Volatile Evolution of Long Non-Coding RNA Repertoire in Retinal Pigment Epithelium: Insights from Comparison of Bovine and Human RNA Expression Profiles

 ,
,  ,
, 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Datasets and Cell Cultures
2.2. Reconstruction and Analysis of Expression of Novel Bovine Long Non-Coding RNAs (lncRNAs)
2.3. Reconstruction of Putative Orthologous Regions in the Human Genome
2.4. Analysis of Expression of Putative Human lncRNA Genes
2.5. Confirmation of lncRNA Transcript Sequences
3. Results
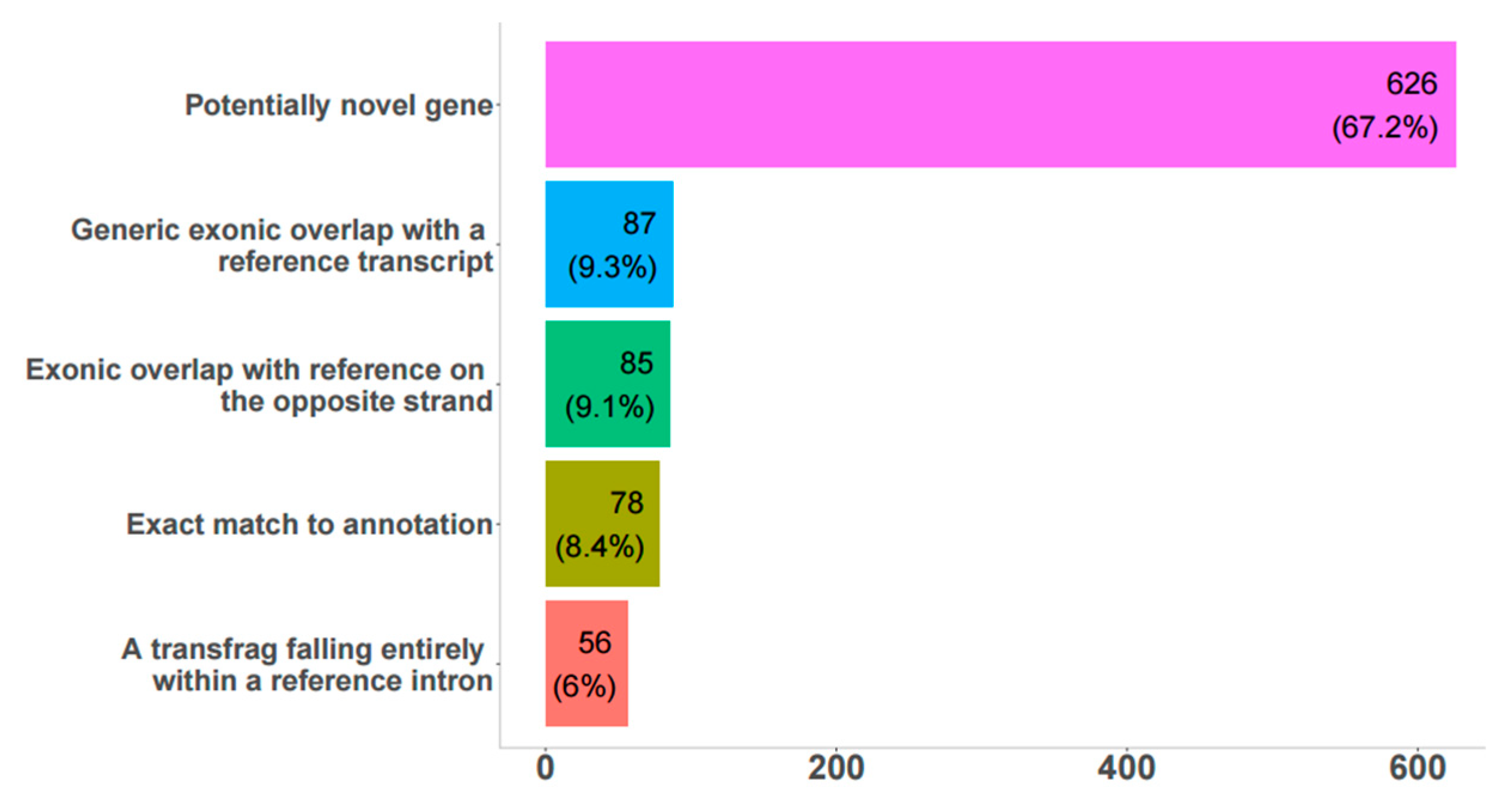
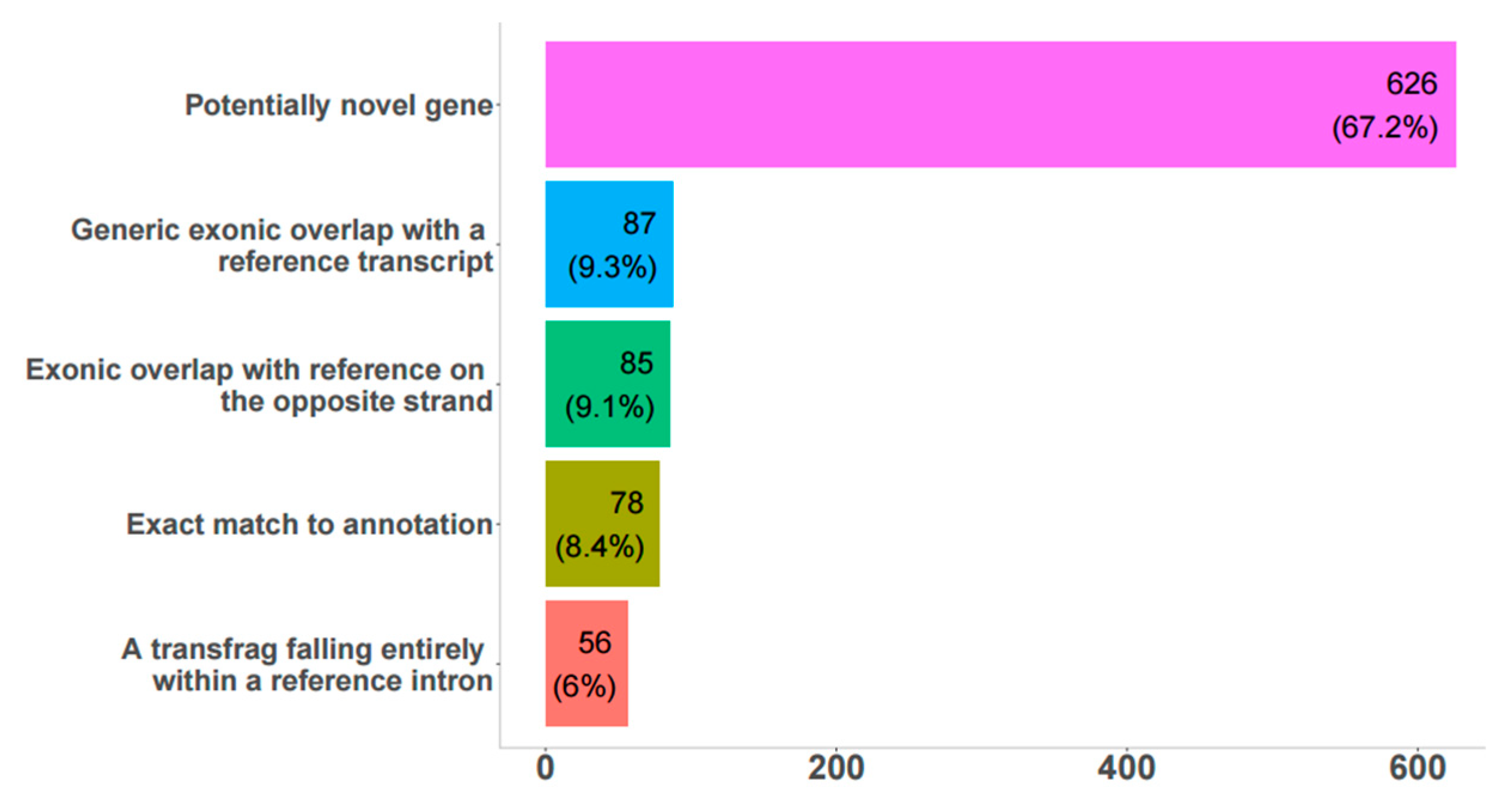
3.1. Reconstruction and Analysis of Expression of Novel Bovine lncRNAs
3.2. Reconstruction and Analysis of Expression of Putative Orthologous Human lncRNA Genes
3.3. Experimental Verification of Novel Bovine and Human lncRNAs.
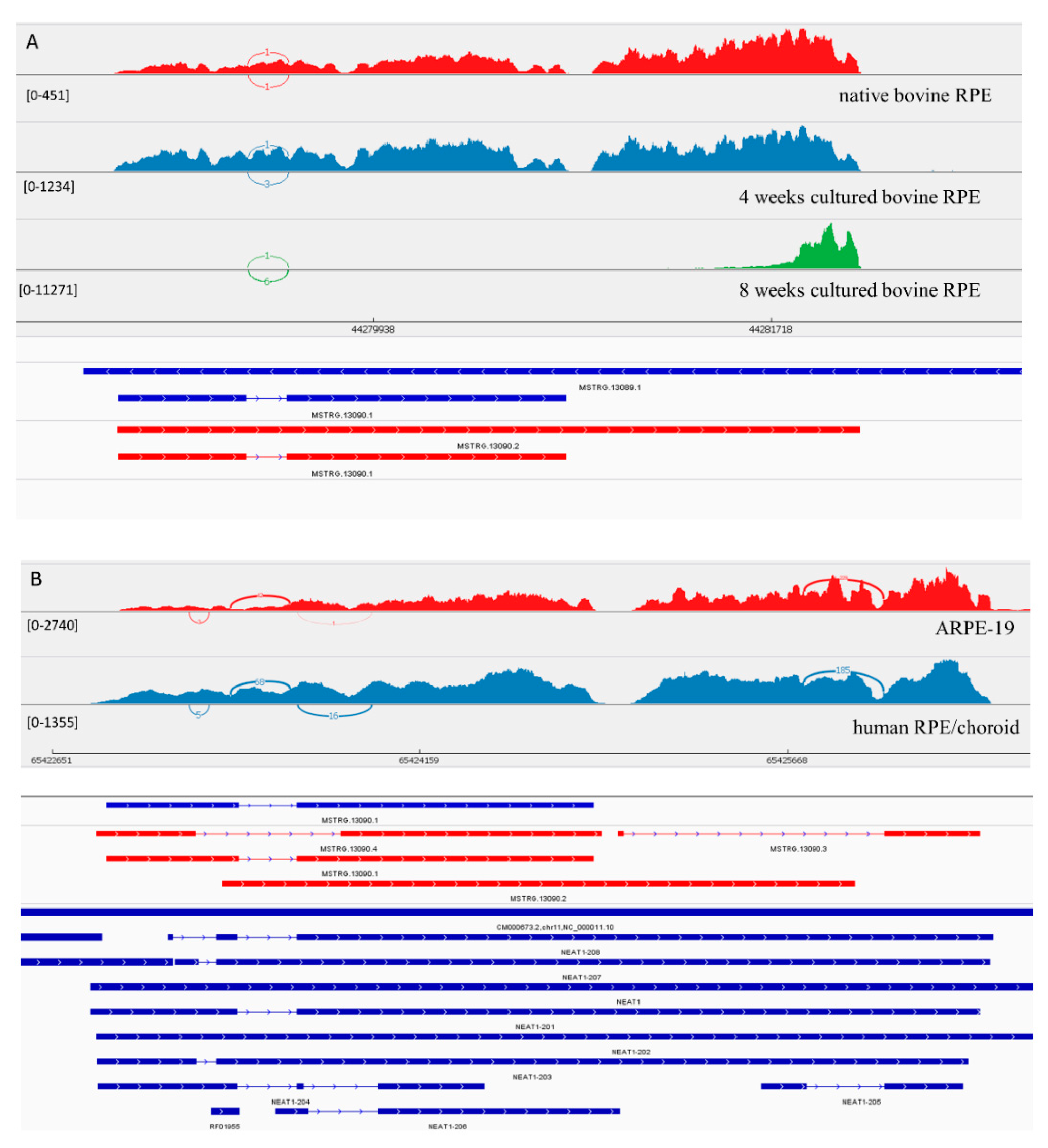
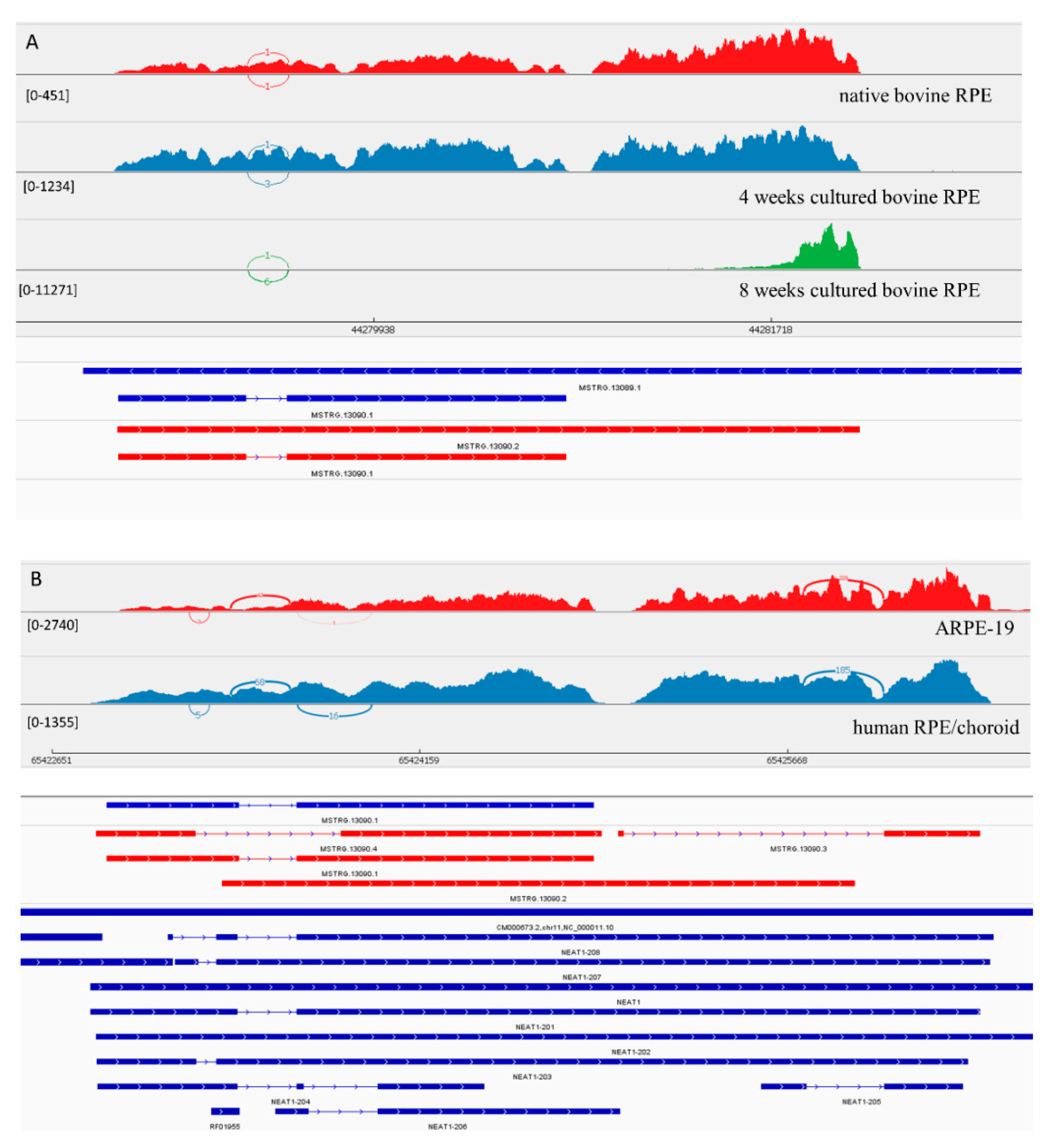
3.3.1. LncRNA Gene: NEAT1
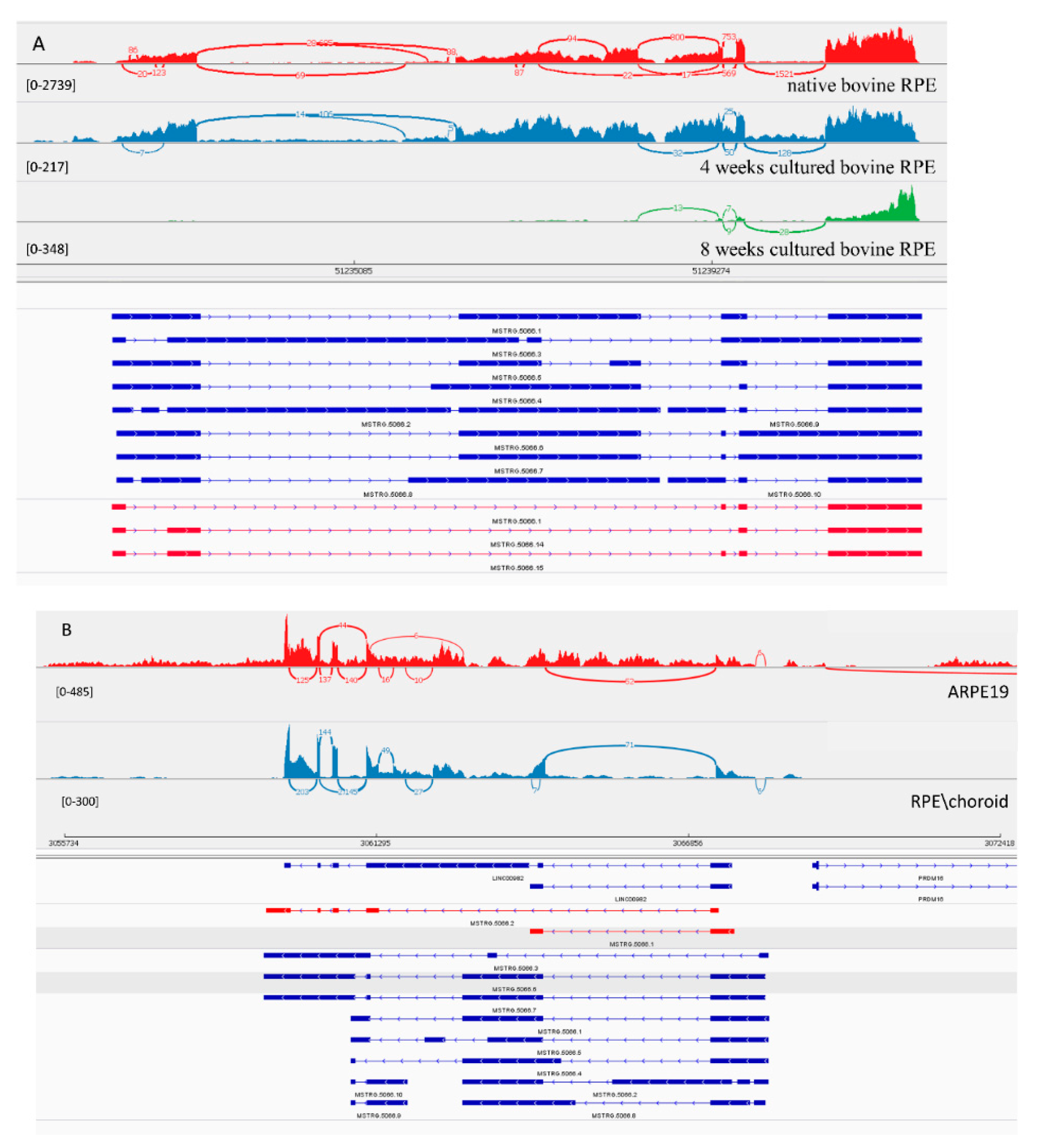
3.3.2. LncRNA Gene: MSTRG.5066
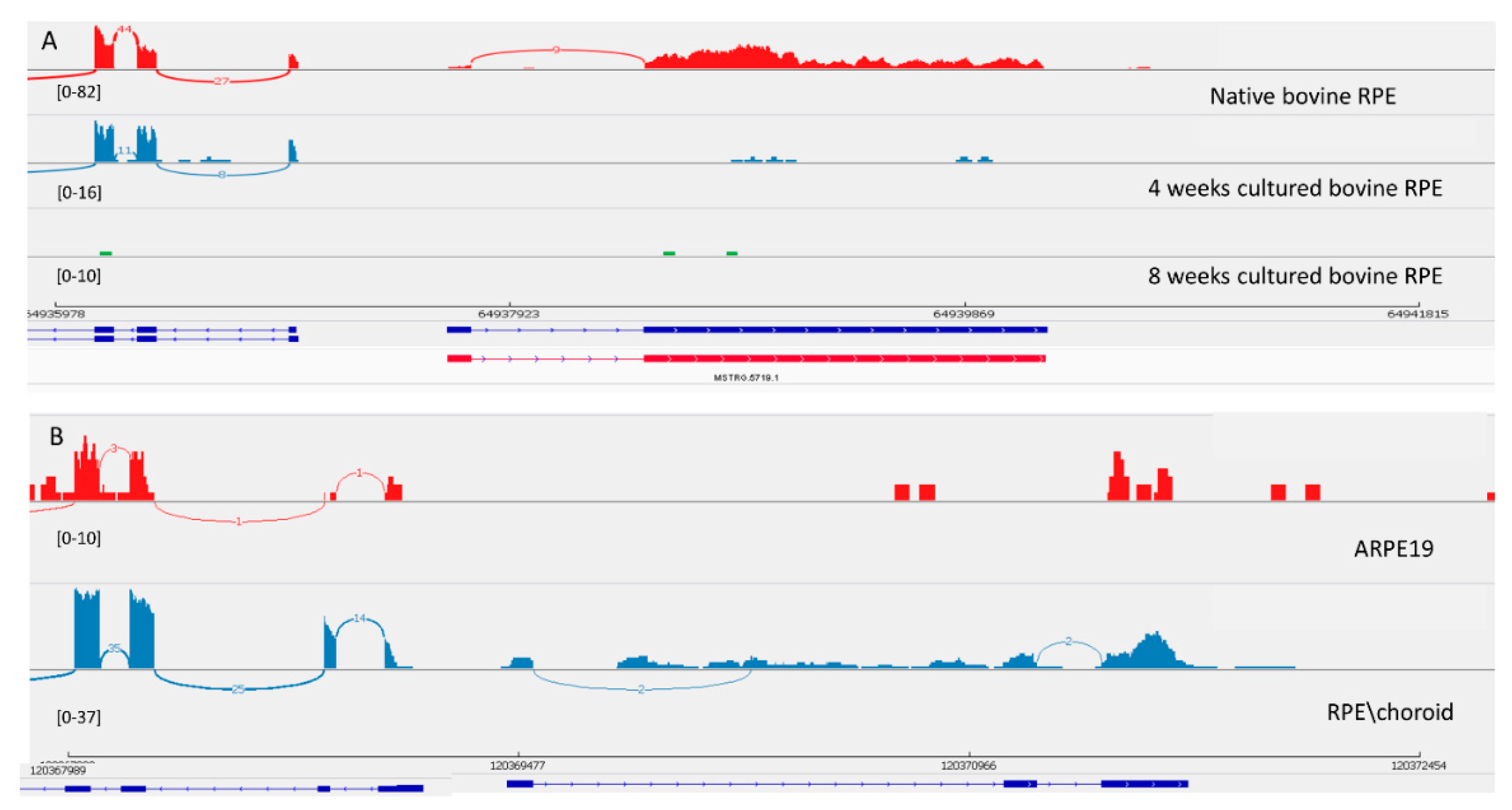
3.3.3. LncRNA Gene: MSTRG.5719
3.3.4. LncRNA Gene: MSTRG.1909 (Linc1833)
3.3.5. LncRNA Gene: MSTRG.2517 (Linc00094)
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [PubMed]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011, 25, 1915–1927. [Google Scholar] [CrossRef] [PubMed]
- Bertone, P.; Stolc, V.; Royce, T.E.; Rozowsky, J.S.; Urban, A.E.; Zhu, X.; Rinn, J.L.; Tongprasit, W.; Samanta, M.; Weissman, S.; et al. Global identification of human transcribed sequences with genome tiling arrays. Science 2004, 306, 2242–2246. [Google Scholar] [CrossRef] [PubMed]
- Mattick, J.S.; Makunin, I.V. Non-coding RNA. Hum. Mol. Genet. 2006, 15, R17–R29. [Google Scholar] [CrossRef]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Ponjavic, J.; Ponting, C.P.; Lunter, G. Functionality or transcriptional noise? Evidence for selection within long noncoding RNAs. Genome Res. 2007, 17, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Ponting, C.P.; Belgard, T.G. Transcribed dark matter: Meaning or myth? Hum. Mol. Genet. 2010, 19, R162–R168. [Google Scholar] [CrossRef] [PubMed]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and functions of long noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Rivea Morales, D.; Thomas, K.; Presser, A.; Bernstein, B.E.; van Oudenaarden, A.; et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–11672. [Google Scholar] [CrossRef]
- Robinson, R. Dark matter transcripts: Sound and fury, signifying nothing? PLoS Biol. 2010, 8, e1000370. [Google Scholar] [CrossRef]
- Van Bakel, H.; Hughes, T.R. Establishing legitimacy and function in the new transcriptome. Brief Funct. Genom. Proteom. 2009, 8, 424–436. [Google Scholar] [CrossRef] [PubMed]
- Marques, A.C.; Ponting, C.P. Catalogues of mammalian long noncoding RNAs: Modest conservation and incompleteness. Genome Biol. 2009, 10, R124. [Google Scholar] [CrossRef] [PubMed]
- Siepel, A.; Bejerano, G.; Pedersen, J.S.; Hinrichs, A.S.; Hou, M.; Rosenbloom, K.; Clawson, H.; Spieth, J.; Hillier, L.W.; Richards, S.; et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005, 15, 1034–1050. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef]
- Managadze, D.; Rogozin, I.B.; Chernikova, D.; Shabalina, S.A.; Koonin, E.V. Negative correlation between expression level and evolutionary rate of long intergenic noncoding RNAs. Genome Biol. Evol. 2011, 3, 1390–1404. [Google Scholar] [CrossRef] [PubMed]
- Huttenhofer, A.; Vogel, J. Experimental approaches to identify non-coding RNAs. Nucl. Acids Res. 2006, 34, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Amaral, P.P.; Dinger, M.E.; Mattick, J.S. Non-coding RNAs in homeostasis, disease and stress responses: An evolutionary perspective. Brief Funct. Genom. 2013, 12, 254–278. [Google Scholar] [CrossRef]
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C.; et al. The transcriptional landscape of the mammalian genome. Science 2005, 309, 1559–1563. [Google Scholar]
- Liu, J.; Gough, J.; Rost, B. Distinguishing protein-coding from non-coding RNAs through support vector machines. PLoS Genet 2006, 2, e29. [Google Scholar] [CrossRef]
- Ji, Z.; Song, R.; Regev, A.; Struhl, K. Many lncRNAs, 5′UTRs, and pseudogenes are translated and some are likely to express functional proteins. Elife 2015, 4, e08890. [Google Scholar] [CrossRef]
- Managadze, D.; Lobkovsky, A.E.; Wolf, Y.I.; Shabalina, S.A.; Rogozin, I.B.; Koonin, E.V. The vast, conserved mammalian lincRNome. PLoS Comput. Biol. 2013, 9, e1002917. [Google Scholar] [CrossRef] [PubMed]
- Chodroff, R.A.; Goodstadt, L.; Sirey, T.M.; Oliver, P.L.; Davies, K.E.; Green, E.D.; Molnar, Z.; Ponting, C.P. Long noncoding RNA genes: Conservation of sequence and brain expression among diverse amniotes. Genome Biol. 2010, 11, R72. [Google Scholar] [CrossRef]
- Necsulea, A.; Soumillon, M.; Warnefors, M.; Liechti, A.; Daish, T.; Zeller, U.; Baker, J.C.; Grutzner, F.; Kaessmann, H. The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature 2014, 505, 635–640. [Google Scholar] [CrossRef]
- Ulitsky, I.; Shkumatava, A.; Jan, C.H.; Sive, H.; Bartel, D.P. Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell 2011, 147, 1537–1550. [Google Scholar] [CrossRef] [PubMed]
- Washietl, S.; Kellis, M.; Garber, M. Evolutionary dynamics and tissue specificity of human long noncoding RNAs in six mammals. Genome Res. 2014, 24, 616–628. [Google Scholar] [CrossRef] [PubMed]
- Chernikova, D.; Managadze, D.; Glazko, G.V.; Makalowski, W.; Rogozin, I.B. Conservation of the Exon-Intron Structure of Long Intergenic Non-Coding RNA Genes in Eutherian Mammals. Life (Basel) 2016, 6, 27. [Google Scholar] [CrossRef] [PubMed]
- Kapusta, A.; Feschotte, C. Volatile evolution of long noncoding RNA repertoires: Mechanisms and biological implications. Trends Genet. 2014, 30, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Mustafi, D.; Kevany, B.M.; Bai, X.; Maeda, T.; Sears, J.E.; Khalil, A.M.; Palczewski, K. Evolutionarily conserved long intergenic non-coding RNAs in the eye. Hum. Mol. Genet. 2013, 22, 2992–3002. [Google Scholar] [CrossRef]
- Au, E.D.; Fernandez-Godino, R.; Kaczynksi, T.J.; Sousa, M.E.; Farkas, M.H. Characterization of lincRNA expression in the human retinal pigment epithelium and differentiated induced pluripotent stem cells. PLoS ONE 2017, 12, e0183939. [Google Scholar] [CrossRef]
- Wan, P.; Su, W.; Zhuo, Y. Precise long non-coding RNA modulation in visual maintenance and impairment. J. Med. Genet. 2017, 54, 450–459. [Google Scholar] [CrossRef]
- Rapicavoli, N.A.; Poth, E.M.; Blackshaw, S. The long noncoding RNA RNCR2 directs mouse retinal cell specification. BMC Dev. Biol. 2010, 10, 49. [Google Scholar] [CrossRef] [PubMed]
- Krol, J.; Krol, I.; Alvarez, C.P.; Fiscella, M.; Hierlemann, A.; Roska, B.; Filipowicz, W. A network comprising short and long noncoding RNAs and RNA helicase controls mouse retina architecture. Nat. Commun. 2015, 6, 7305. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Yang, S.; Zhou, Z.; Zhao, X.; Zhong, J.; Reinach, P.S.; Yan, D. The Long Noncoding RNA Landscape of the Mouse Eye. Invest. Ophthalmol. Vis. Sci. 2017, 58, 6308–6317. [Google Scholar] [CrossRef] [PubMed]
- Samuel, W.; Jaworski, C.; Postnikova, O.A.; Kutty, R.K.; Duncan, T.; Tan, L.X.; Poliakov, E.; Lakkaraju, A.; Redmond, T.M. Appropriately differentiated ARPE-19 cells regain phenotype and gene expression profiles similar to those of native RPE cells. Mol. Vis. 2017, 23, 60–89. [Google Scholar] [PubMed]
- Whitmore, S.S.; Wagner, A.H.; DeLuca, A.P.; Drack, A.V.; Stone, E.M.; Tucker, B.A.; Zeng, S.; Braun, T.A.; Mullins, R.F.; Scheetz, T.E. Transcriptomic analysis across nasal, temporal, and macular regions of human neural retina and RPE/choroid by RNA-Seq. Exp. Eye Res. 2014, 129, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Wucher, V.; Legeai, F.; Hedan, B.; Rizk, G.; Lagoutte, L.; Leeb, T.; Jagannathan, V.; Cadieu, E.; David, A.; Lohi, H.; et al. FEELnc: A tool for long non-coding RNA annotation and its application to the dog transcriptome. Nucl. Acids Res. 2017, 45, e57. [Google Scholar] [CrossRef]
- Goecks, J.; Nekrutenko, A.; Taylor, J.; Galaxy, T. Galaxy: A comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010, 11, R86. [Google Scholar] [CrossRef]
- Prakash, A.; Tompa, M. Measuring the accuracy of genome-size multiple alignments. Genome Biol. 2007, 8, R124. [Google Scholar] [CrossRef]
- Rogozin, I.B.; D’Angelo, D.; Milanesi, L. Protein-coding regions prediction combining similarity searches and conservative evolutionary properties of protein-coding sequences. Gene 1999, 226, 129–137. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D.; Cieply, B.; Carstens, R.; Ramamurthy, V.; Stoilov, P. The Musashi 1 controls the splicing of photoreceptor-specific exons in the vertebrate retina. PLoS Genet. 2016, 12, e1006256. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, J.N.; Ensminger, A.W.; Clemson, C.M.; Lynch, C.R.; Lawrence, J.B.; Chess, A. A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genom. 2007, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Johnsson, P.; Lipovich, L.; Grander, D.; Morris, K.V. Evolutionary conservation of long non-coding RNAs; sequence, structure, function. Biochim. Biophys. Acta 2014, 1840, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.B.; Johnston, R.L.; Inostroza-Ponta, M.; Fox, A.H.; Fortini, E.; Moscato, P.; Dinger, M.E.; Mattick, J.S. Genome-wide analysis of long noncoding RNA stability. Genome Res. 2012, 22, 885–898. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Rinn, J.L. Modular regulatory principles of large non-coding RNAs. Nature 2012, 482, 339–346. [Google Scholar] [CrossRef]
- Novikova, I.V.; Hennelly, S.P.; Sanbonmatsu, K.Y. Structural architecture of the human long non-coding RNA, steroid receptor RNA activator. Nucl. Acids Res. 2012, 40, 5034–5051. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Set | Reads/Library | Description | Publication |
|---|---|---|---|
| Nasal, temporal, and macular regions of human retinal pigment epithelium (RPE)/choroid | 2 × 100 bp Paired-end stranded | Retina and RPE/choroid from the temporal, macular, and nasal regions of four clinically normal human donor eyes | Whitmore et al. 2014 [35] |
| Human ARPE-19 cells | 2 × 50 bp Paired end rRNA depleted | ARPE-19 cells cultured for 4 months | Samuel et al. 2017 [34] |
| Bovine RPE cells | 2 × 50 bp Paired end rRNA depleted | Fresh native bovine RPE; bovine RPE cultured for 4 and 8 weeks | This study |
| Native Retina/RPE | Bovine 4 Weeks Culture/Fresh RPE | Bovine 8 Weeks Culture/Fresh RPE | ARPE19 Cells 4D/4M | Human | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene Name | Base Mean | Fold Change | padj | Base Mean | Fold Change | padj | Base Mean | Fold Change | padj | Base Mean | Fold Change | padj | Annotation |
| MSTRG.13090 | 6175.61 | 2.43 | 7.45 × 10−31 | 9016.25 | 1.87 | 8.01 × 10−15 | 3028.53 | 1.42 | 1.01 × 10−3 | 0.64 | 0.12 | NEAT1 | |
| MSTRG.13096 | 5.99 | 1.64 | 2.18 × 10−3 | 47.69 | 2.79 | 2.44 × 10−6 | 333.50 | 6.99 | 2.04 × 10−43 | 0.64 | 1.31 | NEAT1 | |
| MSTRG.5066 | 2160.63 | 7.28 | 1.38 × 10−49 | 73.54 | −3.38 | 1.15 × 10−11 | 30.64 | −3.65 | 5.59 × 10−6 | 166.77 | −4.29 | 9.70 × 10−42 | linc00982 |
| MSTRG.5821 | 5222.03 | −0.52 | 4.94 × 10−4 | 351.11 | −0.21 | 5.20 × 10−1 | 111.81 | −1.34 | 3.96 × 10−4 | 2399.04 | −0.22 | 6.12 × 10−3 | TUG1 |
| MSTRG.2517 | 212.94 | −2.91 | 5.43 × 10−5 | 555.27 | −1.79 | 9.19 × 10−10 | 239.46 | −1.70 | 5.02 × 10−10 | 1087.27 | −0.15 | 2.24 × 10−1 | LINC000094 |
| MSTRG.1397 | 55.94 | 0.21 | 6.69 × 10−1 | 5.07 | −4.88 | 1.55 × 10−3 | 18.75 | −5.98 | 3.84 × 10−6 | 15.11 | −2.33 | 5.31 × 10−3 | OTX2-AS1 |
| MSTRG.1909 | 130.71 | 0.29 | 2.35 × 10−1 | 17.39 | −1.98 | 3.23 × 10−2 | 28.53 | −4.03 | 4.62 × 10−6 | 45.83 | 7.83 | 4.05 × 10−13 | LINC01833 |
| MSTRG.14528 | 599.24 | −1.30 | 7.77 × 10−16 | 153.68 | −1.22 | 9.62 × 10−5 | 88.45 | −0.08 | 8.86 × 10−1 | 0.00 | MAGI2-AS | ||
| MSTRG.5719 | 236.41 | −3.93 | 6.49 × 10−20 | 357.87 | −5.51 | 5.83 × 10−46 | 151.93 | −5.61 | 5.85 × 10−20 | 0.00 | RPS27P25-202 | ||
| MSTRG.19295 | 56.36 | 0.08 | 7.79 × 10−1 | 674.42 | 1.22 | 1.00 × 10−3 | 20.14 | 2.93 | 2.25 × 10−5 | 0.50 | 0.48 | FTX | |
| MSTRG.9718 | 54.45 | −0.61 | 6.41 × 10−1 | 76.35 | 5.31 | 6.25 × 10−7 | 2239.60 | −12.44 | 1.47 × 10−42 | MEG3 | |||
| MSTRG.18250 | 23.79 | −0.25 | 8.50 × 10−1 | 215.43 | 2.81 | 5.71 × 10−12 | 0.00 | ZFAS1 | |||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Postnikova, O.A.; Rogozin, I.B.; Samuel, W.; Nudelman, G.; Babenko, V.N.; Poliakov, E.; Redmond, T.M. Volatile Evolution of Long Non-Coding RNA Repertoire in Retinal Pigment Epithelium: Insights from Comparison of Bovine and Human RNA Expression Profiles. Genes 2019, 10, 205. https://doi.org/10.3390/genes10030205
Postnikova OA, Rogozin IB, Samuel W, Nudelman G, Babenko VN, Poliakov E, Redmond TM. Volatile Evolution of Long Non-Coding RNA Repertoire in Retinal Pigment Epithelium: Insights from Comparison of Bovine and Human RNA Expression Profiles. Genes. 2019; 10(3):205. https://doi.org/10.3390/genes10030205
Chicago/Turabian StylePostnikova, Olga A., Igor B. Rogozin, William Samuel, German Nudelman, Vladimir N. Babenko, Eugenia Poliakov, and T. Michael Redmond. 2019. "Volatile Evolution of Long Non-Coding RNA Repertoire in Retinal Pigment Epithelium: Insights from Comparison of Bovine and Human RNA Expression Profiles" Genes 10, no. 3: 205. https://doi.org/10.3390/genes10030205
APA StylePostnikova, O. A., Rogozin, I. B., Samuel, W., Nudelman, G., Babenko, V. N., Poliakov, E., & Redmond, T. M. (2019). Volatile Evolution of Long Non-Coding RNA Repertoire in Retinal Pigment Epithelium: Insights from Comparison of Bovine and Human RNA Expression Profiles. Genes, 10(3), 205. https://doi.org/10.3390/genes10030205