Beyond Biodiversity: Can Environmental DNA (eDNA) Cut It as a Population Genetics Tool?

 ,
,  , ,
, ,
Abstract
:1. Introduction
Why Use eDNA over Direct Sampling for Population Genetics?
2. Environmental DNA for Population Genetics
3. Challenges Facing Environmental DNA Population Genetics
4. Further Developments and Future Technologies
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lenormand, T. Gene flow and the limits to natural selection. Trends Ecol. Evol. 2002, 17, 183–189. [Google Scholar] [CrossRef]
- McGuire, J.M.; Scribner, K.T.; Congdon, J.D. Spatial aspects of movements, mating patterns, and nest distributions influence gene flow among population subunits of Blanding’s turtles (Emydoidea blandingii). Conserv. Genet. 2013, 14, 1029–1042. [Google Scholar] [CrossRef]
- Epps, C.W.; Palsbøll, P.J.; Wehausen, J.D.; Roderick, G.K.; Ramey, R.R.; McCullough, D.R. Highways block gene flow and cause a rapid decline in genetic diversity of desert bighorn sheep. Ecol. Lett. 2005, 8, 1029–1038. [Google Scholar] [CrossRef]
- McGuigan, K. Studying phenotypic evolution using multivariate quantitative genetics. Mol. Ecol. 2006, 15, 883–896. [Google Scholar] [CrossRef] [PubMed]
- Lande, R.; Arnold, S.J. The measurement of selection on correlated characters. Evolution. 1983, 37, 1210–1226. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-I. The genic view of the process of speciation. J. Evol. Biol. 2001, 14, 851–865. [Google Scholar] [CrossRef]
- Keller, L.F.; Waller, D.M. Inbreeding effects in wild populations. Trends Ecol. Evol. 2002, 17, 230–241. [Google Scholar] [CrossRef]
- Keller, L.F. Inbreeding and its fitness effects in an insular population of song sparrows (Melospiza melodia). Evolution. 1998, 52, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Mills, L.S.; Smouse, P.E. Demographic consequences of inbreeding in remnant populations. Am. Nat. 1994, 144, 412–431. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A.; Shapiro, B.; Pybus, O.G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 2005, 22, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Dussex, N.; Robertson, B.C.; Salis, A.T.; Kalinin, A.; Best, H.; Gemmell, N.J. Low spatial genetic differentiation associated with rapid recolonization in the New Zealand fur seal Arctocephalus forsteri. J. Hered. 2016, 107, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, M.L. Minimum population sizes for species conservation. Bioscience 1981, 31, 131–134. [Google Scholar] [CrossRef]
- Gemmell, N.J.; Burg, T.M.; Boyd, I.L.; Amos, W. Low reproductive success in territorial male Antarctic fur seals (Arctocephalus gazella) suggests the existence of alternative mating strategies. Mol. Ecol. 2001, 10, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Erwin, T.L. Beetles and other insects of tropical forest canopies at Manaus, Brazil, sampled by insecticidal fogging. Trop. Rain For. Ecol. Manag. 1983, 2, 59–75. [Google Scholar]
- Bearzi, G. First report of a common dolphin (Delphinus delphis) death following penetration of a biopsy dart. J. Cetacean Res. Manag. 2000, 2, 217–221. [Google Scholar]
- Cassone, B.J.; Boulding, E.G. Genetic structure and phylogeography of the lined shore crab, Pachygrapsus crassipes, along the northeastern and western Pacific coasts. Mar. Biol. 2006, 149, 213–226. [Google Scholar] [CrossRef]
- Romero, L.M.; Reed, J.M. Collecting baseline corticosterone samples in the field: is under 3 min good enough? Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2005, 140, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, T.W.; Janečka, J.E. Applications and techniques for non-invasive faecal genetics research in felid conservation. Eur. J. Wildl. Res. 2013, 59, 1–16. [Google Scholar] [CrossRef]
- Jerde, C.L.; Mahon, A.R.; Chadderton, W.L.; Lodge, D.M. “Sight-unseen” detection of rare aquatic species using environmental DNA. Conserv. Lett. 2011, 4, 150–157. [Google Scholar] [CrossRef]
- Greenbaum, E. A practical and safe method for the capture and restraint of venomous snakes in the field. Bull. Chic. Herpetol. Soc 2003, 38, 1–3. [Google Scholar]
- Ivanyi, C.; Altimari, W. Venomous reptile bites in academic research. Herpetol. Rev. 2004, 35, 49–50. [Google Scholar]
- Sigsgaard, E.E.; Carl, H.; Møller, P.R.; Thomsen, P.F. Monitoring the near-extinct European weather loach in Denmark based on environmental DNA from water samples. Biol. Conserv. 2015, 183, 46–52. [Google Scholar] [CrossRef]
- Bailey, L.L.; Mackenzie, D.I.; Nichols, J.D. Advances and applications of occupancy models. Methods Ecol. Evol. 2014, 5, 1269–1279. [Google Scholar] [CrossRef]
- MacKenzie, D.I.; Nichols, J.D.; Sutton, N.; Kawanishi, K.; Bailey, L.L. Improving inferences in population studies of rare species that are detected imperfectly. Ecology 2005, 86, 1101–1113. [Google Scholar] [CrossRef]
- Gu, W.; Swihart, R.K. Absent or undetected? Effects of non-detection of species occurrence on wildlife–habitat models. Biol. Conserv. 2004, 116, 195–203. [Google Scholar] [CrossRef]
- Lugg, W.H.; Griffiths, J.; van Rooyen, A.R.; Weeks, A.R.; Tingley, R. Optimal survey designs for environmental DNA sampling. Methods Ecol. Evol. 2018, 9, 1049–1059. [Google Scholar] [CrossRef]
- Parsons, K.M.; Dallas, J.F.; Claridge, D.E.; Durban, J.W.; Balcomb, K.C.; Thompson, P.M.; Noble, L.R. Amplifying dolphin mitochondrial DNA from faecal plumes. Mol. Ecol. 1999, 8, 1766–1768. [Google Scholar] [CrossRef] [PubMed]
- Frère, C.H.; Krzyszczyk, E.; Patterson, E.M.; Hunter, S.; Ginsburg, A.; Mann, J. Thar she blows! A novel method for DNA collection from cetacean blow. PLoS One 2010, 5, e12299. [Google Scholar] [CrossRef] [PubMed]
- Pierszalowski, S.P.; Ferrari, M.; Glockner-Ferrari, D.; Mizroch, S.; Clapham, P.J.; Dickerson, B.R. Investigating the feasibility of using DNA from sloughed skin for individual identification and kinship analysis in humpback whales (Megaptera novaeangliae). Mar. Mammal Sci. 2013, 29, 533–541. [Google Scholar] [CrossRef]
- Swanson, B.J.; Kelly, B.P.; Maddox, C.K.; Moran, J.R. Shed skin as a source of DNA for genotyping seals. Mol. Ecol. Notes 2006, 6, 1006–1009. [Google Scholar] [CrossRef]
- Meekan, M.; Austin, C.M.; Tan, M.H.; Wei, N.-W.V.; Miller, A.; Pierce, S.J.; Rowat, D.; Stevens, G.; Davies, T.K.; Ponzo, A.; et al. iDNA at Sea: Recovery of whale shark (Rhincodon typus) mitochondrial DNA sequences from the whale shark copepod (Pandarus rhincodonicus) confirms global population structure. Front. Mar. Sci. 2017, 4, 420. [Google Scholar] [CrossRef]
- Gottelli, D.; Wang, J.; Bashir, S.; Durant, S.M. Genetic analysis reveals promiscuity among female cheetahs. Proc. Biol. Sci. 2007, 274, 1993–2001. [Google Scholar] [CrossRef] [PubMed]
- Parsons, K.M.; Durban, J.W.; Claridge, D.E.; Balcomb, K.C.; Noble, L.R.; Thompson, P.M. Kinship as a basis for alliance formation between male bottlenose dolphins, Tursiops truncatus, in the Bahamas. Anim. Behav. 2003, 66, 185–194. [Google Scholar] [CrossRef]
- Parsons, K.M.; Durban, J.W.; Claridge, D.E.; Herzing, D.L.; Balcomb, K.C.; Noble, L.R. Population genetic structure of coastal bottlenose dolphins (Tursiops truncatus) in the northern Bahamas. Mar. Mamm. Sci. 2006, 22, 276–298. [Google Scholar] [CrossRef]
- Lukacs, P.M.; Burnham, K.P. Review of capture-recapture methods applicable to noninvasive genetic sampling. Mol. Ecol. 2005, 14, 3909–3919. [Google Scholar] [CrossRef] [PubMed]
- Ruell, E.W.; Riley, S.P.D.; Douglas, M.R.; Pollinger, J.P.; Crooks, K.R. Estimating bobcat population sizes and densities in a fragmented urban landscape using noninvasive capture–recapture sampling. J. Mammal. 2009, 90, 129–135. [Google Scholar] [CrossRef]
- Mondol, S.; Ullas Karanth, K.; Samba Kumar, N.; Gopalaswamy, A.M.; Andheria, A.; Ramakrishnan, U. Evaluation of non-invasive genetic sampling methods for estimating tiger population size. Biol. Conserv. 2009, 142, 2350–2360. [Google Scholar] [CrossRef]
- Janečka, J.E.; Munkhtsog, B.; Jackson, R.M.; Naranbaatar, G.; Mallon, D.P.; Murphy, W.J. Comparison of noninvasive genetic and camera-trapping techniques for surveying snow leopards. J. Mammal. 2011, 92, 771–783. [Google Scholar] [CrossRef]
- Sharma, R.; Stuckas, H.; Bhaskar, R.; Khan, I.; Goyal, S.P.; Tiedemann, R. Genetically distinct population of Bengal tiger (Panthera tigris tigris) in Terai Arc Landscape (TAL) of India. Mamm. Biol. 2011, 76, 484–490. [Google Scholar] [CrossRef]
- Taberlet, P.; Bonin, A.; Zinger, L.; Coissac, E. Environmental DNA: For Biodiversity Research and Monitoring; Oxford University Press: Oxford, UK, 2018; ISBN 0198767226. [Google Scholar]
- Gillett, R.M.; White, B.N.; Rolland, R.M. Quantification and genetic profiling of DNA isolated from free-floating feces of the North Atlantic right whale (Eubalaena glacialis). Mar. Mamm. Sci. 2008, 24, 341–355. [Google Scholar] [CrossRef]
- Aylward, M.L.; Sullivan, A.P.; Perry, G.H.; Johnson, S.E.; Louis, E.E. An environmental DNA sampling method for aye-ayes from their feeding traces. Ecol. Evol. 2018, 8, 9229–9240. [Google Scholar] [CrossRef] [PubMed]
- Monge, O.; Dumas, D.; Baus, I. Environmental DNA from avian residual saliva in fruits and its potential uses in population genetics. Conserv. Genet. Resour. 2018, 1–9. [Google Scholar] [CrossRef]
- Perez, I.; Geffen, E.; Mokady, O. Critically endangered Arabian leopards Panthera pardus nimr in Israel: estimating population parameters using molecular scatology. Oryx 2006, 40, 295–301. [Google Scholar] [CrossRef]
- Farrell, L.E.; Roman, J.; Sunquist, M.E. Dietary separation of sympatric carnivores identified by molecular analysis of scats. Mol. Ecol. 2000, 9, 1583–1590. [Google Scholar] [CrossRef] [PubMed]
- Janečka, J.E.; Jackson, R.; Yuquang, Z.; Diqiang, L.; Munkhtsog, B.; Buckley-Beason, V.; Murphy, W.J. Population monitoring of snow leopards using noninvasive collection of scat samples: a pilot study. Anim. Conserv. 2008, 11, 401–411. [Google Scholar] [CrossRef]
- Borthakur, U.; Barman, R.D.; Das, C.; Basumatary, A.; Talukdar, A.; Ahmed, M.F.; Talukdar, B.K.; Bharali, R. Noninvasive genetic monitoring of tiger (Panthera tigris tigris) population of Orang National Park in the Brahmaputra floodplain, Assam, India. Eur. J. Wildl. Res. 2011, 57, 603–613. [Google Scholar] [CrossRef]
- Russello, M.A.; Gladyshev, E.; Miquelle, D.; Caccone, A. Potential genetic consequences of a recent bottleneck in the Amur tiger of the Russian far east. Conserv. Genet. 2004, 5, 707–713. [Google Scholar] [CrossRef]
- Smith, O.; Wang, J. When can noninvasive samples provide sufficient information in conservation genetics studies? Mol. Ecol. Resour. 2014, 14, 1011–1023. [Google Scholar] [CrossRef] [PubMed]
- Ernest, H.B.; Boyce, W.M.; Bleich, V.C.; May, B.; Stiver, S.J.; Torres, S.G. Genetic structure of mountain lion (Puma concolor) populations in California. Conserv. Genet. 2003, 4, 353–366. [Google Scholar] [CrossRef]
- Haag, T.; Santos, A.S.; Sana, D.A.; Morato, R.G.; Cullen JR, L.; Cranshaw JR, P.G.; De Angelo, C.; Di Bitetti, M.S.; Salzano, F.M.; Eizirik, E. The effect of habitat fragmentation on the genetic structure of a top predator: loss of diversity and high differentiation among remnant populations of Atlantic Forest jaguars (Panthera onca). Mol. Ecol. 2010, 19, 4906–4921. [Google Scholar] [CrossRef] [PubMed]
- Goricki, Š.; Stankovic, D.; Snoj, A.; Kuntner, M.; Jeffery, W.R.; Trontelj, P.; Pavic, M.; Grizelj, Z.; Naparus-Aljancic, M.; Aljancic, G. Environmental DNA in subterranean biology: Range extension and taxonomic implications for Proteus. Sci. Rep. 2017, 7, 45054. [Google Scholar] [CrossRef] [PubMed]
- Stat, M.; John, J.; DiBattista, J.D.; Newman, S.J.; Bunce, M.; Harvey, E.S. Combined use of eDNA metabarcoding and video surveillance for the assessment of fish biodiversity. Conserv. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Boussarie, G.; Bakker, J.; Wangensteen, O.S.; Mariani, S.; Bonnin, L.; Juhel, J.-B.; Kiszka, J.J.; Kulbicki, M.; Manel, S.; Robbins, W.D.; et al. Environmental DNA illuminates the dark diversity of sharks. Sci. Adv. 2018, 4, eaap9661. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, P.F.; Møller, P.R.; Sigsgaard, E.E.; Knudsen, S.W.; Jørgensen, O.A.; Willerslev, E. Environmental DNA from seawater samples correlate with trawl catches of Subarctic, deepwater fishes. PLoS One 2016, 11, e0165252. [Google Scholar] [CrossRef] [PubMed]
- Smart, A.S.; Weeks, A.R.; van Rooyen, A.R.; Moore, A.; McCarthy, M.A.; Tingley, R. Assessing the cost-efficiency of environmental DNA sampling. Methods Ecol. Evol. 2016, 7, 1291–1298. [Google Scholar] [CrossRef]
- Port, J.A.; O’Donnell, J.L.; Romero-Maraccini, O.C.; Leary, P.R.; Litvin, S.Y.; Nickols, K.J.; Yamahara, K.M.; Kelly, R.P. Assessing vertebrate biodiversity in a kelp forest ecosystem using environmental DNA. Mol. Ecol. 2016, 25, 527–541. [Google Scholar] [CrossRef] [PubMed]
- Rees, H.C.; Bishop, K.; Middleditch, D.J.; Patmore, J.R.M.; Maddison, B.C.; Gough, K.C. The application of eDNA for monitoring of the great crested newt in the UK. Ecol. Evol. 2014, 4, 4023–4032. [Google Scholar] [CrossRef] [PubMed]
- Ardura, A.; Zaiko, A.; Martinez, J.L.; Samulioviene, A.; Semenova, A.; Garcia-Vazquez, E. eDNA and specific primers for early detection of invasive species – A case study on the bivalve Rangia cuneata, currently spreading in Europe. Mar. Environ. Res. 2015, 112, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Woodley, N.E.; Janzen, D.H.; Hallwachs, W.; Hebert, P.D.N. DNA barcodes reveal cryptic host-specificity within the presumed polyphagous members of a genus of parasitoid flies (Diptera: Tachinidae). Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 3657–3662. [Google Scholar] [CrossRef] [PubMed]
- Uthicke, S.; Lamare, M.; Doyle, J.R. eDNA detection of corallivorous seastar (Acanthaster cf. solaris) outbreaks on the Great Barrier Reef using digital droplet PCR. Coral Reefs 2018, 37, 1229–1239. [Google Scholar] [CrossRef]
- Yamanaka, H.; Minamoto, T. The use of environmental DNA of fishes as an efficient method of determining habitat connectivity. Ecol. Indic. 2016, 62, 147–153. [Google Scholar] [CrossRef]
- Gargan, L.M.; Morato, T.; Pham, C.K.; Finarelli, J.A.; Carlsson, J.E.L.; Carlsson, J. Development of a sensitive detection method to survey pelagic biodiversity using eDNA and quantitative PCR: a case study of devil ray at seamounts. Mar. Biol. 2017, 164, 112. [Google Scholar] [CrossRef]
- McKelvey, K.S.; Young, M.K.; Knotek, W.L.; Carim, K.J.; Wilcox, T.M.; Padgett-Stewart, T.M.; Schwartz, M.K. Sampling large geographic areas for rare species using environmental DNA: A study of bull trout Salvelinus confluentus occupancy in western Montana. J. Fish Biol. 2016, 88, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, T.M.; McKelvey, K.S.; Young, M.K.; Sepulveda, A.J.; Shepard, B.B.; Jane, S.F.; Whiteley, A.R.; Lowe, W.H.; Schwartz, M.K. Understanding environmental DNA detection probabilities: A case study using a stream-dwelling char Salvelinus fontinalis. Biol. Conserv. 2016, 194, 209–216. [Google Scholar] [CrossRef]
- Snyder, D.E. Invited overview: Conclusions from a review of electrofishing and its harmful effects on fish. Rev. Fish Biol. Fish. 2004, 13, 445–453. [Google Scholar] [CrossRef]
- Kim, P.; Kim, D.; Yoon, T.J.; Shin, S. Early detection of marine invasive species, Bugula neritina (Bryozoa: Cheilostomatida), using species-specific primers and environmental DNA analysis in Korea. Mar. Environ. Res. 2018, 139, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sigsgaard, E.E.; Nielsen, I.B.; Bach, S.S.; Lorenzen, E.D.; Robinson, D.P.; Knudsen, S.W.; Pedersen, M.W.; Al Jaidah, M.; Orlando, L.; Willerslev, E.; et al. Population characteristics of a large whale shark aggregation inferred from seawater environmental DNA. Nat. Ecol. Evol. 2016, 1, 0004. [Google Scholar] [CrossRef] [PubMed]
- Parsons, K.M.; Everett, M.; Dahlheim, M.; Park, L. Water, water everywhere: environmental DNA can unlock population structure in elusive marine species. R. Soc. Open Sci. 2018, 5, 180537. [Google Scholar] [CrossRef] [PubMed]
- Elbrecht, V.; Vamos, E.E.; Steinke, D.; Leese, F. Assessing intraspecific genetic diversity from community DNA metabarcoding data. PeerJ 2018, 6, e4644. [Google Scholar] [CrossRef] [PubMed]
- Baker, C.S.; Steel, D.; Nieukirk, S.; Klinck, H. Environmental DNA (eDNA) from the wake of the whales: droplet digital PCR for detection and species identification. Front. Mar. Sci. 2018, 5, 133. [Google Scholar] [CrossRef]
- Stat, M.; Huggett, M.J.; Bernasconi, R.; DiBattista, J.D.; Berry, T.E.; Newman, S.J.; Harvey, E.S.; Bunce, M. Ecosystem biomonitoring with eDNA: metabarcoding across the tree of life in a tropical marine environment. Sci. Rep. 2017, 7, 12240. [Google Scholar] [CrossRef] [PubMed]
- Ruru, J.; O’Lyver, P.B.; Scott, N.; Edmunds, D. Reversing the decline in New Zealand’s biodiversity: empowering Māori within reformed conservation law. Policy Q. 2017, 13. [Google Scholar] [CrossRef]
- Tipa, G.; Nelson, K. Eco-cultural restoration across multiple spatial scales: A New Zealand case study. Water Hist. 2017, 9, 87–106. [Google Scholar] [CrossRef]
- Witmer, G.W. Wildlife population monitoring: some practical considerations. Wildl. Res. 2005, 32, 259. [Google Scholar] [CrossRef]
- Bland, L.M.; Collen, B.; Orme, C.D.L.; Bielby, J. Predicting the conservation status of data-deficient species. Conserv. Biol. 2015, 29, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Rees, H.C.; Baker, C.A.; Gardner, D.S.; Maddison, B.C.; Gough, K.C. The detection of great crested newts year round via environmental DNA analysis. BMC Res. Notes 2017, 10, 327. [Google Scholar] [CrossRef] [PubMed]
- Grillas, C.; Ugnon-Coussioz, E.; Priol, P.; Renet, J. Low occurrence of the great crested newt Triturus cristatus at the limits of its range: an alarming preliminary study. Herpetol. Bull. 2018, 145, 14–18. [Google Scholar]
- Ishige, T.; Miya, M.; Ushio, M.; Sado, T.; Ushioda, M.; Maebashi, K.; Yonechi, R.; Lagan, P.; Matsubayashi, H. Tropical-forest mammals as detected by environmental DNA at natural saltlicks in Borneo. Biol. Conserv. 2017, 210, 281–285. [Google Scholar] [CrossRef]
- Allendorf, F.W. Genetics and the conservation of natural populations: allozymes to genomes. Mol. Ecol. 2017, 26, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Dejean, T.; Valentini, A.; Miquel, C.; Taberlet, P.; Bellemain, E.; Miaud, C. Improved detection of an alien invasive species through environmental DNA barcoding: The example of the American bullfrog Lithobates catesbeianus. J. Appl. Ecol. 2012, 49, 953–959. [Google Scholar] [CrossRef]
- Peñarrubia, L.; Alcaraz, C.; bij de Vaate, A.; Sanz, N.; Pla, C.; Vidal, O.; Viñas, J. Validated methodology for quantifying infestation levels of dreissenid mussels in environmental DNA (eDNA) samples. Sci. Rep. 2016, 6, 39067. [Google Scholar] [CrossRef] [PubMed]
- Gingera, T.D.; Steeves, T.B.; Boguski, D.A.; Whyard, S.; Li, W.; Docker, M.F. Detection and identification of lampreys in Great Lakes streams using environmental DNA. J. Gt. Lakes Res. 2016, 42, 649–659. [Google Scholar] [CrossRef]
- Goldstien, S.J.; Schiel, D.R.; Gemmell, N.J. Regional connectivity and coastal expansion: Differentiating pre-border and post-border vectors for the invasive tunicate Styela clava. Mol. Ecol. 2010, 19, 874–885. [Google Scholar] [CrossRef] [PubMed]
- Blackman, R.C.; Hänfling, B.; Lawson-Handley, L. The use of environmental DNA as an early warning tool in the detection of new freshwater invasive non-native species. CAB Rev. 2018, 13, 1–15. [Google Scholar] [CrossRef]
- Pimentel, D.; Zuniga, R.; Morrison, D. Update on the environmental and economic costs associated with alien-invasive species in the United States. Ecol. Econ. 2005, 52, 273–288. [Google Scholar] [CrossRef]
- Murray, D.C.; Coghlan, M.L.; Bunce, M. From benchtop to desktop: Important considerations when designing amplicon sequencing workflows. PLoS One 2015, 10, e0124671. [Google Scholar] [CrossRef] [PubMed]
- Nichols, R.V.; Cromsigt, J.P.G.M.; Spong, G. DNA left on browsed twigs uncovers bite-scale resource use patterns in European ungulates. Oecologia 2015, 178, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Bohmann, K.; Schnell, I.B.; Gilbert, M.T.P. When bugs reveal biodiversity. Mol. Ecol. 2013, 22, 909–911. [Google Scholar] [CrossRef] [PubMed]
- Calvignac-Spencer, S.; Leendertz, F.H.; Gilbert, M.T.P.; Schubert, G. An invertebrate stomach’s view on vertebrate ecology: Certain invertebrates could be used as “vertebrate samplers” and deliver DNA-based information on many aspects of vertebrate ecology. BioEssays 2013. [Google Scholar] [CrossRef] [PubMed]
- Schnell, I.B.; Sollmann, R.; Calvignac-Spencer, S.; Siddall, M.E.; Yu, D.W.; Wilting, A.; Gilbert, M.T.P. iDNA from terrestrial haematophagous leeches as a wildlife surveying and monitoring tool - prospects, pitfalls and avenues to be developed. Front. Zool. 2015, 12, 24. [Google Scholar] [CrossRef] [PubMed]
- Jombart, T. adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [PubMed]
- Corander, J.; Waldmann, P.; Marttinen, P.; Sillanpää, M.J. BAPS 2: Enhanced possibilities for the analysis of genetic population structure. Bioinformatics 2004, 20, 2363–2369. [Google Scholar] [CrossRef] [PubMed]
- McKelvey, K.S.; Schwartz, M.K. Genetic errors associated with population estimation using non-invasive molecular tagging: problems and new solutions. J. Wildl. Manage. 2004, 68, 439–448. [Google Scholar] [CrossRef]
- Oliver, A.K.; Brown, S.P.; Callaham, M.A.; Jumpponen, A. Polymerase matters: Non-proofreading enzymes inflate fungal community richness estimates by up to 15%. Fungal Ecol. 2015, 15, 86–89. [Google Scholar] [CrossRef]
- Cooke, T.F.; Yee, M.C.; Muzzio, M.; Sockell, A.; Bell, R.; Cornejo, O.E.; Kelley, J.L.; Bailliet, G.; Bravi, C.M.; Bustamante, C.D.; et al. GBStools: A statistical method for estimating allelic dropout in reduced representation sequencing data. PLoS Genet. 2016, 12, e1005631. [Google Scholar] [CrossRef] [PubMed]
- Ficetola, G.F.; Taberlet, P.; Coissac, E. How to limit false positives in environmental DNA and metabarcoding? Mol. Ecol. Resour. 2016, 16, 604–607. [Google Scholar] [CrossRef] [PubMed]
- Alberdi, A.; Aizpurua, O.; Gilbert, M.T.P.; Bohmann, K. Scrutinizing key steps for reliable metabarcoding of environmental samples. Methods Ecol. Evol. 2018, 9, 134–147. [Google Scholar] [CrossRef]
- Yamamoto, S.; Masuda, R.; Sato, Y.; Sado, T.; Araki, H.; Kondoh, M.; Minamoto, T.; Miya, M. Environmental DNA metabarcoding reveals local fish communities in a species-rich coastal sea. Sci. Rep. 2017, 7, 40368. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, V.G. Pitfalls in relative abundance estimation using eDNA metabarcoding. Mol. Ecol. Resour. 2018, 18, 923–926. [Google Scholar] [CrossRef]
- Fernandez, S.; Sandin, M.M.; Beaulieu, P.G.; Clusa, L.; Martinez, J.L.; Ardura, A.; García-Vázquez, E. Environmental DNA for freshwater fish monitoring: insights for conservation within a protected area. PeerJ 2018, 6, e4486. [Google Scholar] [CrossRef] [PubMed]
- Fernández, S.; Rodríguez, S.; Martínez, J.L.; Borrell, Y.J.; Ardura, A.; García-Vázquez, E. Evaluating freshwater macroinvertebrates from eDNA metabarcoding: A river Nalón case study. PLoS One 2018, 13, e0201741. [Google Scholar] [CrossRef] [PubMed]
- Eisenstein, M. Microbiology: making the best of PCR bias. Nat. Methods 2018, 15, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Elbrecht, V.; Leese, F. Can DNA-based ecosystem assessments quantify species abundance? Testing primer bias and biomass-sequence relationships with an innovative metabarcoding protocol. PLoS One 2015, 10, e0130324. [Google Scholar] [CrossRef] [PubMed]
- Raemy, M.; Ursenbacher, S. Detection of the European pond turtle (Emys orbicularis) by environmental DNA: Is eDNA adequate for reptiles? Amphibia-Reptilia 2018, 39, 135–143. [Google Scholar] [CrossRef]
- Levi, T.; Allen, J.M.; Bell, D.; Joyce, J.; Russell, J.R.; Tallmon, D.A.; Vulstek, S.C.; Yang, C.; Yu, D.W. Environmental DNA for the enumeration and management of Pacific salmon. Mol. Ecol. Resour. 2018, 394445. [Google Scholar] [CrossRef] [PubMed]
- Vasselon, V.; Bouchez, A.; Rimet, F.; Jacquet, S.; Trobajo, R.; Corniquel, M.; Tapolczai, K.; Domaizon, I. Avoiding quantification bias in metabarcoding: Application of a cell biovolume correction factor in diatom molecular biomonitoring. Methods Ecol. Evol. 2018, 9, 1060–1069. [Google Scholar] [CrossRef]
- Klymus, K.E.; Richter, C.A.; Chapman, D.C.; Paukert, C. Quantification of eDNA shedding rates from invasive bighead carp Hypophthalmichthys nobilis and silver carp Hypophthalmichthys molitrix. Biol. Conserv. 2015, 183, 77–84. [Google Scholar] [CrossRef]
- Dunn, N.; Priestley, V.; Herraiz, A.; Arnold, R.; Savolainen, V. Behavior and season affect crayfish detection and density inference using environmental DNA. Ecol. Evol. 2017, 7, 7777–7785. [Google Scholar] [CrossRef] [PubMed]
- Collins, R.A.; Wangensteen, O.S.; O’Gorman, E.J.; Mariani, S.; Sims, D.W.; Genner, M.J. Persistence of environmental DNA in marine systems. Commun. Biol. 2018, 1, 185. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.; Martín, P.; Albarrán, C.; García, P.; García, O.; Fernández De Simón, L.; García-Hirschfeld, J.; Sancho, M.; De La Rúa, C.; Fernández-Piqueras, J. Real-time PCR designs to estimate nuclear and mitochondrial DNA copy number in forensic and ancient DNA studies. Forensic Sci. Int. 2004, 139, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.M.; Kemp, B.M. Rescue PCR: Reagent-rich PCR recipe improves amplification of degraded DNA extracts. J. Archaeol. Sci. Reports 2017, 11, 683–694. [Google Scholar] [CrossRef]
- Gill, P.; Curran, J.; Elliot, K. A graphical simulation model of the entire DNA process associated with the analysis of short tandem repeat loci. Nucleic Acids Res. 2005, 33, 632–643. [Google Scholar] [CrossRef] [PubMed]
- Furlan, E.M.; Gleeson, D.; Hardy, C.M.; Duncan, R.P. A framework for estimating the sensitivity of eDNA surveys. Mol. Ecol. Resour. 2016, 16, 641–654. [Google Scholar] [CrossRef] [PubMed]
- Hunter, M.E.; Dorazio, R.M.; Butterfield, J.S.S.; Meigs-Friend, G.; Nico, L.G.; Ferrante, J.A. Detection limits of quantitative and digital PCR assays and their influence in presence-absence surveys of environmental DNA. Mol. Ecol. Resour. 2017, 17, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Zhan, A.; Gao, Y.; Zhang, L.; Haffner, G.D.; MacIsaac, H.J. Early detection of a highly invasive bivalve based on environmental DNA (eDNA). Biol. Invasions 2018, 20, 437–447. [Google Scholar] [CrossRef]
- Ratnasingham, S.; Hebert, P.D.N. bold: The Barcode of Life Data System (http://www.barcodinglife.org). Mol. Ecol. Notes 2007, 7, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Hebert, P.D.N.; Cywinska, A.; Ball, S.L.; DeWaard, J.R. Biological identifications through DNA barcodes. Proc. R. Soc. Lond. B Biol. Sci. 2003, 270, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Bogenhagen, D.; Clayton, D.A. The number of mitochondrial deoxyribonucleic acid genomes in mouse L and human HeLa cells. Quantitative isolation of mitochondrial deoxyribonucleic acid. J. Biol. Chem. 1974, 249, 7991–7995. [Google Scholar] [PubMed]
- Taberlet, P.; Luikart, G.; Waits, L.P. Noninvasive genetic sampling: Look before you leap. Trends Ecol. Evol. 1999, 14, 323–327. [Google Scholar] [CrossRef]
- Thomsen, P.F.; Willerslev, E. Environmental DNA - An emerging tool in conservation for monitoring past and present biodiversity. Biol. Conserv. 2015, 183, 4–18. [Google Scholar] [CrossRef]
- Turner, C.R.; Barnes, M.A.; Xu, C.C.Y.; Jones, S.E.; Jerde, C.L.; Lodge, D.M. Particle size distribution and optimal capture of aqueous macrobial eDNA. Methods Ecol. Evol. 2014, 5, 676–684. [Google Scholar] [CrossRef]
- Nukazawa, K.; Hamasuna, Y.; Suzuki, Y. Simulating the advection and degradation of the environmental DNA of common carp along a river. Environ. Sci. Technol. 2018, 52, 10562–10570. [Google Scholar] [CrossRef] [PubMed]
- Bylemans, J.; Furlan, E.M.; Gleeson, D.M.; Hardy, C.M.; Duncan, R.P. Does size matter? An experimental evaluation of the relative abundance and decay rates of aquatic eDNA. Environ. Sci. Technol. 2018, 52, 6408–6416. [Google Scholar] [CrossRef] [PubMed]
- Dejean, T.; Valentini, A.; Duparc, A.; Pellier-Cuit, S.; Pompanon, F.; Taberlet, P.; Miaud, C. Persistence of environmental DNA in freshwater ecosystems. PLoS One 2011, 6, e23398. [Google Scholar] [CrossRef] [PubMed]
- Deiner, K.; Renshaw, M.A.; Li, Y.; Olds, B.P.; Lodge, D.M.; Pfrender, M.E. Long-range PCR allows sequencing of mitochondrial genomes from environmental DNA. Methods Ecol. Evol. 2017, 8, 1888–1898. [Google Scholar] [CrossRef]
- Rubinoff, D.; Cameron, S.; Will, K. A genomic perspective on the shortcomings of mitochondrial DNA for “barcoding” identification. J. Hered. 2006, 97, 581–594. [Google Scholar] [CrossRef] [PubMed]
- Hurst, G.D.D.; Jiggins, F.M. Problems with mitochondrial DNA as a marker in population, phylogeographic and phylogenetic studies: The effects of inherited symbionts. Proc. R. Soc. B Biol. Sci. 2005, 272, 1525–1534. [Google Scholar] [CrossRef] [PubMed]
- Rubinoff, D. Utility of mitochondrial DNA barcodes in species conservation. Conserv. Biol. 2006, 20, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Carranza, J.; Salinas, M.; de Andrés, D.; Pérez-González, J. Iberian red deer: paraphyletic nature at mtDNA but nuclear markers support its genetic identity. Ecol. Evol. 2016, 6, 905–922. [Google Scholar] [CrossRef] [PubMed]
- Pratlong, M.; Rancurel, C.; Pontarotti, P.; Aurelle, D. Monophyly of Anthozoa (Cnidaria): why do nuclear and mitochondrial phylogenies disagree? Zool. Scr. 2017, 46, 363–371. [Google Scholar] [CrossRef]
- Weigand, H.; Weiss, M.; Cai, H.; Li, Y.; Yu, L.; Zhang, C.; Leese, F. Deciphering the origin of mito-nuclear discordance in two sibling caddisfly species. Mol. Ecol. 2017, 26, 5705–5715. [Google Scholar] [CrossRef] [PubMed]
- Haran, J.; Koutroumpa, F.; Magnoux, E.; Roques, A.; Roux, G. Ghost mtDNA haplotypes generated by fortuitous NUMTs can deeply disturb infra-specific genetic diversity and phylogeographic pattern. J. Zool. Syst. Evol. Res. 2015, 53, 109–115. [Google Scholar] [CrossRef]
- Gaziev, A.I.; Shaikhaev, G.O. Nuclear mitochondrial pseudogenes. Mol. Biol. 2010, 44, 358–368. [Google Scholar] [CrossRef]
- Hazkani-Covo, E.; Zeller, R.M.; Martin, W. Molecular poltergeists: Mitochondrial DNA copies (numts) in sequenced nuclear genomes. PLoS Genet. 2010, 6, e1000834. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Buhay, J.E.; Whiting, M.F.; Crandall, K.A. Many species in one: DNA barcoding overestimates the number of species when nuclear mitochondrial pseudogenes are coamplified. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 13486–13491. [Google Scholar] [CrossRef] [PubMed]
- Thalmann, O.; Hebler, J.; Poinar, H.N.; Pääbo, S.; Vigilant, L. Unreliable mtDNA data due to nuclear insertions: a cautionary tale from analysis of humans and other great apes. Mol. Ecol. 2004, 13, 321–335. [Google Scholar] [CrossRef] [PubMed]
- Knapp, S.M.; Craig, B.A.; Waits, L.P. Incorporating genotyping error into non-invasive DNA-based mark–recapture population estimates. J. Wildl. Manage. 2009, 73, 598–604. [Google Scholar] [CrossRef]
- Mills, L.S.; Citta, J.J.; Lair, K.P.; Schwartz, M.K.; Tallmon, D.A. Estimating animal abundance using noninvasive DNA sampling: promise and pitfalls. Ecol. Appl. 2000, 10, 283–294. [Google Scholar] [CrossRef]
- Waits, L.P.; Luikart, G.; Taberlet, P. Estimating the probability of identity among genotypes in natural populations: cautions and guidelines. Mol. Ecol. 2001, 10, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Sloane, M.A.; Sunnucks, P.; Alpers, D.; Beheregaray, L.B.; Taylor, A.C. Highly reliable genetic identification of individual northern hairy-nosed wombats from single remotely collected hairs: a feasible censusing method. Mol. Ecol. 2000, 9, 1233–1240. [Google Scholar] [CrossRef] [PubMed]
- Mondol, S.; R, N.; Athreya, V.; Sunagar, K.; Selvaraj, V.M.; Ramakrishnan, U. A panel of microsatellites to individually identify leopards and its application to leopard monitoring in human dominated landscapes. BMC Genet. 2009, 10, 79. [Google Scholar] [CrossRef] [PubMed]
- Jeunen, G.-J.; Knapp, M.; Spencer, H.G.; Taylor, H.R.; Lamare, M.D.; Stat, M.; Bunce, M.; Gemmell, N.J. Species-level biodiversity assessment using marine environmental DNA metabarcoding requires protocol optimization and standardization. Ecol. Evol. 2019, 9, 1323–1335. [Google Scholar] [CrossRef] [PubMed]
- Jeunen, G.-J.; Knapp, M.; Spencer, H.G.; Lamare, M.D.; Taylor, H.R.; Stat, M.; Bunce, M.; Gemmell, N.J. Environmental DNA (eDNA) metabarcoding reveals strong discrimination among diverse marine habitats connected by water movement. Mol. Ecol. Resour. 2019, 19, 326–438. [Google Scholar] [CrossRef] [PubMed]
- Cutler, D.J.; Jensen, J.D. To pool, or not to pool? Genetics 2010, 186, 41–43. [Google Scholar] [CrossRef] [PubMed]
- Barnes, M.A.; Turner, C.R. The ecology of environmental DNA and implications for conservation genetics. Conserv. Genet. 2016, 17, 1–17. [Google Scholar] [CrossRef]
- Morin, P.A.; Luikart, G.; Wayne, R.K. SNPs in ecology, evolution and conservation. Trends Ecol. Evol. 2004, 19, 208–216. [Google Scholar] [CrossRef]
- Fuentes-Pardo, A.P.; Ruzzante, D.E. Whole-genome sequencing approaches for conservation biology: Advantages, limitations and practical recommendations. Mol. Ecol. 2017, 26, 5369–5406. [Google Scholar] [CrossRef] [PubMed]
- Davey, J.W.; Blaxter, M.L. RADSeq: next-generation population genetics. Brief. Funct. Genomics 2010, 9, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Schlötterer, C.; Tobler, R.; Kofler, R.; Nolte, V. Sequencing pools of individuals-mining genome-wide polymorphism data without big funding. Nat. Rev. Genet. 2014, 15, 749–763. [Google Scholar] [CrossRef] [PubMed]
- Luca, F.; Hudson, R.R.; Witonsky, D.B.; Di Rienzo, A. A reduced representation approach to population genetic analyses and applications to human evolution. Genome Res. 2011, 21, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Scholl, U.I.; Ji, W.; Liu, T.; Tikhonova, I.R.; Zumbo, P.; Nayir, A.; Bakkaloğlu, A.; Ozen, S.; Sanjad, S.; et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 19096–19101. [Google Scholar] [CrossRef] [PubMed]
- Koziol, A.; Stat, M.; Simpson, T.; Jarman, S.; DiBattista, J.D.; Harvey, E.S.; Marnane, M.; McDonald, J.; Bunce, M. Environmental DNA metabarcoding studies are critically affected by substrate selection. Mol. Ecol. Resour. 2018. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, C.S.; Turner, C.R.; Deiner, K.; Klymus, K.E.; Thomsen, P.F.; Murphy, M.A.; Spear, S.F.; McKee, A.; Oyler-McCance, S.J.; Cornman, R.S.; et al. Critical considerations for the application of environmental DNA methods to detect aquatic species. Methods Ecol. Evol. 2016, 7, 1299–1307. [Google Scholar] [CrossRef]
- Kalisky, T.; Quake, S.R. Single-cell genomics. Nat. Methods 2011, 8, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Macaulay, I.C.; Ponting, C.P.; Voet, T. Single-cell multiomics: multiple measurements from single cells. Trends Genet. 2017, 33, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Lan, F.; Demaree, B.; Ahmed, N.; Abate, A.R. Single-cell genome sequencing at ultra-high-throughput with microfluidic droplet barcoding. Nat. Biotechnol. 2017, 35, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, M.; Sciambi, A.; Treusch, S.; Durruthy-Durruthy, R.; Gokhale, K.; Jacob, J.; Chen, T.X.; Geis, J.A.; Oldham, W.; Matthews, J.; et al. High-throughput single-cell DNA sequencing of acute myeloid leukemia tumors with droplet microfluidics. Genome Res. 2018, 28, 1345–1352. [Google Scholar] [CrossRef] [PubMed]
- Mason, O.U.; Hazen, T.C.; Borglin, S.; Chain, P.S.G.; Dubinsky, E.A.; Fortney, J.L.; Han, J.; Holman, H.-Y.N.; Hultman, J.; Lamendella, R.; et al. Metagenome, metatranscriptome and single-cell sequencing reveal microbial response to Deepwater Horizon oil spill. ISME J. 2012, 6, 1715–1727. [Google Scholar] [CrossRef] [PubMed]
- Yuan, G.-C.; Cai, L.; Elowitz, M.; Enver, T.; Fan, G.; Guo, G.; Irizarry, R.; Kharchenko, P.; Kim, J.; Orkin, S.; et al. Challenges and emerging directions in single-cell analysis. Genome Biol. 2017, 18, 84. [Google Scholar] [CrossRef] [PubMed]
- Tanay, A.; Regev, A. Scaling single-cell genomics from phenomenology to mechanism. Nature 2017, 541, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-Z.; Adalsteinsson, V.A.; Francis, J.; Cornils, H.; Jung, J.; Maire, C.; Ligon, K.L.; Meyerson, M.; Love, J.C. Calibrating genomic and allelic coverage bias in single-cell sequencing. Nat. Commun. 2015, 6, 6822. [Google Scholar] [CrossRef] [PubMed]
- Ficetola, G.F.; Pansu, J.; Bonin, A.; Coissac, E.; Giguet-Covex, C.; De Barba, M.; Gielly, L.; Lopes, C.M.; Boyer, F.; Pompanon, F.; et al. Replication levels, false presences and the estimation of the presence/absence from eDNA metabarcoding data. Mol. Ecol. Resour. 2015, 15, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Creer, S.; Deiner, K.; Frey, S.; Porazinska, D.; Taberlet, P.; Thomas, W.K.; Potter, C.; Bik, H.M. The ecologist’s field guide to sequence-based identification of biodiversity. Methods Ecol. Evol. 2016, 7, 1008–1018. [Google Scholar] [CrossRef]
- Nathan, L.M.; Simmons, M.; Wegleitner, B.J.; Jerde, C.L.; Mahon, A.R. Quantifying environmental DNA signals for aquatic invasive species across multiple detection platforms. Environ. Sci. Technol. 2014, 48, 12800–12806. [Google Scholar] [CrossRef] [PubMed]
- Doi, H.; Uchii, K.; Takahara, T.; Matsuhashi, S.; Yamanaka, H.; Minamoto, T. Use of droplet digital PCR for estimation of fish abundance and biomass in environmental DNA surveys. PLoS One 2015, 10, e0122763. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, M.L.; Buenrostro, J.D.; Valdiosera, C.; Schroeder, H.; Allentoft, M.E.; Sikora, M.; Rasmussen, M.; Gravel, S.; Guillén, S.; Nekhrizov, G.; et al. Pulling out the 1%: whole-genome capture for the targeted enrichment of ancient dna sequencing libraries. Am. J. Hum. Genet. 2013, 93, 852–864. [Google Scholar] [CrossRef] [PubMed]
- Bleidorn, C. Third generation sequencing: Technology and its potential impact on evolutionary biodiversity research. Syst. Biodivers. 2016, 14, 1–8. [Google Scholar] [CrossRef]
- Günther, B.; Knebelsberger, T.; Neumann, H.; Laakmann, S.; Martínez Arbizu, P. Metabarcoding of marine environmental DNA based on mitochondrial and nuclear genes. Sci. Rep. 2018, 8, 14822. [Google Scholar] [CrossRef] [PubMed]
- Schlötterer, C.; Kofler, R.; Versace, E.; Tobler, R.; Franssen, S.U. Combining experimental evolution with next-generation sequencing: a powerful tool to study adaptation from standing genetic variation. Heredity. 2015, 114, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Hivert, V.; Leblois, R.; Petit, E.J.; Gautier, M.; Vitalis, R. Measuring genetic differentiation from pool-seq data. Genetics 2018, 210, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Neethiraj, R.; Hornett, E.A.; Hill, J.A.; Wheat, C.W. Investigating the genomic basis of discrete phenotypes using a Pool-seq-only approach: New insights into the genetics underlying colour variation in diverse taxa. Mol. Ecol. 2017, 26, 4990–5002. [Google Scholar] [CrossRef] [PubMed]
- Soubrier, J.; Gower, G.; Chen, K.; Richards, S.M.; Llamas, B.; Mitchell, K.J.; Ho, S.Y.W.; Kosintsev, P.; Lee, M.S.Y.; Baryshnikov, G.; et al. Early cave art and ancient DNA record the origin of European bison. Nat. Commun. 2016, 7, 13158. [Google Scholar] [CrossRef] [PubMed]
- Cahill, J.A.; Stirling, I.; Kistler, L.; Salamzade, R.; Ersmark, E.; Fulton, T.L.; Stiller, M.; Green, R.E.; Shapiro, B. Genomic evidence of geographically widespread effect of gene flow from polar bears into brown bears. Mol. Ecol. 2015, 24, 1205–1217. [Google Scholar] [CrossRef] [PubMed]
- Wangensteen, O.S.; Cebrian, E.; Palacín, C.; Turon, X. Under the canopy: Community-wide effects of invasive algae in Marine Protected Areas revealed by metabarcoding. Mar. Pollut. Bull. 2018, 127, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Korpelainen, H.; Pietiläinen, M. Biodiversity of pollen in indoor air samples as revealed by DNA metabarcoding. Nord. J. Bot. 2017, 35, 602–608. [Google Scholar] [CrossRef]
- Pochon, X.; Zaiko, A.; Fletcher, L.M.; Laroche, O.; Wood, S.A. Wanted dead or alive? Using metabarcoding of environmental DNA and RNA to distinguish living assemblages for biosecurity applications. PLoS One 2017, 12, e0187636. [Google Scholar] [CrossRef] [PubMed]
- Zaiko, A.; Pochon, X.; Garcia-Vazquez, E.; Olenin, S.; Wood, S.A. Advantages and limitations of environmental DNA/RNA tools for marine biosecurity: Management and surveillance of non-indigenous species. Front. Mar. Sci. 2018, 5, 322. [Google Scholar] [CrossRef]
- Laroche, O.; Wood, S.A.; Tremblay, L.A.; Lear, G.; Ellis, J.I.; Pochon, X. Metabarcoding monitoring analysis: the pros and cons of using co-extracted environmental DNA and RNA data to assess offshore oil production impacts on benthic communities. PeerJ 2017, 5, e3347. [Google Scholar] [CrossRef] [PubMed]
- Pochon, X.; Wood, S.A.; Keeley, N.B.; Lejzerowicz, F.; Esling, P.; Drew, J.; Pawlowski, J. Accurate assessment of the impact of salmon farming on benthic sediment enrichment using foraminiferal metabarcoding. Mar. Pollut. Bull. 2015, 100, 370–382. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.S.; Swenson, W. Community genetics and community selection. Ecology 2003, 84, 586–588. [Google Scholar] [CrossRef]
- Bálint, M.; Pfenninger, M.; Grossart, H.-P.; Taberlet, P.; Vellend, M.; Leibold, M.A.; Englund, G.; Bowler, D. Environmental DNA time series in ecology. Trends Ecol. Evol. 2018, 33, 945–957. [Google Scholar] [CrossRef] [PubMed]
- Bohan, D.A.; Vacher, C.; Tamaddoni-Nezhad, A.; Raybould, A.; Dumbrell, A.J.; Woodward, G. Next-generation global biomonitoring: Large-scale, automated reconstruction of ecological networks. Trends Ecol. Evol. 2017, 32, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Thyssen, M.; Tarran, G.A.; Zubkov, M.V.; Holland, R.J.; Gregori, G.; Burkill, P.H.; Denis, M. The emergence of automated high-frequency flow cytometry: revealing temporal and spatial phytoplankton variability. J. Plankton Res. 2007, 30, 333–343. [Google Scholar] [CrossRef]
- Mills, A.M.; Thurber, B.G.; Mackenzie, S.A.; Taylor, P.D. Passerines use nocturnal flights for landscape-scale movements during migration stopover. Condor 2011, 113, 597–607. [Google Scholar] [CrossRef]
- Magurran, A.E.; Baillie, S.R.; Buckland, S.T.; Dick, J.M.; Elston, D.A.; Scott, E.M.; Smith, R.I.; Somerfield, P.J.; Watt, A.D. Long-term datasets in biodiversity research and monitoring: assessing change in ecological communities through time. Trends Ecol. Evol. 2010, 25, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Heupel, M.R.; Semmens, J.M.; Hobday, A.J. Automated acoustic tracking of aquatic animals: scales, design and deployment of listening station arrays. Mar. Freshw. Res. 2006, 57, 113. [Google Scholar] [CrossRef]
- Liu, S.; Wang, X.; Xie, L.; Tan, M.; Li, Z.; Su, X.; Zhang, H.; Misof, B.; Kjer, K.M.; Tang, M.; et al. Mitochondrial capture enriches mito-DNA 100 fold, enabling PCR-free mitogenomics biodiversity analysis. Mol. Ecol. Resour. 2016, 16, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, T.M.; Zarn, K.E.; Piggott, M.P.; Young, M.K.; McKelvey, K.S.; Schwartz, M.K. Capture enrichment of aquatic environmental DNA: A first proof of concept. Mol. Ecol. Resour. 2018, 18, 1392–1401. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, M.; Shimabukuro, H.; Hori, M.; Yoshida, G.; Terada, T.; Miyajima, T. Quantitative real-time polymerase chain reaction (PCR) and droplet digital PCR duplex assays for detecting Zostera marina DNA in coastal sediments. Limnol. Oceanogr. Methods 2018. [Google Scholar] [CrossRef]
- Miotke, L.; Lau, B.T.; Rumma, R.T.; Ji, H.P. High sensitivity detection and quantitation of DNA copy number and single nucleotide variants with single color droplet digital PCR. Anal. Chem. 2014, 86, 2618–2624. [Google Scholar] [CrossRef] [PubMed]
- Doi, H.; Takahara, T.; Minamoto, T.; Matsuhashi, S.; Uchii, K.; Yamanaka, H. Droplet digital polymerase chain reaction (PCR) outperforms real-time PCR in the detection of environmental DNA from an invasive fish species. Environ. Sci. Technol. 2015, 49, 5601–5608. [Google Scholar] [CrossRef] [PubMed]
- Hindson, C.M.; Chevillet, J.R.; Briggs, H.A.; Gallichotte, E.N.; Ruf, I.K.; Hindson, B.J.; Vessella, R.L.; Tewari, M. Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat. Methods 2013, 10, 1003–1005. [Google Scholar] [CrossRef] [PubMed]
- Skelly, D.A.; Johansson, M.; Madeoy, J.; Wakefield, J.; Akey, J.M. A powerful and flexible statistical framework for testing hypotheses of allele-specific gene expression from RNA-seq data. Genome Res. 2011, 21, 1728–1737. [Google Scholar] [CrossRef] [PubMed]
- Kamoroff, C.; Goldberg, C.S. An issue of life or death: using eDNA to detect viable individuals in wilderness restoration. Freshw. Sci. 2018, 37, 685–696. [Google Scholar] [CrossRef]
- Sims, D.; Sudbery, I.; Ilott, N.E.; Heger, A.; Ponting, C.P. Sequencing depth and coverage: key considerations in genomic analyses. Nat. Rev. Genet. 2014, 15, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Sboner, A.; Mu, X.; Greenbaum, D.; Auerbach, R.K.; Gerstein, M.B. The real cost of sequencing: higher than you think! Genome Biol. 2011, 12, 125. [Google Scholar] [CrossRef] [PubMed]
- Bik, H.M. Let’s rise up to unite taxonomy and technology. PLOS Biol. 2017, 15, e2002231. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, G.W.; Freckleton, R.P. Declines in the numbers of amateur and professional taxonomists: implications for conservation. Anim. Conserv. 2002, 5, 245–249. [Google Scholar] [CrossRef]
- Bearhop, S.; Adams, C.E.; Waldron, S.; Fuller, R.A.; Macleod, H. Determining trophic niche width: A novel approach using stable isotope analysis. J. Anim. Ecol. 2004, 73, 1007–1012. [Google Scholar] [CrossRef]
- McKinney, M.A.; Arukwe, A.; De Guise, S.; Martineau, D.; Béland, P.; Dallaire, A.; Lair, S.; Lebeuf, M.; Letcher, R.J. Characterization and profiling of hepatic cytochromes P450 and phase II xenobiotic-metabolizing enzymes in beluga whales (Delphinapterus leucas) from the St. Lawrence River Estuary and the Canadian Arctic. Aquat. Toxicol. 2004, 69, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Polich, R.L. Stress hormone levels in a freshwater turtle from sites differing in human activity. Conserv. Physiol. 2016, 4, cow016. [Google Scholar] [CrossRef] [PubMed]
- Greig, D.J.; Mashburn, K.L.; Rutishauser, M.; Gulland, F.M.D.; Williams, T.M.; Atkinson, S. Seasonal changes in circulating progesterone and estrogen concentrations in the California sea lion (Zalophus californianus). J. Mammal. 2007, 88, 67–72. [Google Scholar] [CrossRef]
- Skoglund, P.; Posth, C.; Sirak, K.; Spriggs, M.; Valentin, F.; Bedford, S.; Clark, G.R.; Reepmeyer, C.; Petchey, F.; Fernandes, D.; et al. Genomic insights into the peopling of the Southwest Pacific. Nature 2016, 538, 510–513. [Google Scholar] [CrossRef] [PubMed]
- Schuenemann, V.J.; Avanzi, C.; Krause-Kyora, B.; Seitz, A.; Herbig, A.; Inskip, S.; Bonazzi, M.; Reiter, E.; Urban, C.; Dangvard Pedersen, D.; et al. Ancient genomes reveal a high diversity of Mycobacterium leprae in medieval Europe. PLoS Pathog. 2018, 14, e1006997. [Google Scholar] [CrossRef] [PubMed]
- Fellows Yates, J.A.; Drucker, D.G.; Reiter, E.; Heumos, S.; Welker, F.; Münzel, S.C.; Wojtal, P.; Lázničková-Galetová, M.; Conard, N.J.; Herbig, A.; et al. Central European Woolly Mammoth population dynamics: Insights from late Pleistocene mitochondrial genomes. Sci. Rep. 2017, 7, 17714. [Google Scholar] [CrossRef] [PubMed]
- Pimm, S.L.; Russell, G.J.; Gittleman, J.L.; Brooks, T.M. The future of biodiversity. Science 1995, 269, 347–350. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.; Faurby, S.; Svenning, J.-C. Mammal diversity will take millions of years to recover from the current biodiversity crisis. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 11262–11267. [Google Scholar] [CrossRef] [PubMed]
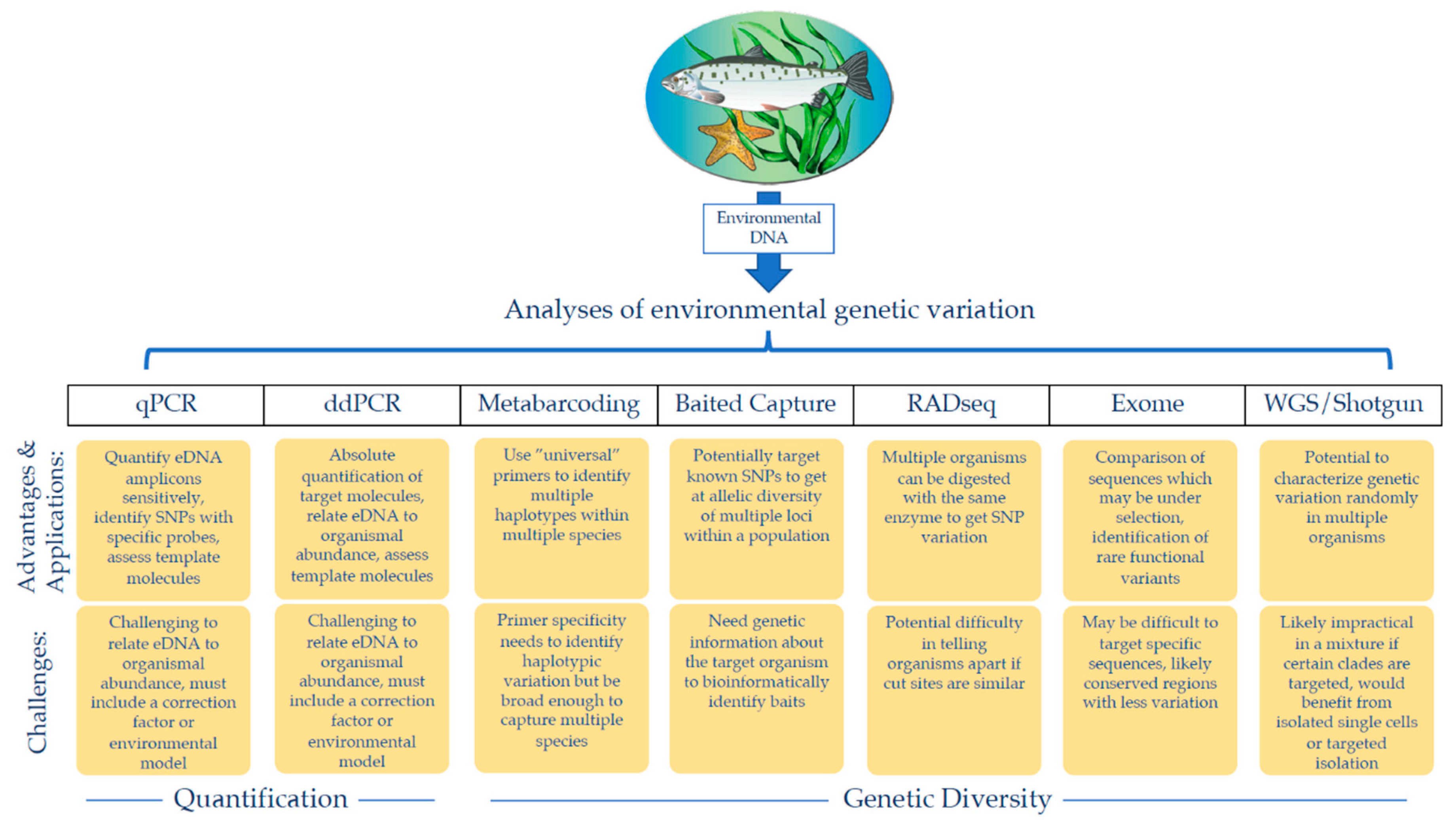

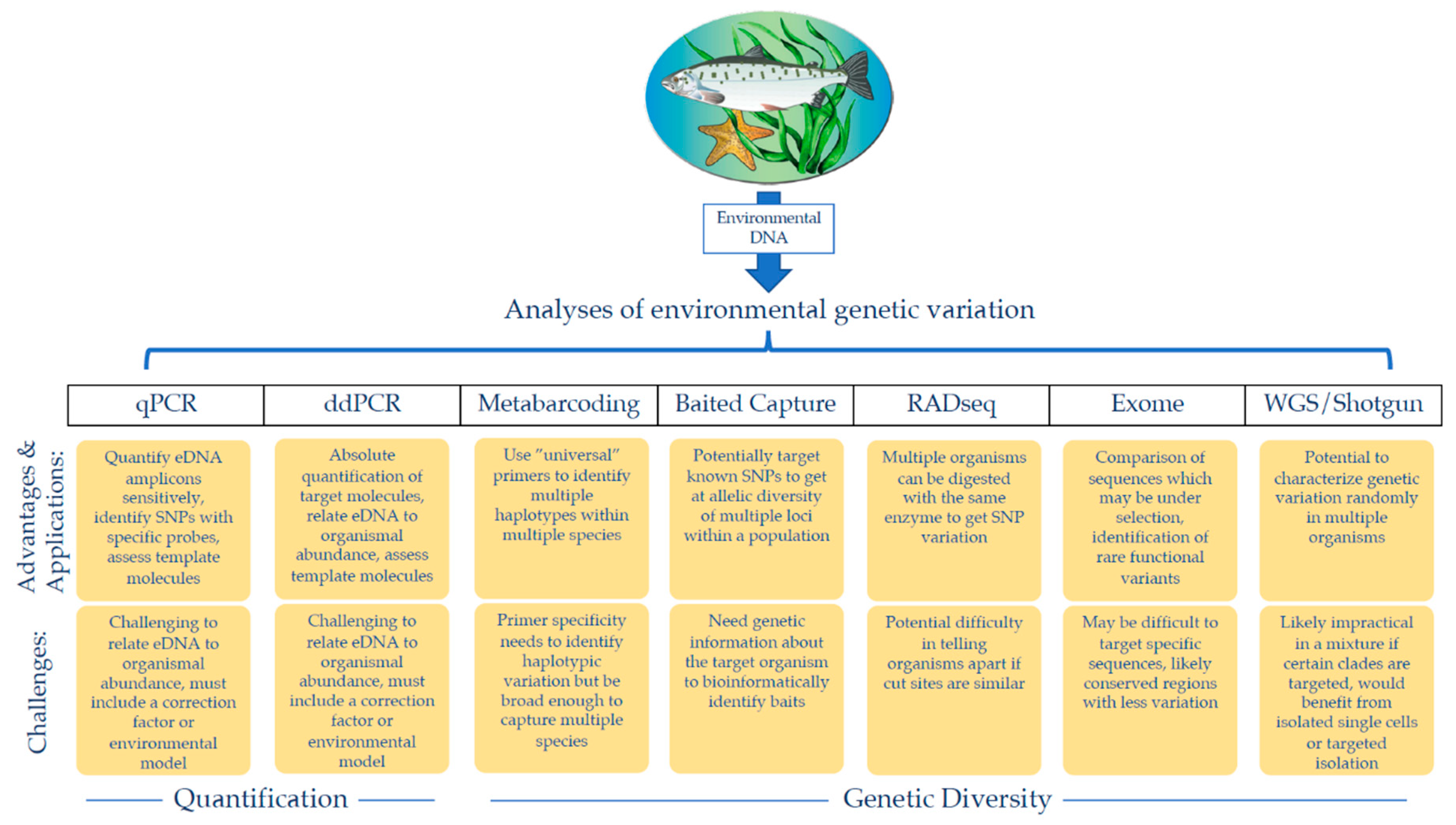{kind=link}
| Challenges | |||||||
|---|---|---|---|---|---|---|---|
| Abundance | Allelic Drop-Out | Bioinformatic Challenges | Identifying Individuals | Long-Term Datasets | Obtaining Nuclear Markers | ||
| Tools and Techniques | Automated sampling | Reduce spatial and temporal variance, especially for difficult-to-sample areas. Standardized deployment may help detect abundance changes in regular intervals [183,184]. | Document individual presence repeated through time [185]. | Precise, standardized capture across time and space [186,187]. | |||
| Baited capture methodology | Baited capture may reduce PCR amplification needed, reflecting true abundance ratios better. [188]. | Targeted capture of specific allelic variation [189]. | Identification of specific SNPs in a population. | Capture of specific allelic variation across time. | Target of nuDNA, especially SNP markers [189]. | ||
| Droplet digital PCR | Absolute quantification of target molecules [61,165,190]. | Sensitively amplify different allelic variation, could reduce drop-out [191]. | Provides absolute quantification of specific molecule abundance [192,193]. | Perhaps amplify and quantify single-cell eDNA. | Sensitively quantify changes in target molecules over time. | Amplify and quantify nuclear marker loci. | |
| eRNA | Increase temporal resolution [177]. | Increase temporal resolution of expressed alleles. | Identify allele-specific expression at the population level [194]. | Detection of live individuals [195]. | Examine expressed gene changes within a population or community [178,180]. | Increased temporal resolution of nuclear genetic variation [194,195]. | |
| Increased sequencing depth | Could increase detection chance of low copy number alleles [196]. Pool-seq may aid in elucidating allelic variation of large samples. | Could increase chance of detecting genetic diversity in replicates, perhaps allows for stricter filtering. | Could increase confidence in detection of individuals, especially if using single-cell techniques. | More robust datasets may show change throughout time at a finer scale. | Increased probability of detecting rare alleles. | ||
| Increased sequencing read length (nanopore, long-range mtDNA) | Capture of long reads or mtDNA genomes, see which alleles are linked [126] | Longer reads may help compile individuals’ mtGenomes [126]. | Links SNPs to form genomic or mtDNA haplotypes. | May see recombination patterns through time. | Increased genomic coverage [126]. | ||
| Single-cell sequencing | Approximation of unique individuals per sample assuming different genomes. | May have some allelic dropout if depth of sequencing is low [162]. | Identification of individuals allows for information to be analyzed with traditional population-genetics methodology. | Identify individuals based on cell genome [156]. | Identify changes in individual presence. | Target of nuDNA, perhaps even able to aid in sequencing of whole genome [155]. | |
| “Universal” primers specific enough for intraspecific variation | Alleles of multiple species identified in the same sample with same primer [72]. | Possibility to identify multiple individuals of multiple species if individuals can be sorted. | Multiple species targeted for community composition snapshots [70]. | Possibility to target nuclear markers in multiple species in the same sample. | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adams, C.I.M.; Knapp, M.; Gemmell, N.J.; Jeunen, G.-J.; Bunce, M.; Lamare, M.D.; Taylor, H.R. Beyond Biodiversity: Can Environmental DNA (eDNA) Cut It as a Population Genetics Tool? Genes 2019, 10, 192. https://doi.org/10.3390/genes10030192
Adams CIM, Knapp M, Gemmell NJ, Jeunen G-J, Bunce M, Lamare MD, Taylor HR. Beyond Biodiversity: Can Environmental DNA (eDNA) Cut It as a Population Genetics Tool? Genes. 2019; 10(3):192. https://doi.org/10.3390/genes10030192
Chicago/Turabian StyleAdams, Clare I.M., Michael Knapp, Neil J. Gemmell, Gert-Jan Jeunen, Michael Bunce, Miles D. Lamare, and Helen R. Taylor. 2019. "Beyond Biodiversity: Can Environmental DNA (eDNA) Cut It as a Population Genetics Tool?" Genes 10, no. 3: 192. https://doi.org/10.3390/genes10030192
APA StyleAdams, C. I. M., Knapp, M., Gemmell, N. J., Jeunen, G.-J., Bunce, M., Lamare, M. D., & Taylor, H. R. (2019). Beyond Biodiversity: Can Environmental DNA (eDNA) Cut It as a Population Genetics Tool? Genes, 10(3), 192. https://doi.org/10.3390/genes10030192