ProtParCon: A Framework for Processing Molecular Data and Identifying Parallel and Convergent Amino Acid Replacements



Abstract
1. Introduction
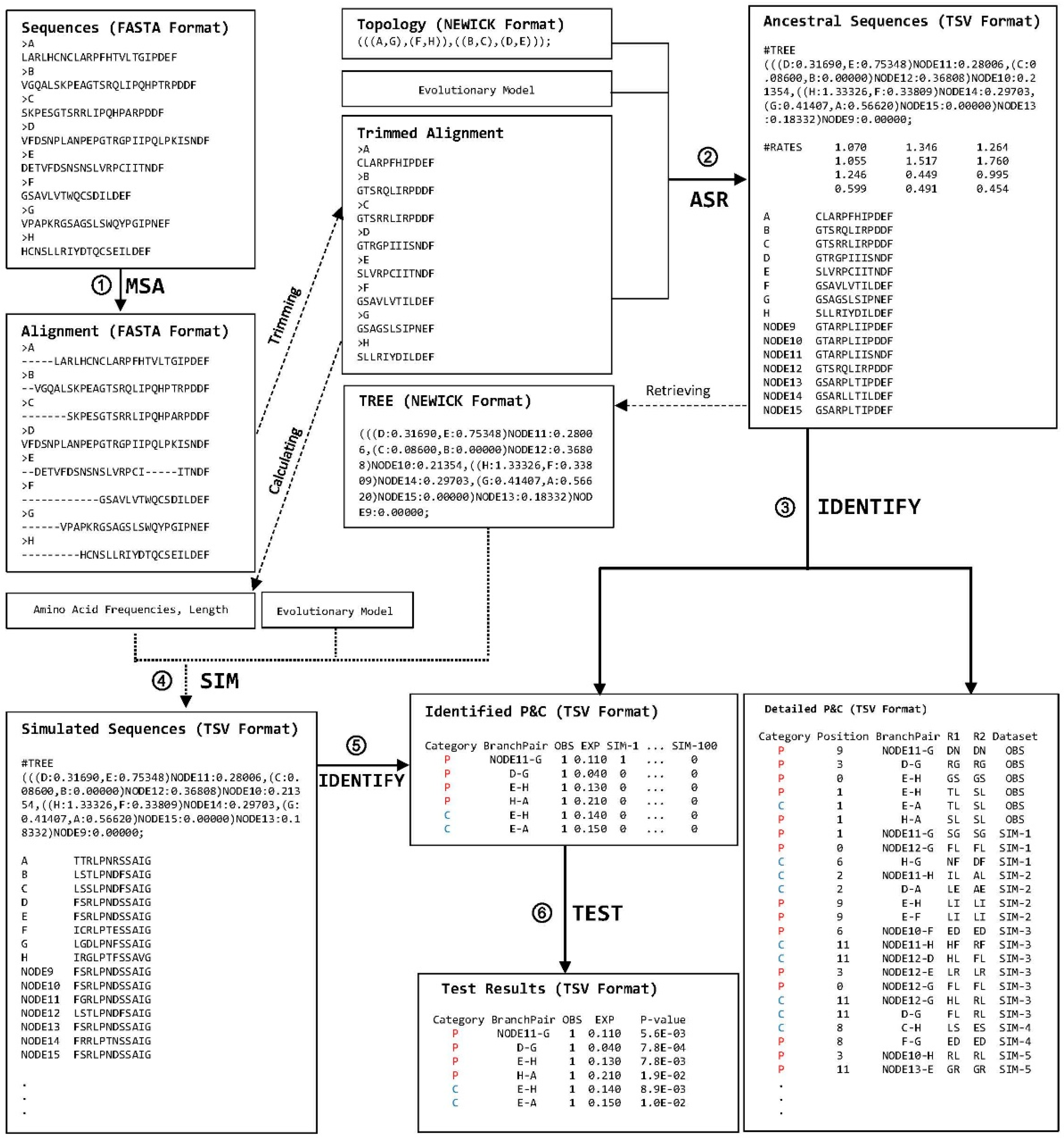
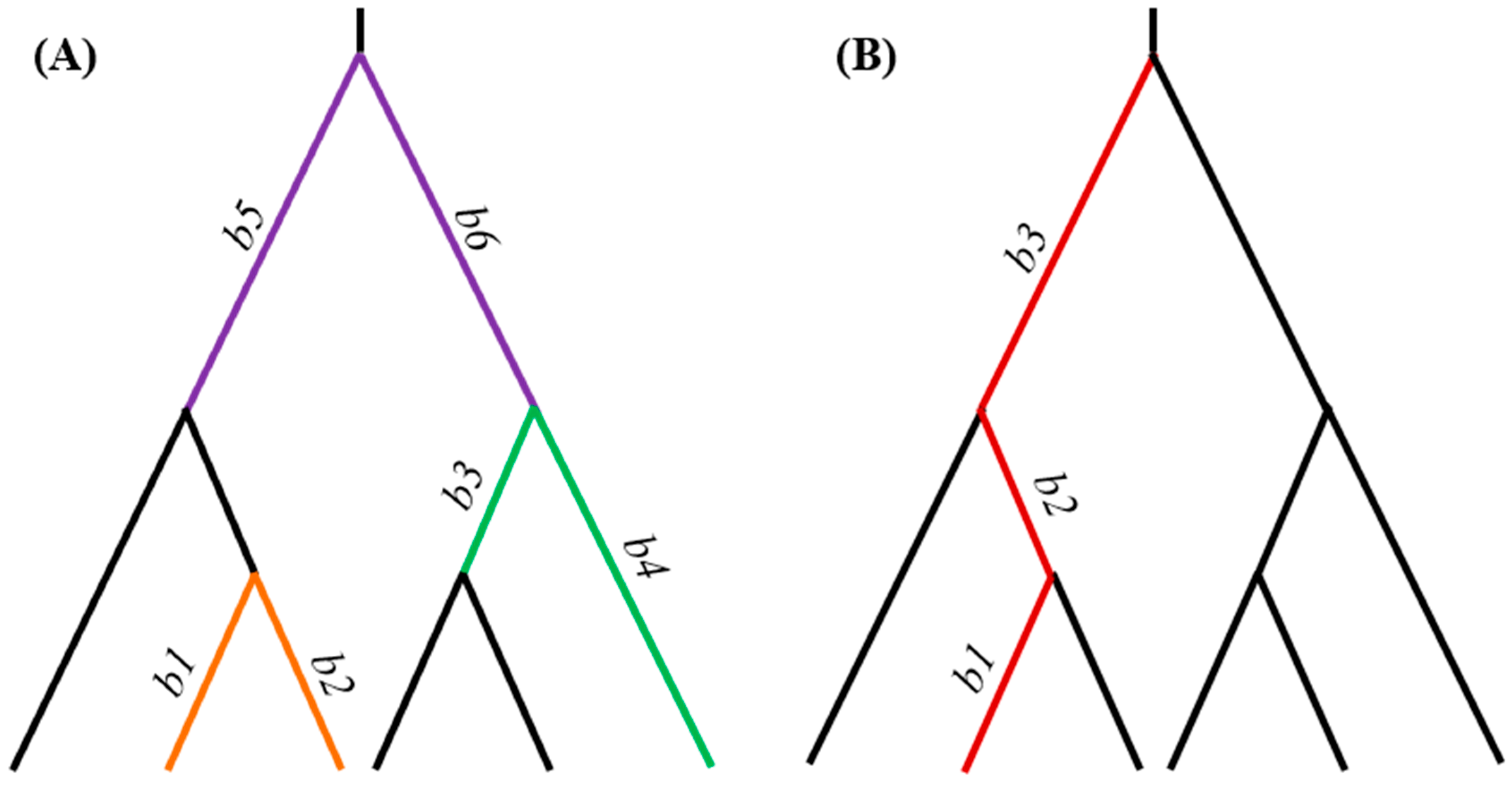
2. Description of the Computational Steps
2.1. Implementation of ProtParCon
2.2. Package Validation
3. Case Study: Parallel and Convergent Amino Acid Replacements in Lysozyme C Sequences
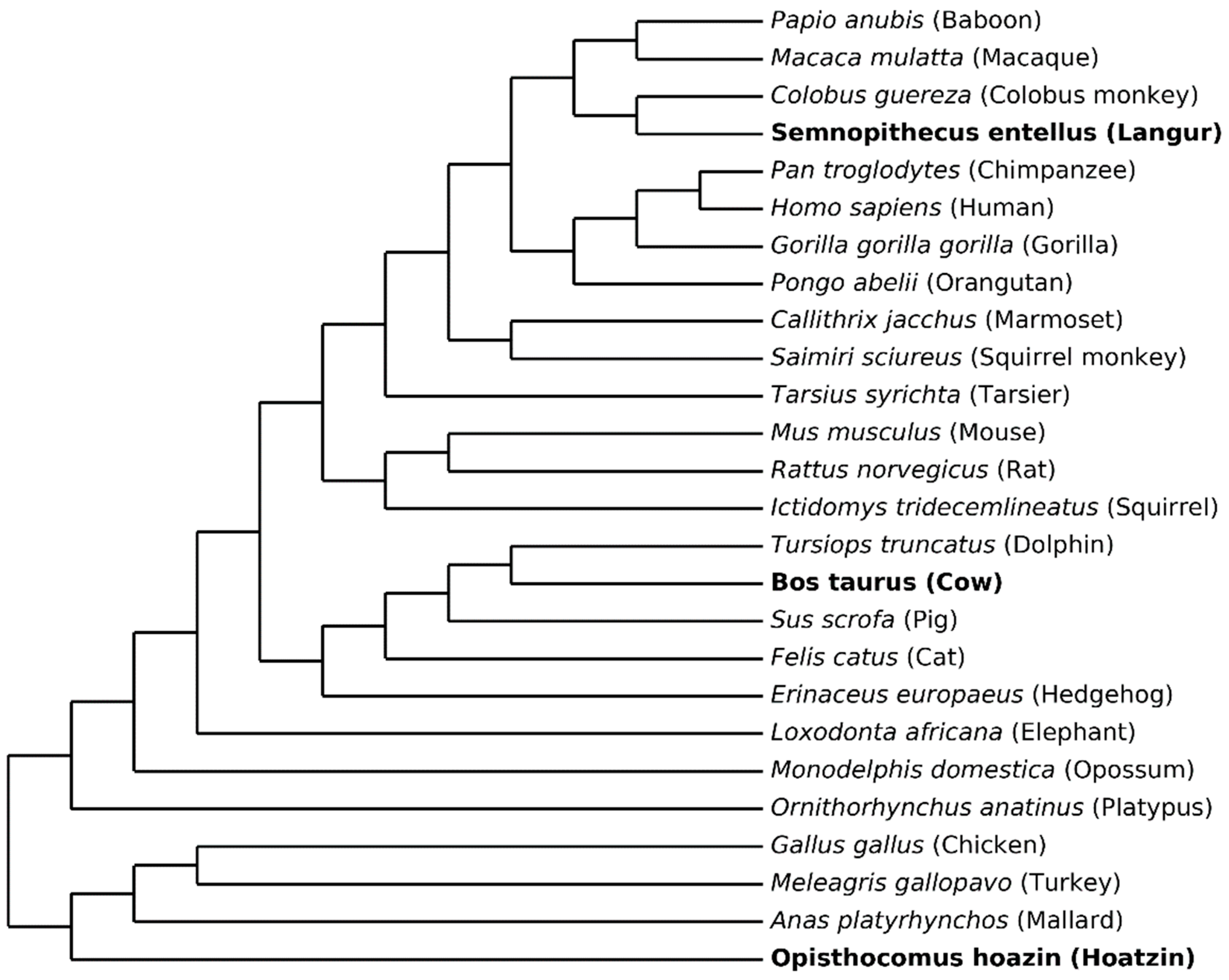
3.1. Data
3.2. Parallel and Convergent Amino Acid Replacements in Lysozyme c
3.3. Results and Tentative Conclusions
4. Discussion
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Nei, M. DNA Polymorphism and Adaptive Evolution, Plant Population Genetics, Breeding and Genetic Resources; Sinauer Associates Inc.: Sunderland, MA, USA, 1990; pp. 128–142. [Google Scholar]
- Pagel, M.D.; Harvey, P.H. Comparative methods for examining adaptation depend on evolutionary models. Folia Primatol. 1989, 53, 203–220. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Kumar, S. Detection of convergent and parallel evolution at the amino acid sequence level. Mol. Biol. Evol. 1997, 14, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Graur, D. Molecular and Genome Evolution; Sinauer Associates Inc.: Sunderland, MA, USA, 2016. [Google Scholar]
- Zou, Z.; Zhang, J. Are Convergent and Parallel Amino Acid Substitutions in Protein Evolution More Prevalent Than Neutral Expectations? Mol. Biol. Evol. 2015, 32, 2085–2096. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Tommaso, P.; Moretti, S.; Xenarios, I.; Orobitg, M.; Montanyola, A.; Chang, J.M.; Taly, J.F.; Notredame, C. T-Coffee: A web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. 2011, 39, W13–W17. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2-Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Delsuc, F.; Dufayard, J.F.; Gascuel, O. Estimating maximum likelihood phylogenies with PhyML. Methods Mol. Biol. 2009, 537, 113–137. [Google Scholar] [PubMed]
- Rambaut, A.; Grassly, N.C. Seq-Gen: An application for the Monte Carlo simulation of DNA sequence evolution along phylogenetic trees. Comput. Appl. Biosci. 1997, 13, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Shimodaira, H. An approximately unbiased test of phylogenetic tree selection. Syst. Biol. 2002, 51, 492–508. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.W.; Hahn, M.W. Determining the null model for detecting adaptive convergence from genomic data: A case study using echolocating mammals. Mol. Biol. Evol. 2015, 32, 1232–1236. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.B.; Schilling, J.W.; Wilson, A.C. Adaptive Evolution in the Stomach Lysozymes of Foregut Fermenters. Nature 1987, 330, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Kornegay, J.R.; Schilling, J.W.; Wilson, A.C. Molecular adaptation of a leaf-eating bird: Stomach lysozyme of the hoatzin. Mol. Biol. Evol. 1994, 11, 921–928. [Google Scholar] [PubMed]
- Irwin, D.M. Molecular evolution of ruminant lysozymes. EXS 1996, 75, 347–361. [Google Scholar] [PubMed]
- Consortium, T.U. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar]
- Prasad, A.B.; Marc, W.; Allard, D. Confirming the phylogeny of mammals by use of large comparative sequence data sets. Mol. Biol. Evol. 2008, 25, 1795–1808. [Google Scholar] [CrossRef] [PubMed]
- Irwin, D.M.; Biegel, J.M.; Stewart, C.B. Evolution of the mammalian lysozyme gene family. BMC Evol. Biol. 2011, 11, 166. [Google Scholar] [CrossRef] [PubMed]
- Esselstyn, J.A.; Oliveros, C.H.; Swanson, M.T.; Faircloth, B.C. Investigating difficult nodes in the placental mammal tree with expanded taxon sampling and thousands of ultraconserved elements. Genome Biol. Evol. 2017, 9, 2308–2321. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, E.D.; Mirarab, S.; Aberer, A.J.; Li, B.; Houde, P.; Li, C.; Ho, S.Y.; Faircloth, B.C.; Nabholz, B.; Howard, J.T.; et al. Whole-genome analyses resolve early branches in the tree of life of modern birds. Science 2014, 346, 1320–1331. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Bioinformatics 1992, 8, 275–282. [Google Scholar] [CrossRef]
- Yuan, F.; Nguyen, H.; Graur, D. A new null model for detecting adaptive parallelism and convergence in proteins. J. Mol. Evol. under review.
- Hughes, A.L.; Packer, B.; Welch, R.; Bergen, A.W.; Chanock, S.J.; Yeager, M. Widespread purifying selection at polymorphic sites in human protein-coding loci. Proc. Natl. Acad. Sci. USA 2003, 100, 15754–15757. [Google Scholar] [CrossRef] [PubMed]
- Landan, G.; Graur, D. Heads or tails: A simple reliability check for multiple sequence alignments. Mol. Biol. Evol. 2007, 24, 1380–1383. [Google Scholar] [CrossRef] [PubMed]
- Sela, I.; Ashkenazy, H.; Katoh, K.; Pupko, T. GUIDANCE2: Accurate detection of unreliable alignment regions accounting for the uncertainty of multiple parameters. Nucl. Acids Res. 2015, 43, 7–14. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Functions | Description | Supported Programs |
|---|---|---|
| oma | For OMA orthology database | N/A |
| msa | For multiple sequence alignment | MUSCLE [6] MAFFT [7] Clustal Omega [8] T-COFFEE [9] |
| asr | For ancestral states reconstruction | CODEML 1 RAxML [10] |
| mlt | For maximum-likelihood tree inference | FastTree [11] IQ-TREE [12] RAxML PhyML [13] |
| aut | For topology test 2 | IQ-TREE |
| sim | For protein sequence simulation | EVOLVER 1 Seq-Gen [14] |
| imc | P&C identification 3 | N/A |
| Branch Pair | Parallel Replacement | Convergent Replacement | ||||
|---|---|---|---|---|---|---|
| Obs. | Exp. | p-Value | Obs. | Exp. | p-Value | |
| Cow-Langur | 0 | 0.43 | 0.6505 | 1 | 0.35 | 0.0487 * |
| Cow-Hoatzin | 8 | 1.51 | 0.0000 ** | 1 | 0.78 | 0.184 |
| Langur-Hoatzin | 2 | 0.52 | 0.0159 * | 1 | 0.32 | 0.0415 * |
| Cow-Squirrel | 1 | 0.98 | 0.2569 | 0 | 0.23 | 0.7945 |
| Cow-Mouse | 2 | 1.96 | 0.3125 | 0 | 0.57 | 0.5655 |
| Cow-Tarsier | 2 | 1.51 | 0.1937 | 1 | 0.5 | 0.0902 |
| Cow-Colobus | 1 | 0.34 | 0.0462 * | 0 | 0.17 | 0.8437 |
| Hoatzin-Mallard | 1 | 0.61 | 0.1252 | 0 | 0.43 | 0.6505 |
| Hoatzin-Chicken | 1 | 0.35 | 0.0487 * | 0 | 0.27 | 0.7634 |
| Hoatzin-Turkey | 1 | 0.34 | 0.0462 * | 0 | 0.23 | 0.7945 |
| Hoatzin-Opossum | 7 | 3.08 | 0.0137 * | 0 | 0.14 | 0.8694 |
| Hoatzin-Elephant | 4 | 1.43 | 0.0155 * | 0 | 0.26 | 0.7711 |
| Hoatzin-Hedgehog | 1 | 1.6 | 0.5249 | 0 | 0.46 | 0.6313 |
| Hoatzin-Cat | 7 | 1.98 | 0.0010 ** | 1 | 0.73 | 0.1663 |
| Hoatzin-Pig | 4 | 2.1 | 0.0621 | 2 | 0.64 | 0.0273 * |
| Hoatzin-Dolphin | 1 | 0.94 | 0.2422 | 0 | 0.42 | 0.657 |
| Hoatzin-Squirrel | 1 | 1.22 | 0.6554 | 0 | 0.35 | 0.7047 |
| Hoatzin-Mouse | 5 | 2.62 | 0.0505 | 0 | 0.72 | 0.4868 |
| Hoatzin-Rat | 7 | 1.88 | 0.0007 ** | 1 | 0.59 | 0.1186 |
| Hoatzin-Tarsier | 5 | 2.02 | 0.0173 * | 0 | 0.74 | 0.4771 |
| Hoatzin-Colobus | 1 | 0.38 | 0.0563 | 0 | 0.28 | 0.7558 |
| Langur-Mouse | 1 | 0.57 | 0.1121 | 0 | 0.28 | 0.7558 |
| Langur-Rat | 1 | 0.55 | 0.1057 | 0 | 0.3 | 0.7408 |
| Langur-Tarsier | 2 | 0.48 | 0.0129 * | 0 | 0.27 | 0.7634 |
| Langur-Pig | 1 | 0.46 | 0.0783 | 0 | 0.29 | 0.7483 |
| Langur-Cat | 1 | 0.38 | 0.0563 | 0 | 0.27 | 0.7634 |
| Cat-Pig | 5 | 2.87 | 0.0714 | 0 | 0.00 | N/A |
| Cat-Cow | 5 | 1.5 | 0.0045 ** | 0 | 0.42 | 0.657 |
| Cat-Squirrel | 1 | 1.67 | 0.5026 | 0 | 0.11 | 0.8958 |
| Cat-Mouse | 4 | 2.21 | 0.0736 | 0 | 0.36 | 0.6977 |
| Cat-Rat | 2 | 1.88 | 0.2909 | 0 | 0.25 | 0.7788 |
| Cat-Tarsier | 3 | 2.03 | 0.1483 | 0 | 0.32 | 0.7261 |
| Cat-Colobus | 2 | 0.43 | 0.0096 ** | 0 | 0.16 | 0.8521 |
| Chicken-Elephant | 1 | 0.19 | 0.0159 * | 0 | 0.20 | 0.8187 |
| Chicken-Mouse | 0 | 0.31 | 0.7334 | 1 | 0.22 | 0.0209 * |
| Dolphin-Squirrel | 1 | 0.77 | 0.1805 | 0 | 0.13 | 0.8781 |
| Dolphin-Rat | 1 | 0.96 | 0.2495 | 0 | 0.31 | 0.7334 |
| Dolphin-Tarsier | 2 | 0.98 | 0.0767 | 0 | 0.28 | 0.7558 |
| Elephant-Hedgehog | 1 | 1.48 | 0.5645 | 0 | 0.17 | 0.8437 |
| Elephant-Cat | 2 | 1.54 | 0.2013 | 0 | 0.11 | 0.8958 |
| Elephant-Pig | 2 | 1.71 | 0.2454 | 1 | 0.11 | 0.0056 ** |
| Elephant-Cow | 1 | 1.02 | 0.7284 | 0 | 0.28 | 0.7558 |
| Elephant-Squirrel | 1 | 1.31 | 0.6233 | 0 | 0.03 | 0.9704 |
| Elephant-Mouse | 2 | 1.64 | 0.2270 | 0 | 0.15 | 0.8607 |
| Elephant-Rat | 2 | 1.59 | 0.2141 | 0 | 0.12 | 0.8869 |
| Elephant-Tarsier | 1 | 1.50 | 0.5578 | 0 | 0.19 | 0.827 |
| Elephant-Marmoset | 1 | 0.31 | 0.0392 * | 0 | 0.08 | 0.9231 |
| Hedgehog-Cat | 3 | 2.39 | 0.2192 | 0 | 0.00 | N/A |
| Hedgehog-Pig | 5 | 2.75 | 0.0608 | 0 | 0.00 | N/A |
| Hedgehog-Dolphin | 3 | 0.93 | 0.0150 * | 0 | 0.32 | 0.7261 |
| Hedgehog-Cow | 2 | 1.52 | 0.1962 | 0 | 0.38 | 0.6839 |
| Hedgehog-Squirrel | 3 | 1.58 | 0.0761 | 0 | 0.08 | 0.9231 |
| Hedgehog-Mouse | 5 | 2.37 | 0.0339 * | 0 | 0.36 | 0.6977 |
| Hedgehog-Rat | 4 | 1.91 | 0.0449 * | 0 | 0.33 | 0.7189 |
| Hedgehog-Tarsier | 3 | 1.94 | 0.1322 | 0 | 0.29 | 0.7483 |
| Hedgehog-Colobus | 2 | 0.37 | 0.0064 ** | 0 | 0.21 | 0.8106 |
| Mallard-Platypus | 1 | 0.47 | 0.0812 | 0 | 0.38 | 0.6839 |
| Mallard-Mouse | 1 | 0.62 | 0.1285 | 0 | 0.41 | 0.6637 |
| Mallard-Rat | 1 | 0.40 | 0.0616 | 0 | 0.46 | 0.6313 |
| Opossum-Elephant | 3 | 1.31 | 0.0441 * | 0 | 0.24 | 0.7866 |
| Opossum-Hedgehog | 1 | 1.68 | 0.4995 | 0 | 0.53 | 0.5886 |
| Opossum-Cat | 3 | 1.67 | 0.0888 | 0 | 0.51 | 0.6005 |
| Opossum-Pig | 3 | 1.90 | 0.1253 | 0 | 0.54 | 0.5827 |
| Opossum-Cow | 0 | 1.35 | 0.2592 | 1 | 0.48 | 0.0842 |
| Opossum-Squirrel | 2 | 1.05 | 0.0897 | 0 | 0.33 | 0.7189 |
| Opossum-Mouse | 4 | 1.72 | 0.0309 * | 0 | 0.59 | 0.5543 |
| Opossum-Rat | 5 | 1.66 | 0.0072 ** | 0 | 0.68 | 0.5066 |
| Opossum-Tarsier | 2 | 1.74 | 0.2534 | 0 | 0.45 | 0.6376 |
| Opossum-Marmoset | 1 | 0.35 | 0.0487 * | 0 | 0.17 | 0.8437 |
| Platypus-Hedgehog | 2 | 1.15 | 0.1099 | 0 | 0.38 | 0.6839 |
| Platypus-Pig | 2 | 1.45 | 0.1787 | 0 | 0.45 | 0.6376 |
| Platypus-Dolphin | 1 | 0.61 | 0.1252 | 0 | 0.36 | 0.6977 |
| Platypus-Squirrel | 1 | 0.85 | 0.2093 | 0 | 0.33 | 0.7189 |
| Platypus-Mouse | 1 | 1.51 | 0.5545 | 1 | 0.45 | 0.0754 |
| Platypus-Rat | 2 | 1.45 | 0.1787 | 0 | 0.47 | 0.625 |
| Platypus-Tarsier | 1 | 1.19 | 0.6662 | 0 | 0.48 | 0.6188 |
| Pig-Cow | 1 | 1.83 | 0.4540 | 0 | 0.32 | 0.7261 |
| Pig-Squirrel | 1 | 1.51 | 0.5545 | 0 | 0.10 | 0.9048 |
| Pig-Mouse | 4 | 2.36 | 0.0909 | 1 | 0.34 | 0.0462 * |
| Pig-Rat | 6 | 2.23 | 0.0080 ** | 0 | 0.35 | 0.7047 |
| Pig-Tarsier | 3 | 2.29 | 0.1986 | 0 | 0.24 | 0.7866 |
| Rat-Tarsier | 5 | 1.87 | 0.0123 * | 0 | 0.36 | 0.6977 |
| Squirrel-Mouse | 1 | 1.53 | 0.5478 | 0 | 0.13 | 0.8781 |
| Squirrel-Tarsier | 1 | 1.48 | 0.5645 | 0 | 0.20 | 0.8187 |
| Squirrel-Marmoset | 1 | 0.34 | 0.0462 * | 0 | 0.10 | 0.9048 |
| Mouse-Tarsier | 3 | 2.23 | 0.1866 | 0 | 0.34 | 0.7118 |
| Mouse-Marmoset | 1 | 0.37 | 0.0537 | 0 | 0.22 | 0.8025 |
| Mouse-Colobus | 1 | 0.39 | 0.0589 | 0 | 0.18 | 0.8353 |
| Tarsier-Colobus | 1 | 0.42 | 0.0670 | 0 | 0.16 | 0.8521 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, F.; Nguyen, H.; Graur, D. ProtParCon: A Framework for Processing Molecular Data and Identifying Parallel and Convergent Amino Acid Replacements. Genes 2019, 10, 181. https://doi.org/10.3390/genes10030181
Yuan F, Nguyen H, Graur D. ProtParCon: A Framework for Processing Molecular Data and Identifying Parallel and Convergent Amino Acid Replacements. Genes. 2019; 10(3):181. https://doi.org/10.3390/genes10030181
Chicago/Turabian StyleYuan, Fei, Hoa Nguyen, and Dan Graur. 2019. "ProtParCon: A Framework for Processing Molecular Data and Identifying Parallel and Convergent Amino Acid Replacements" Genes 10, no. 3: 181. https://doi.org/10.3390/genes10030181
APA StyleYuan, F., Nguyen, H., & Graur, D. (2019). ProtParCon: A Framework for Processing Molecular Data and Identifying Parallel and Convergent Amino Acid Replacements. Genes, 10(3), 181. https://doi.org/10.3390/genes10030181