the Differentiation of PSCs
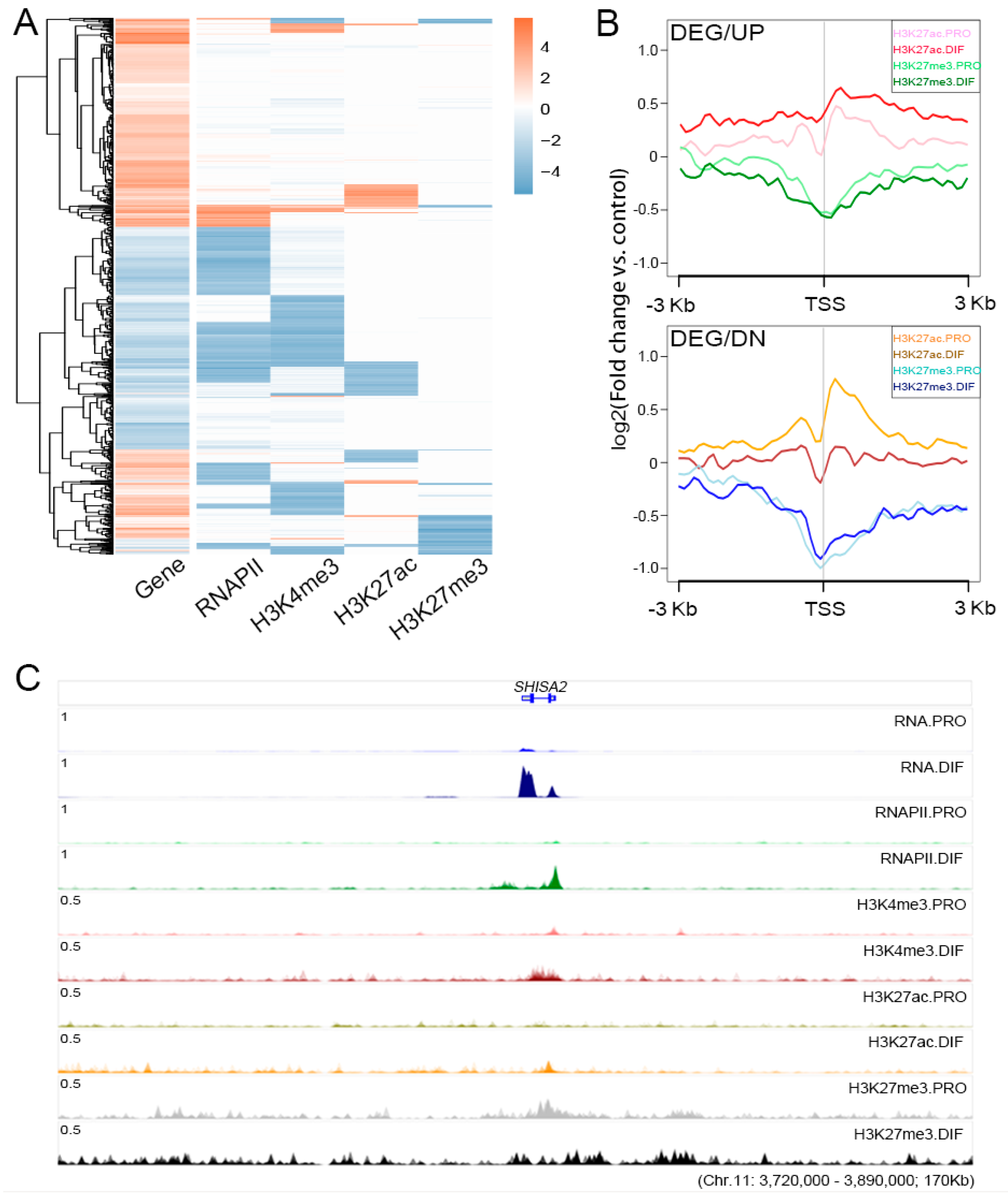
3.4. The Regulation Roles of Epigenetic Modification
3.5. The Roles of H3K27me3 During Cell Differentiation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Luo, W.; Chen, S.; Cheng, D.; Wang, L.; Li, Y.; Ma, X.; Song, X.; Liu, X.; Li, W.; Liang, J.; et al. Genome-wide association study of porcine hematological parameters in a Large White x Minzhu F2 resource population. Int. J. Biol. Sci. 2012, 8, 870–881. [Google Scholar] [CrossRef] [PubMed]
- Douglas, W.R. Of pigs and men and research: A review of applications and analogies of the pig, Sus scrofa, in human medical research. Space Life Sci. 1972, 3, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Boonsanay, V.; Zhang, T.; Georgieva, A.; Kostin, S.; Qi, H.; Yuan, X.; Zhou, Y.; Braun, T. Regulation of Skeletal Muscle Stem Cell Quiescence by Suv4-20h1-Dependent Facultative Heterochromatin Formation. Cell Stem Cell 2016, 18, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Wang, F.; Liu, Y.; Li, S.; Zhou, G.; Hu, P. Characterization and isolation of highly purified porcine satellite cells. Cell Death Discov. 2017, 3, 17003. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X.; Dumont, N.A.; Rudnicki, M.A. Muscle stem cells at a glance. J. Cell Sci. 2014, 127, 4543–4548. [Google Scholar] [CrossRef] [PubMed]
- Segales, J.; Perdiguero, E.; Munoz-Canoves, P. Epigenetic control of adult skeletal muscle stem cell functions. FEBS J. 2015, 282, 1571–1588. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Park, S.; Scheffler, J.M.; Kuang, S.; Grant, A.L.; Gerrard, D.E. Porcine satellite cells are restricted to a phenotype resembling their muscle origin. J. Anim. Sci. 2013, 91, 4684–4691. [Google Scholar] [CrossRef]
- Barreiro, E.; Tajbakhsh, S. Epigenetic regulation of muscle development. J. Muscle Res. Cell Motil. 2017, 38, 31–35. [Google Scholar] [CrossRef]
- Baquero-Perez, B.; Kuchipudi, S.V.; Nelli, R.K.; Chang, K.C. A simplified but robust method for the isolation of avian and mammalian muscle satellite cells. BMC Cell Biol. 2012, 13, 16. [Google Scholar] [CrossRef]
- Zammit, P.S. Function of the myogenic regulatory factors Myf5, MyoD, Myogenin and MRF4 in skeletal muscle, satellite cells and regenerative myogenesis. Semin. Cell Dev. Biol. 2017, 72, 19–32. [Google Scholar] [CrossRef]
- Liu, L.; Cheung, T.H.; Charville, G.W.; Hurgo, B.M.; Leavitt, T.; Shih, J.; Brunet, A.; Rando, T.A. Chromatin modifications as determinants of muscle stem cell quiescence and chronological aging. Cell Rep. 2013, 4, 189–204. [Google Scholar] [CrossRef]
- Sebastian, S.; Sreenivas, P.; Sambasivan, R.; Cheedipudi, S.; Kandalla, P.; Pavlath, G.K.; Dhawan, J. MLL5, a trithorax homolog, indirectly regulates H3K4 methylation, represses cyclin A2 expression, and promotes myogenic differentiation. Proc. Natl. Acad. Sci. USA 2009, 106, 4719–4724. [Google Scholar] [CrossRef]
- Asp, P.; Blum, R.; Vethantham, V.; Parisi, F.; Micsinai, M.; Cheng, J.; Bowman, C.; Kluger, Y.; Dynlacht, B.D. Genome-wide remodeling of the epigenetic landscape during myogenic differentiation. Proc. Natl. Acad. Sci. USA 2011, 108, E149–E158. [Google Scholar] [CrossRef]
- Dilworth, F.J.; Blais, A. Epigenetic regulation of satellite cell activation during muscle regeneration. Stem Cell Res. Ther. 2011, 2, 18. [Google Scholar] [CrossRef]
- Consalvi, S.; Brancaccio, A.; Dall’Agnese, A.; Puri, P.L.; Palacios, D. Praja1 E3 ubiquitin ligase promotes skeletal myogenesis through degradation of EZH2 upon p38α activation. Nat. Commun. 2017, 8, 13956. [Google Scholar] [CrossRef]
- Faralli, H.; Wang, C.; Nakka, K.; Benyoucef, A.; Sebastian, S.; Zhuang, L.; Chu, A.; Palii, C.G.; Liu, C.; Camellato, B.; et al. UTX demethylase activity is required for satellite cell-mediated muscle regeneration. J. Clin. Investig. 2016, 126, 1555–1565. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, G.; Ruan, X.; Auerbach, R.K.; Sandhu, K.S.; Zheng, M.; Wang, P.; Poh, H.M.; Goh, Y.; Lim, J.; Zhang, J.; et al. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell 2012, 148, 84–98. [Google Scholar] [CrossRef]
- Yu, G.C.; Wang, L.G.; Han, Y.Y.; He, Q.Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Walter, W.; Sanchez-Cabo, F.; Ricote, M. GOplot: An R package for visually combining expression data with functional analysis. Bioinformatics 2015, 31, 2912–2914. [Google Scholar] [CrossRef]
- The R Project for Statistical Computing. Available online: http://www.r-project.org/ (accessed on 11 March 2019).
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, F.; Ryan, D.P.; Gruning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dundar, F.; Manke, T. deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef]
- Yu, G.C.; Wang, L.G.; He, Q.Y. ChIPseeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015, 31, 2382–2383. [Google Scholar] [CrossRef] [PubMed]
- Ryall, J.G. Metabolic reprogramming as a novel regulator of skeletal muscle development and regeneration. FEBS J. 2013, 280, 4004–4013. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, C.; Liu, X.; Kuang, S. Shisa2 regulates the fusion of muscle progenitors. Stem Cell Res. 2018, 31, 31–41. [Google Scholar] [CrossRef]
- Jiao, Y.R.; Huang, B.; Chen, Y.; Hong, G.L.; Xu, J.; Hu, C.Y.; Wang, C. Integrated Analyses Reveal Overexpressed Notch1 Promoting Porcine Satellite Cells’ Proliferation through Regulating the Cell Cycle. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.Z.; Garry, D.J. Muscle stem cells in development, regeneration, and disease. Gene Dev. 2006, 20, 1692–1708. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.G.; Kim, J.; Yuan, X.J.; Braun, T. Epigenetic Modifications of Stem Cells A Paradigm for the Control of Cardiac Progenitor Cells. Circ. Res. 2011, 109, 1067–1081. [Google Scholar] [CrossRef] [PubMed]
- Agger, K.; Cloos, P.A.C.; Christensen, J.; Pasini, D.; Rose, S.; Rappsilber, J.; Issaeva, I.; Canaani, E.; Salcini, A.E.; Helin, K. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 2007, 449, 731–734. [Google Scholar] [CrossRef]
- Woodhouse, S.; Pugazhendhi, D.; Brien, P.; Pell, J.M. Ezh2 maintains a key phase of muscle satellite cell expansion but does not regulate terminal differentiation. J. Cell Sci. 2013, 126, 565–579. [Google Scholar] [CrossRef]
- Seenundun, S.; Rampalli, S.; Liu, Q.C.; Aziz, A.; Palii, C.; Hong, S.H.; Blais, A.; Brand, M.; Ge, K.; Dilworth, F.J. UTX mediates demethylation of H3K27me3 at muscle-specific genes during myogenesis. EMBO J. 2010, 29, 1401–1411. [Google Scholar] [CrossRef]
- Caretti, G.; Di Padova, M.; Micales, B.; Lyons, G.E.; Sartorelli, V. The Polycomb Ezh2 methyltransferase regulates muscle gene expression and skeletal muscle differentiation. Gene Dev. 2004, 18, 2627–2638. [Google Scholar] [CrossRef]
- Liu, L.; Rando, T.A. UTX in muscle regeneration - the right dose and the right time. J. Clin. Investig. 2016, 126, 1233–1235. [Google Scholar] [CrossRef] [PubMed]
- Chakroun, I.; Yang, D.B.; Girgis, J.; Gunasekharan, A.; Phenix, H.; Kaern, M.; Blais, A. Genome-wide association between Six4, MyoD, and the histone demethylase Utx during myogenesis. FASEB J. 2015, 29, 4738–4755. [Google Scholar] [CrossRef] [PubMed]
- Davis, F.P.; Eddy, S.R. Transcription Factors That Convert Adult Cell Identity Are Differentially Polycomb Repressed. PLoS ONE 2013, 8, e63407. [Google Scholar] [CrossRef]
- Li, X.; Wang, J.Q.; Jiang, Z.; Guo, F.; Soloway, P.D.; Zhao, R.Q. Role of PRDM16 and its PR domain in the epigenetic regulation of myogenic and adipogenic genes during transdifferentiation of C2C12 cells. Gene 2015, 570, 191–198. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Sun, Y.; Ren, R.; Xie, J.; Tian, X.; Zhao, S.; Li, X.; Cao, J. H3K27me3 Depletion during Differentiation Promotes Myogenic Transcription in Porcine Satellite Cells. Genes 2019, 10, 231. https://doi.org/10.3390/genes10030231
Wang S, Sun Y, Ren R, Xie J, Tian X, Zhao S, Li X, Cao J. H3K27me3 Depletion during Differentiation Promotes Myogenic Transcription in Porcine Satellite Cells. Genes. 2019; 10(3):231. https://doi.org/10.3390/genes10030231
Chicago/Turabian StyleWang, Sheng, Yan Sun, Ruimin Ren, Junhui Xie, Xiaohuan Tian, Shuhong Zhao, Xinyun Li, and Jianhua Cao. 2019. "H3K27me3 Depletion during Differentiation Promotes Myogenic Transcription in Porcine Satellite Cells" Genes 10, no. 3: 231. https://doi.org/10.3390/genes10030231
APA StyleWang, S., Sun, Y., Ren, R., Xie, J., Tian, X., Zhao, S., Li, X., & Cao, J. (2019). H3K27me3 Depletion during Differentiation Promotes Myogenic Transcription in Porcine Satellite Cells. Genes, 10(3), 231. https://doi.org/10.3390/genes10030231