Transcriptome Profiling and Molecular Therapeutic Advances in Cystic Fibrosis: Recent Insights


Abstract
1. Introduction: Cystic Fibrosis
2. Transcriptome Profiling in Cystic Fibrosis
3. Cystic Fibrosis-Associated Genes and Their Pathways
3.1. Signal Transduction Pathways
3.2. Immune System Pathways
4. Molecular Advances in Developing Therapeutics for Cystic Fibrosis
4.1. Gene Replacement Therapy and Gene Editing
4.2. Cystic Fibrosis-Associated Molecules: Small and Large Molecular Targets for Treating Cystic Fibrosis
5. Emerging Targets for Transcriptome Profiling
5.1. Non-Coding RNAs
5.2. Alternate Splicing and Cystic Fibrosis-Relevant Transcript Isoforms
6. Challenges for Cystic Fibrosis Transcriptomics Studies
6.1. Lack of Useful Model Systems
6.2. Bulk Transcriptome Profiling
7. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Madacsy, T.; Pallagi, P.; Maleth, J. Cystic fibrosis of the pancreas: The role of CFTR channel in the regulation of intracellular Ca(2+) signaling and mitochondrial function in the exocrine pancreas. Front. Physiol. 2018, 9, 1585. [Google Scholar] [CrossRef] [PubMed]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.L.; et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- Veit, G.; Avramescu, R.G.; Chiang, A.N.; Houck, S.A.; Cai, Z.; Peters, K.W.; Hong, J.S.; Pollard, H.B.; Guggino, W.B.; Balch, W.E.; et al. From CFTR biology toward combinatorial pharmacotherapy: Expanded classification of cystic fibrosis mutations. Mol. Biol. Cell 2016, 27, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Fajac, I.; Wainwright, C.E. New treatments targeting the basic defects in cystic fibrosis. Presse Med. 2017, 46, E165–E175. [Google Scholar] [CrossRef] [PubMed]
- Carithers, L.J.; Moore, H.M. The genotype-tissue expression (GTEx) project. Biopreserv. Biobank. 2015, 13, 307–308. [Google Scholar] [CrossRef] [PubMed]
- Burney, T.J.; Davies, J.C. Gene therapy for the treatment of cystic fibrosis. Appl. Clin. Genet. 2012, 5, 29–36. [Google Scholar] [PubMed]
- Cooney, A.L.; McCray, P.B., Jr.; Sinn, P.L. Cystic fibrosis gene therapy: Looking back, looking forward. Genes (Basel) 2018, 9, 538. [Google Scholar] [CrossRef] [PubMed]
- Mekus, F.; Ballmann, M.; Bronsveld, I.; Bijman, J.; Veeze, H.; Tummler, B. Categories of ΔF508 homozygous cystic fibrosis twin and sibling pairs with distinct phenotypic characteristics. Twin Res. 2000, 3, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Corvol, H.; Blackman, S.M.; Boelle, P.Y.; Gallins, P.J.; Pace, R.G.; Stonebraker, J.R.; Accurso, F.J.; Clement, A.; Collaco, J.M.; Dang, H.; et al. Genome-wide association meta-analysis identifies five modifier loci of lung disease severity in cystic fibrosis. Nat. Commun. 2015, 6, 8382. [Google Scholar] [CrossRef] [PubMed]
- Kormann, M.S.D.; Dewerth, A.; Eichner, F.; Baskaran, P.; Hector, A.; Regamey, N.; Hartl, D.; Handgretinger, R.; Antony, J.S. Transcriptomic profile of cystic fibrosis patients identifies type I interferon response and ribosomal stalk proteins as potential modifiers of disease severity. PLoS ONE 2017, 12, e0183526. [Google Scholar] [CrossRef] [PubMed]
- Worgall, S.; Heguy, A.; Luettich, K.; O’Connor, T.P.; Harvey, B.G.; Quadri, L.E.; Crystal, R.G. Similarity of gene expression patterns in human alveolar macrophages in response to Pseudomonas aeruginosa and Burkholderia cepacia. Infect. Immun. 2005, 73, 5262–5268. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.M.; Merlo, C.A.; Reynolds, J.B.; Zeitlin, P.L.; Garcia, J.G.; Guggino, W.B.; Boyle, M.P. Respiratory epithelial gene expression in patients with mild and severe cystic fibrosis lung disease. Am. J. Respir. Cell Mol. Biol. 2006, 35, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Verhaeghe, C.; Remouchamps, C.; Hennuy, B.; Vanderplasschen, A.; Chariot, A.; Tabruyn, S.P.; Oury, C.; Bours, V. Role of IKK and ERK pathways in intrinsic inflammation of cystic fibrosis airways. Biochem. Pharmacol. 2007, 73, 1982–1994. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.M.; Hurd, H.; Wu, Y.; Martino, M.E.; Jones, L.; Brighton, B.; Boucher, R.C.; O’Neal, W.K. Azithromycin treatment alters gene expression in inflammatory, lipid metabolism, and cell cycle pathways in well-differentiated human airway epithelia. PLoS ONE 2009, 4, e5806. [Google Scholar] [CrossRef] [PubMed]
- Ogilvie, V.; Passmore, M.; Hyndman, L.; Jones, L.; Stevenson, B.; Wilson, A.; Davidson, H.; Kitchen, R.R.; Gray, R.D.; Shah, P.; et al. Differential global gene expression in cystic fibrosis nasal and bronchial epithelium. Genomics 2011, 98, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Hampton, T.H.; Ballok, A.E.; Bomberger, J.M.; Rutkowski, M.R.; Barnaby, R.; Coutermarsh, B.; Conejo-Garcia, J.R.; O’Toole, G.A.; Stanton, B.A. Does the ΔF508-CFTR mutation induce a proinflammatory response in human airway epithelial cells? Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L509–L518. [Google Scholar] [CrossRef] [PubMed]
- Levy, H.; Wang, X.; Kaldunski, M.; Jia, S.; Kramer, J.; Pavletich, S.J.; Reske, M.; Gessel, T.; Yassai, M.; Quasney, M.W.; et al. Transcriptional signatures as a disease-specific and predictive inflammatory biomarker for type 1 diabetes. Genes Immun. 2012, 13, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.A.; Sousa, L.; Barreto, C.; Amaral, M.D. Changes in transcriptome of native nasal epithelium expressing F508del-CFTR and intersecting data from comparable studies. Respir. Res. 2013, 14, 38. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.L.; Blohmke, C.J.; Falsafi, R.; Fjell, C.D.; Madera, L.; Turvey, S.E.; Hancock, R.E. Rescue of dysfunctional autophagy attenuates hyperinflammatory responses from cystic fibrosis cells. J. Immunol. 2013, 190, 1227–1238. [Google Scholar] [CrossRef] [PubMed]
- Stanke, F.; van Barneveld, A.; Hedtfeld, S.; Wolfl, S.; Becker, T.; Tummler, B. The CF-modifying gene EHF promotes p.Phe508del-CFTR residual function by altering protein glycosylation and trafficking in epithelial cells. Eur. J. Hum. Genet. 2014, 22, 660–666. [Google Scholar] [CrossRef] [PubMed]
- Chesne, J.; Danger, R.; Botturi, K.; Reynaud-Gaubert, M.; Mussot, S.; Stern, M.; Danner-Boucher, I.; Mornex, J.F.; Pison, C.; Dromer, C.; et al. Systematic analysis of blood cell transcriptome in end-stage chronic respiratory diseases. PLoS ONE 2014, 9, e109291. [Google Scholar] [CrossRef] [PubMed]
- Voisin, G.; Bouvet, G.F.; Legendre, P.; Dagenais, A.; Masse, C.; Berthiaume, Y. Oxidative stress modulates the expression of genes involved in cell survival in ΔF508 cystic fibrosis airway epithelial cells. Physiol. Genomics 2014, 46, 634–646. [Google Scholar] [CrossRef] [PubMed]
- O’Neal, W.K.; Gallins, P.; Pace, R.G.; Dang, H.; Wolf, W.E.; Jones, L.C.; Guo, X.; Zhou, Y.H.; Madar, V.; Huang, J.; et al. Gene expression in transformed lymphocytes reveals variation in endomembrane and HLA pathways modifying cystic fibrosis pulmonary phenotypes. Am. J. Hum. Genet. 2015, 96, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Zeitlin, P.L.; Diener-West, M.; Callahan, K.A.; Lee, S.; Talbot, C.C., Jr.; Pollard, B.; Boyle, M.P.; Lechtzin, N. Digitoxin for airway inflammation in cystic fibrosis: Preliminary assessment of safety, pharmacokinetics, and dose finding. Ann. Am. Thorac. Soc. 2017, 14, 220–229. [Google Scholar] [PubMed]
- Polineni, D.; Dang, H.; Gallins, P.J.; Jones, L.C.; Pace, R.G.; Stonebraker, J.R.; Commander, L.A.; Krenicky, J.E.; Zhou, Y.H.; Corvol, H.; et al. Airway mucosal host defense is key to genomic regulation of cystic fibrosis lung disease severity. Am. J. Respir. Crit. Care Med. 2017, 197, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.; Poppenberg, K.E.; Wong, L.; Chen, Y.; Borowitz, D.; Goetz, D.; Sheehan, D.; Frederick, C.; Tutino, V.M.; Meng, H.; et al. RNA sequencing data from neutrophils of patients with cystic fibrosis reveals potential for developing biomarkers for pulmonary exacerbations. J. Cyst. Fibros. 2018. [Google Scholar] [CrossRef] [PubMed]
- Levy, H.; Jia, S.; Pan, A.; Zhang, X.; Kaldunski, M.L.; Nugent, M.L.; Reske, M.; Feliciano, R.A.; Quintero, D.; Renda, M.M.; et al. Identification of molecular signatures of cystic fibrosis disease status using plasma-based functional genomics. Physiol. Genom. 2019, 51, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Casamassimi, A.; Federico, A.; Rienzo, M.; Esposito, S.; Ciccodicola, A. Transcriptome profiling in human diseases: New advances and perspectives. Int. J. Mol. Sci. 2017, 18, 1652. [Google Scholar] [CrossRef] [PubMed]
- Kasoju, N.; Wang, H.; Zhang, B.; George, J.; Gao, S.; Triffitt, J.T.; Cui, Z.; Ye, H. Transcriptomics of human multipotent mesenchymal stromal cells: Retrospective analysis and future prospects. Biotechnol. Adv. 2017, 35, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Yu, L.; Zou, F.; Hu, H.; Liu, K.; Lin, Z. Gene expression profiling and functional analysis reveals that p53 pathway-related gene expression is highly activated in cancer cells treated by cold atmospheric plasma-activated medium. PeerJ 2017, 5, e3751. [Google Scholar] [CrossRef] [PubMed]
- Westermann, A.J.; Barquist, L.; Vogel, J. Resolving host–pathogen interactions by dual RNA-seq. PLoS Pathogens 2017, 13, e1006033. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.W.; Touchman, J.W.; Blakesley, R.W.; Bouffard, G.G.; Beckstrom-Sternberg, S.M.; Margulies, E.H.; Blanchette, M.; Siepel, A.C.; Thomas, P.J.; McDowell, J.C.; et al. Comparative analyses of multi-species sequences from targeted genomic regions. Nature 2003, 424, 788–793. [Google Scholar] [CrossRef] [PubMed]
- Collawn, J.F.; Matalon, S. CFTR and lung homeostasis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L917–L923. [Google Scholar] [CrossRef] [PubMed]
- Byrne, A.J.; Maher, T.M.; Lloyd, C.M. Pulmonary macrophages: A new therapeutic pathway in fibrosing lung disease? Trends Mol. Med. 2016, 22, 303–316. [Google Scholar] [CrossRef] [PubMed]
- Ratner, D.; Mueller, C. Immune responses in cystic fibrosis: Are they intrinsically defective? Am. J. Respir. Cell Mol. Biol. 2012, 46, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Del Porto, P.; Cifani, N.; Guarnieri, S.; Di Domenico, E.G.; Mariggio, M.A.; Spadaro, F.; Guglietta, S.; Anile, M.; Venuta, F.; Quattrucci, S.; et al. Dysfunctional CFTR alters the bactericidal activity of human macrophages against Pseudomonas aeruginosa. PLoS ONE 2011, 6, e19970. [Google Scholar]
- Ideozu, J.E.; Zhang, X.; Pan, A.; Ashrafi, Z.; Woods, K.J.; Hessner, M.J.; Simpson, P.; Levy, H. Increased expression of plasma-induced ABCC1 mRNA in cystic fibrosis. Int. J. Mol. Sci. 2017, 18, 1752. [Google Scholar] [CrossRef] [PubMed]
- Kiel, C.; Yus, E.; Serrano, L. Engineering signal transduction pathways. Cell 2010, 140, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Reilly, R.; Mroz, M.S.; Dempsey, E.; Wynne, K.; Keely, S.J.; McKone, E.F.; Hiebel, C.; Behl, C.; Coppinger, J.A. Targeting the PI3K/AKT/mTOR signalling pathway in cystic fibrosis. Sci. Rep. 2017, 7, 7642. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Strubberg, A.M.; Liu, J.; Walker, N.M.; Stefanski, C.D.; MacLeod, R.J.; Magness, S.T.; Clarke, L.L. CFTR modulates Wnt/β-catenin signaling and stem cell proliferation in murine intestine. Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 253–271. [Google Scholar] [CrossRef] [PubMed]
- Bodas, M.; Vij, N. The NF-κβ signaling in cystic fibrosis lung disease: Pathophysiology and therapeutic potential. Discov. Med. 2010, 9, 346–356. [Google Scholar] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. Nf-κβ signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Verhaeghe, C.; Delbecque, K.; de Leval, L.; Oury, C.; Bours, V. Early inflammation in the airways of a cystic fibrosis foetus. J. Cyst. Fibros. 2007, 6, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Quon, B.S.; Rowe, S.M. New and emerging targeted therapies for cystic fibrosis. BMJ 2016, 352, i859. [Google Scholar] [CrossRef] [PubMed]
- Schwank, G.; Koo, B.K.; Sasselli, V.; Dekkers, J.F.; Heo, I.; Demircan, T.; Sasaki, N.; Boymans, S.; Cuppen, E.; van der Ent, C.K.; et al. Functional repair of CFTR by CRISPR/cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell 2013, 13, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Roy, B.; Zhao, J.; Yang, C.; Luo, W.; Xiong, T.; Li, Y.; Fang, X.; Gao, G.; Singh, C.O.; Madsen, L.; et al. CRISPR/cascade 9-mediated genome editing-challenges and opportunities. Front. Genet. 2018, 9, 240. [Google Scholar] [CrossRef] [PubMed]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.; Burton, B.; Cao, D.; Neuberger, T.; Turnbull, A.; Singh, A.; Joubran, J.; Hazlewood, A.; et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, vx-770. Proc. Natl. Acad. Sci. USA 2009, 106, 18825–18830. [Google Scholar] [CrossRef] [PubMed]
- Koscielny, G.; An, P.; Carvalho-Silva, D.; Cham, J.A.; Fumis, L.; Gasparyan, R.; Hasan, S.; Karamanis, N.; Maguire, M.; Papa, E.; et al. Open targets: A platform for therapeutic target identification and validation. Nucleic Acids Res. 2017, 45, D985–D994. [Google Scholar] [CrossRef] [PubMed]
- Beringer, P.M.; Owens, H.; Nguyen, A.; Benitez, D.; Rao, A.; D’Argenio, D.Z. Pharmacokinetics of doxycycline in adults with cystic fibrosis. Antimicrob. Agents Chemother. 2012, 56, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Hermes, W.A.; Alvarez, J.A.; Lee, M.J.; Chesdachai, S.; Lodin, D.; Horst, R.; Tangpricha, V. Prospective, randomized, double-blind, parallel-group, comparative effectiveness clinical trial comparing a powder vehicle compound of vitamin d with an oil vehicle compound in adults with cystic fibrosis. J. Parenter. Enteral Nutr. 2017, 41, 952–958. [Google Scholar] [CrossRef] [PubMed]
- Dalakas, M.C. Immunotherapy in myasthenia gravis in the era of biologics. Nat. Rev. Neurol. 2018, 15, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Narozna, B.; Langwinski, W.; Szczepankiewicz, A. Non-coding RNAs in pediatric airway diseases. Genes 2017, 8, 348. [Google Scholar] [CrossRef] [PubMed]
- Glasgow, A.M.A.; De Santi, C.; Greene, C.M. Non-coding RNA in cystic fibrosis. Biochem. Soc. Trans. 2018, 46, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Hammond, S.M. An overview of microRNAs. Adv. Drug Deliv. Rev. 2015, 87, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Croce, C.M. The role of microRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef] [PubMed]
- Stolzenburg, L.R.; Harris, A. The role of microRNAs in chronic respiratory disease: Recent insights. Biol. Chem. 2018, 399, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Oglesby, I.K.; McKiernan, P.J. MiRNA expression in cystic fibrosis bronchial epithelial cells. Methods Mol. Biol. 2017, 1509, 57–69. [Google Scholar] [PubMed]
- Bhattacharyya, S.; Balakathiresan, N.S.; Dalgard, C.; Gutti, U.; Armistead, D.; Jozwik, C.; Srivastava, M.; Pollard, H.B.; Biswas, R. Elevated miR-155 promotes inflammation in cystic fibrosis by driving hyperexpression of interleukin-8. J. Biol. Chem. 2011, 286, 11604–11615. [Google Scholar] [CrossRef] [PubMed]
- Oglesby, I.K.; Bray, I.M.; Chotirmall, S.H.; Stallings, R.L.; O’Neill, S.J.; McElvaney, N.G.; Greene, C.M. MiR-126 is downregulated in cystic fibrosis airway epithelial cells and regulates TOM1 expression. J. Immunol. 2010, 184, 1702–1709. [Google Scholar] [CrossRef] [PubMed]
- Palazzo, A.F.; Lee, E.S. Non-coding RNA: What is functional and what is junk? Front. Genet. 2015, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and functions of long noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.; Liu, G. NcRNA-regulated immune response and its role in inflammatory lung diseases. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L1076–L1087. [Google Scholar] [CrossRef] [PubMed]
- Saayman, S.M.; Ackley, A.; Burdach, J.; Clemson, M.; Gruenert, D.C.; Tachikawa, K.; Chivukula, P.; Weinberg, M.S.; Morris, K.V. Long non-coding RNA BGas regulates the cystic fibrosis transmembrane conductance regulator. Mol. Ther. 2016, 24, 1351–1357. [Google Scholar] [CrossRef] [PubMed]
- McKiernan, P.J.; Molloy, K.; Cryan, S.A.; McElvaney, N.G.; Greene, C.M. Long noncoding RNA are aberrantly expressed in vivo in the cystic fibrosis bronchial epithelium. Int. J. Biochem. Cell Biol. 2014, 52, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Balloy, V.; Koshy, R.; Perra, L.; Corvol, H.; Chignard, M.; Guillot, L.; Scaria, V. Bronchial epithelial cells from cystic fibrosis patients express a specific long non-coding RNA signature upon Pseudomonas aeruginosa infection. Front. Cell Infect. Microbiol. 2017, 7, 218. [Google Scholar] [CrossRef] [PubMed]
- Kamei, S.; Maruta, K.; Fujikawa, H.; Nohara, H.; Ueno-Shuto, K.; Tasaki, Y.; Nakashima, R.; Kawakami, T.; Eto, Y.; Suico, M.A.; et al. Integrative expression analysis identifies a novel interplay between CFTR and linc-SUMF1-2 that involves CF-associated gene dysregulation. Biochem. Biophys. Res. Commun. 2019, 509, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Qian, T.; Wang, X.; Liu, J.; Gu, X. Noncoding RNAs and their potential therapeutic applications in tissue engineering. Engineering 2017, 3, 3–15. [Google Scholar] [CrossRef]
- Fabbri, E.; Tamanini, A.; Jakova, T.; Gasparello, J.; Manicardi, A.; Corradini, R.; Sabbioni, G.; Finotti, A.; Borgatti, M.; Lampronti, I.; et al. A peptide nucleic acid against microRNA miR-145-5p enhances the expression of the cystic fibrosis transmembrane conductance regulator (CFTR) in calu-3 cells. Molecules 2017, 23, 71. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, L.F.; Beazley-Long, N. Alternative RNA splicing: Contribution to pain and potential therapeutic strategy. Drug Discov. Today 2016, 21, 1787–1798. [Google Scholar] [CrossRef] [PubMed]
- Cieply, B.; Carstens, R.P. Functional roles of alternative splicing factors in human disease. Wiley Interdiscip. Rev. RNA 2015, 6, 311–326. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Pham, M.H.C.; Ko, K.S.; Rhee, B.D.; Han, J. Alternative splicing isoforms in health and disease. Pflugers Arch. 2018, 470, 995–1016. [Google Scholar] [CrossRef] [PubMed]
- Niu, L.; Huang, W.; Umbach, D.M.; Li, L. Iuta: A tool for effectively detecting differential isoform usage from RNA-seq data. BMC Genom. 2014, 15, 862. [Google Scholar] [CrossRef] [PubMed]
- Stricker, T.P.; Brown, C.D.; Bandlamudi, C.; McNerney, M.; Kittler, R.; Montoya, V.; Peterson, A.; Grossman, R.; White, K.P. Robust stratification of breast cancer subtypes using differential patterns of transcript isoform expression. PLoS Genet. 2017, 13, e1006589. [Google Scholar] [CrossRef] [PubMed]
- Khatoon, Z.; Figler, B.; Zhang, H.; Cheng, F. Introduction to RNA-seq and its applications to drug discovery and development. Drug Dev. Res. 2014, 75, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Snouwaert, J.N.; Brigman, K.K.; Latour, A.M.; Malouf, N.N.; Boucher, R.C.; Smithies, O.; Koller, B.H. An animal-model for cystic-fibrosis made by gene targeting. Science 1992, 257, 1083–1088. [Google Scholar] [CrossRef] [PubMed]
- Mou, H.; Brazauskas, K.; Rajagopal, J. Personalized medicine for cystic fibrosis: Establishing human model systems. Pediatr. Pulmonol. 2015, 50 (Suppl. 40), S14–S23. [Google Scholar] [CrossRef] [PubMed]
- Harris, A. Towards an ovine model of cystic fibrosis. Hum. Mol. Genet. 1997, 6, 2191–2193. [Google Scholar] [CrossRef] [PubMed]
- Rogers, C.S.; Hao, Y.; Rokhlina, T.; Samuel, M.; Stoltz, D.A.; Li, Y.; Petroff, E.; Vermeer, D.W.; Kabel, A.C.; Yan, Z.; et al. Production of CFTR-null and CFTR-ΔF508 heterozygous pigs by adeno-associated virus-mediated gene targeting and somatic cell nuclear transfer. J. Clin. Invest. 2008, 118, 1571–1577. [Google Scholar] [CrossRef] [PubMed]
- Rosen, B.H.; Chanson, M.; Gawenis, L.R.; Liu, J.H.; Sofoluwe, A.; Zoso, A.; Engelhardt, J.F. Animal and model systems for studying cystic fibrosis. J. Cyst. Fibros. 2018, 17, S28–S34. [Google Scholar] [CrossRef] [PubMed]
- Palatnik, A.; Ye, S.; Kendziorski, C.; Iden, M.; Zigman, J.S.; Hessner, M.J.; Rader, J.S. Identification of a serum-induced transcriptional signature associated with metastatic cervical cancer. PLoS ONE 2017, 12, e0181242. [Google Scholar] [CrossRef] [PubMed]
- Kaldunski, M.; Jia, S.A.; Geoffrey, R.; Basken, J.; Prosser, S.; Kansra, S.; Mordes, J.P.; Lernmark, A.; Wang, X.J.; Hessner, M.J. Identification of a serum-induced transcriptional signature associated with type 1 diabetes in the biobreeding rat. Diabetes 2010, 59, 2375–2385. [Google Scholar] [CrossRef] [PubMed]
- Kanter, I.; Kalisky, T. Single cell transcriptomics: Methods and applications. Front. Oncol. 2015, 5, 53. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Khodadoust, M.S.; Liu, C.L.; Newman, A.M.; Alizadeh, A.A. Profiling tumor infiltrating immune cells with cibersort. Methods Mol. Biol. 2018, 1711, 243–259. [Google Scholar] [PubMed]
- Bolen, C.R.; Uduman, M.; Kleinstein, S.H. Cell subset prediction for blood genomic studies. BMC Bioinform. 2011, 12, 258. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Hu, Z.; Butte, A.J. Xcell: Digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef] [PubMed]
- Plasschaert, L.W.; Zilionis, R.; Choo-Wing, R.; Savova, V.; Knehr, J.; Roma, G.; Klein, A.M.; Jaffe, A.B. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 2018, 560, 377–381. [Google Scholar] [CrossRef] [PubMed]


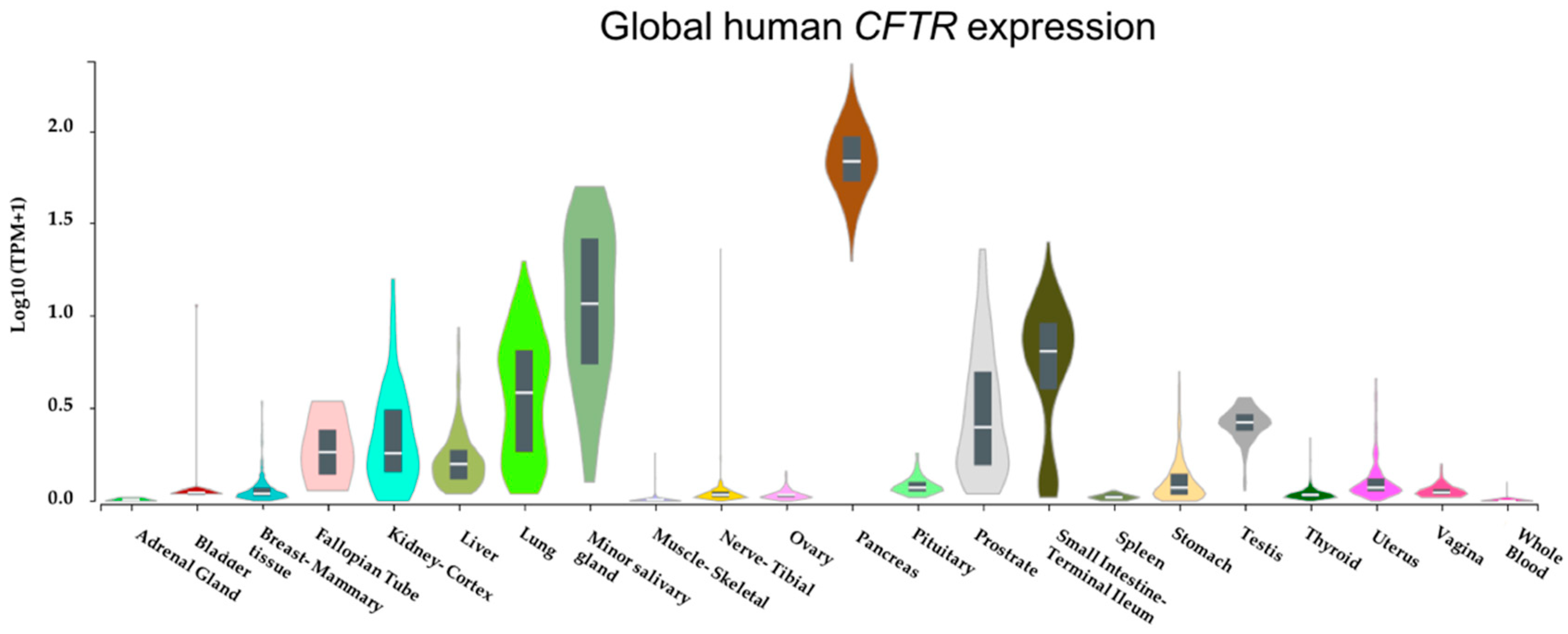{kind=link}
| Year | Focus | Methods | Tissue/Cell | Key Enriched Pathways | Reference |
|---|---|---|---|---|---|
| 2005 | Transcriptional changes induced by CF Pseudomonas aeruginosa and Burkholderia cepacia | Microarray | Alveolar macrophages | Cytokine signaling; NF-κB signaling | [11] |
| 2006 | Mild vs. severe CF lung disease | Microarray | Epithelial | Ubiquitin cycle; lipid metabolism | [12] |
| 2007 | CF vs. healthy controls | Microarray | Epithelial | Activator protein 1 and NF-κB activator pathway | [13] |
| 2009 | Transcriptional changes induced by Azithromycin | Microarray | Epithelial | Lipid/cholesterol biosynthesis; cell division | [14] |
| 2011 | CF vs. non-CF samples | Microarray | Epithelial | Inflammatory response; cell-to-cell signaling; cellular movement | [15] |
| 2012 | Transcriptional changes induced by CF P. aeruginosa | Microarray | Epithelial | TLR signaling; chemokine signaling | [16] |
| 2012 | Transcriptional changes induced by plasma of CF and non-CF | Microarray | PBMCs | Immune response, B- and T-cell activation | [17] |
| 2013 | CF vs. healthy controls | Microarray | Epithelial | Inflammation; defense response | [18] |
| 2013 | Transcriptional changes induced by innate defense regulator 1018 | Microarray | Epithelial, PBMCs | Dysfunctional autophagy, AMPK-Akt signaling | [19] |
| 2013 | Transcriptional changes influenced by a CF modifier gene-EHF | Microarray | Epithelial | Gene regulation; glycosylation of biopolymers | [20] |
| 2014 | Transcriptional changes influenced by CF and other lung diseases | Microarray | Blood, PBMCs | Immune response, leukocyte activation in immune response | [21] |
| 2014 | Transcriptional changes induced by oxidative stress | Microarray | Epithelia | Cell survival; regulation of signal transduction | [22] |
| 2015 | Transcriptional changes influenced by CF | Microarray | Lymphoblasts | Endomembrane function; ER response to stress | [23] |
| 2017 | Mild vs. severe lung phenotype | RNA-Seq | Leukocytes | Type 1 interferon response; protein targeting ER | [10] |
| 2017 | Transcriptional changes induced by digitoxin | Microarray | Epithelial | Inflammatory pathway; immune response | [24] |
| 2018 | Transcriptional changes influenced by genomic variation | RNA-Seq | Epithelial | Inflammation/inflammatory signaling; innate immune response | [25] |
| 2018 | CF before vs. after treatment for exacerbation | RNA-Seq | Blood, neutrophils | Functional enrichment not performed; inflammasome genes | [26] |
| 2018 | Transcriptional changes induced by plasma of CF and its phenotypes | Microarray | PBMCs | E1F2 Signaling, IL-8 signaling, B-cell receptor, production of nitric oxide and oxygen reactive species | [27] |
| Gene | Gene Name | Relevant Pathway(s) | References |
|---|---|---|---|
| Signal Transduction/Transport of Small Molecules | |||
| CDH4 | Cadherin 4 | Wnt signaling | [13] |
| CDH8 | Cadherin 8 | Wnt signaling | [13] |
| CDK6 | Cyclin dependent kinase 6 | PI3K/Akt signaling | [13,27] |
| CHRM3 | Cholinergic receptor muscarinic 3 | Signaling by GPCR, Calcium signaling pathway | [22,27] |
| ESR1 | Estrogen receptor 1 | Signaling by GPCR | [22,27] |
| FGF2 | Fibroblast growth factor 2 | mTOR signaling, PI3K/Akt signaling | [13,27] |
| ITGA4 | Integrin subunit α 4 | PI3K/Akt signaling | [13] |
| ITGA6 | Integrin subunit α 6 | Signaling by GPCR, Wnt signaling | [13,27] |
| JAG1 | Jagged 1 | PI3K/Akt signaling, Signaling by GPCR | [13,27] |
| MMP9 | Matrix metallopeptidase 9 | Signaling by GPCR, Interleukin 4 & 13 signaling | [22] |
| NCF2 | Neutrophil cytosolic factor 2 | Signaling by GPCR | [22,27] |
| NOTCH3 | Notch 3 | PI3K/Akt signaling, Signaling by GPCR | [13,27] |
| NRP2 | Neuropilin 2 | Signaling by GPCR | [13,22,27] |
| PIK3R1 | Phosphoinositide-3-kinase regulatory subunit 1 | mTOR signaling, PI3K/Akt signaling | [22,27] |
| PSMA5 | Proteasome subunit α 5 | Signaling by GPCR, Wnt signaling | [22,27] |
| SFN | Stratifin | mTOR signaling, Signaling by GPCR | [13,27] |
| SFRP1 | Secreted frizzled related protein 1 | Signaling by GPCR, Wnt signaling | [22,27] |
| SOX9 | SRY-box 9 | Signaling by GPCR, Wnt signaling | [13,22,27] |
| STAT1 | Signal transducer and activator of transcription 1 | PI3K/Akt signaling, Signaling by GPCR | [22,27] |
| WNT2B | Wnt family member 2B | Wnt/β-catenin signaling, PI3K/Akt signaling | [13,27] |
| Immune system | |||
| CTSB | Cathepsin B | Innate immune system, Bacterial infections in CF airways | [13] |
| CXCL1 | C-X-C motif chemokine ligand 1 | Cytokines and inflammatory response, immune response | [13,22,27] |
| CXCL10 | C-X-C motif chemokine ligand 10 | Cytokine signaling, Signaling by interleukins | [13,22,27] |
| CXCL8 | C-X-C motif chemokine ligand 8 | NFkB signaling, Innate immune system, Interleukin signaling | [22,27] |
| GHR | Growth hormone receptor | Cytokine signaling, innate immune system | [22] |
| HMOX1 | Heme oxygenase 1 | NFkB signaling, Cytokine signaling; Innate immune system | [13,22] |
| ICAM1 | Intercellular adhesion molecule 1 | Cytokine signaling, Signaling by interleukins | [13,27] |
| IL1A | Interleukin 1 α | NFkB, T-cell receptor signaling, IFN α & β immune response | [13,22,27] |
| IL1B | Interleukin 1 β | Cytokine signaling, immune response, Lung fibrosis | [13,22,27] |
| IL6 | Interleukin 6 | Cytokine signaling, immune response | [13,22,27] |
| ITGA4 | Integrin subunit α 4 | Transcriptional regulation by RUNX3, Generic transcription | [13] |
| LY96 | Lymphocyte antigen 96 | NFkB signaling, Innate immune system | [13,27] |
| MME | Membrane metalloendopeptidase | B-cell development pathways, innate immune system | [13] |
| MMP1 | Matrix metallopeptidase 1 | Cytokine signaling, immune response | [13] |
| NOX4 | NADPH oxidase 4 | Immune cell transmigration, PAK pathway | [13] |
| PTGS2 | Prostaglandin-endoperoxide synthase 2 | NFkB signaling, Cytokine signaling; Innate immune system | [13] |
| RASGRP1 | RAS guanyl releasing protein 1 | T-cell receptor signaling; Cytokine signaling | [13,22,27] |
| SERPINA1 | Serpin family A member 1 | Innate immune system, Lung fibrosis | [13,22,27] |
| TLR4 | Toll like receptor 4 | NFkB signaling, Innate immune system, TREM1 signaling | [13,27] |
| VEGFA | Vascular endothelial growth factor A | Innate immune system, Signaling by interleukins | [13] |
| Drug Name | Phase | Type | Status | Activity | Target | Target Class |
|---|---|---|---|---|---|---|
| Ivacaftor, lumacaftor | 4 | SM | Completed | Agonist | CFTR | Cystic fibrosis transmembrane conductance regulator (CFTR) |
| Doxycycline | 4 | SM | Completed | Antagonist | MMP7 | Metallo protease M10A subfamily |
| Doxycycline | 4 | SM | Completed | Antagonist | MMP1 | Metallo protease M10A subfamily |
| Cholecalciferol | 4 | SM | Completed | Agonist | VDR | Nuclear hormone receptor subfamily 1 group I member 1 |
| Doxycycline | 4 | SM | Completed | Antagonist | MMP13 | Metallo protease M10A subfamily |
| Doxycycline | 4 | SM | Completed | Antagonist | MMP8 | Metallo protease M10A subfamily |
| Somatropin | 3 | Protein | Completed | Agonist | GHR | Membrane receptor |
| Denufosol | 3 | SM | Completed | Agonist | P2RY2 | Purine receptor |
| Insulin glargine | 3 | Protein | Completed | Agonist | INSR | Tyrosine protein kinase InsR family |
| Prednisone | 3 | SM | Recruiting | Agonist | NR3C1 | Nuclear hormone receptor subfamily 3 group C member 1 |
| Tiotropium | 3 | SM | Completed | Antagonist | CHRM3 | Acetylcholine receptor |
| Nitric oxide | 2 | SM | Recruiting | Agonist | GUCY1A2 | Soluble guanylate cyclase |
| PTI-428 | 2 | SM | Completed | Agonist | CFTR | CFTR |
| pGM169/GL67A | 2 | SM | Completed | Agonist | CFTR | CFTR |
| Digitoxin | 2 | SM | Completed | Antagonist | ATP1B3 | Hydrolase |
| Nitric oxide | 2 | SM | Completed | Agonist | GUCY1B1 | Soluble guanylate cyclase |
| Nitric oxide | 2 | SM | Completed | Agonist | GUCY1B2 | Soluble guanylate cyclase |
| Nitric oxide | 2 | SM | Completed | Agonist | GUCY1A1 | Soluble guanylate cyclase |
| Gallium nitrate | 2 | SM | Completed | Antagonist | RRM2 | Enzyme |
| Sildenafil | 2 | SM | Active | Antagonist | PDE5A | Phosphodiesterase 5A |
| Digitoxin | 2 | SM | Completed | Antagonist | ATP1A1 | Hydrolase |
| Omeprazole | 2 | SM | Recruiting | Antagonist | ATP4B | Hydrogen potassium ATPase |
| Miglustat | 2 | SM | Completed | Antagonist | UGCG | Transferase |
| P-1037 | 2 | SM | Completed | Antagonist | SCNN1B | Epithelial sodium channel |
| Gallium nitrate | 2 | SM | Completed | Antagonist | RRM2B | Enzyme |
| Fiboflapon | 2 | SM | Completed | Antagonist | ALOX5AP | Other cytosolic protein |
| Losartan | 2 | SM | Recruiting | Antagonist | AGTR1 | Angiotensin receptor |
| Digitoxin | 2 | SM | Completed | Antagonist | FXYD2 | Hydrolase |
| Digitoxin | 2 | SM | Completed | Antagonist | ATP1A2 | Hydrolase |
| Digitoxin | 2 | SM | Completed | Antagonist | ATP1B2 | Hydrolase |
| Amiloride | 2 | SM | Completed | Antagonist | SCNN1A | Epithelial sodium channel |
| P-1037 | 2 | SM | Completed | Antagonist | SCNN1G | Epithelial sodium channel |
| Digitoxin | 2 | SM | Completed | Antagonist | ATP1A4 | Hydrolase |
| Digitoxin | 2 | SM | Completed | Antagonist | ATP1A3 | Hydrolase |
| Digitoxin | 2 | SM | Completed | Antagonist | ATP1B1 | Hydrolase |
| Gallium nitrate | 2 | SM | Completed | Antagonist | RRM1 | Enzyme |
| Simvastatin | 1 | SM | Completed | Antagonist | HMGCR | Oxidoreductase |
| Hydroxychloroquine | 1 | SM | Completed | Antagonist | TLR9 | Toll-like and Il-1 receptors |
| Amelubant | 1 | SM | Completed | Antagonist | LTB4R | Leukotriene receptor |
| Pioglitazone | 1 | SM | Completed | Agonist | PPARG | Nuclear hormone receptor subfamily 1 group C member 3 |
| Interferon γ-1b | 1 | Protein | Completed | Agonist | IFNGR2 | Membrane receptor |
| Hydroxychloroquine | 1 | SM | Completed | Antagonist | TLR7 | Toll-like and Il-1 receptors |
| Interferon γ-1b | 1 | Protein | Completed | Agonist | IFNGR1 | Membrane receptor |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ideozu, J.E.; Zhang, X.; McColley, S.; Levy, H. Transcriptome Profiling and Molecular Therapeutic Advances in Cystic Fibrosis: Recent Insights. Genes 2019, 10, 180. https://doi.org/10.3390/genes10030180
Ideozu JE, Zhang X, McColley S, Levy H. Transcriptome Profiling and Molecular Therapeutic Advances in Cystic Fibrosis: Recent Insights. Genes. 2019; 10(3):180. https://doi.org/10.3390/genes10030180
Chicago/Turabian StyleIdeozu, Justin E., Xi Zhang, Susanna McColley, and Hara Levy. 2019. "Transcriptome Profiling and Molecular Therapeutic Advances in Cystic Fibrosis: Recent Insights" Genes 10, no. 3: 180. https://doi.org/10.3390/genes10030180
APA StyleIdeozu, J. E., Zhang, X., McColley, S., & Levy, H. (2019). Transcriptome Profiling and Molecular Therapeutic Advances in Cystic Fibrosis: Recent Insights. Genes, 10(3), 180. https://doi.org/10.3390/genes10030180