A 24 h Age Difference Causes Twice as Much Gene Expression Divergence as 100 Generations of Adaptation to a Novel Environment

 , , , , and
, , , , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. RNA-Seq Common Garden Experiment
2.2. RNA-Seq Data Processing and Analysis
2.3. GO Enrichment Analysis
2.4. Comparison to Gene Expression Changes Associated with Aging
2.5. Availibility of Data and Materials
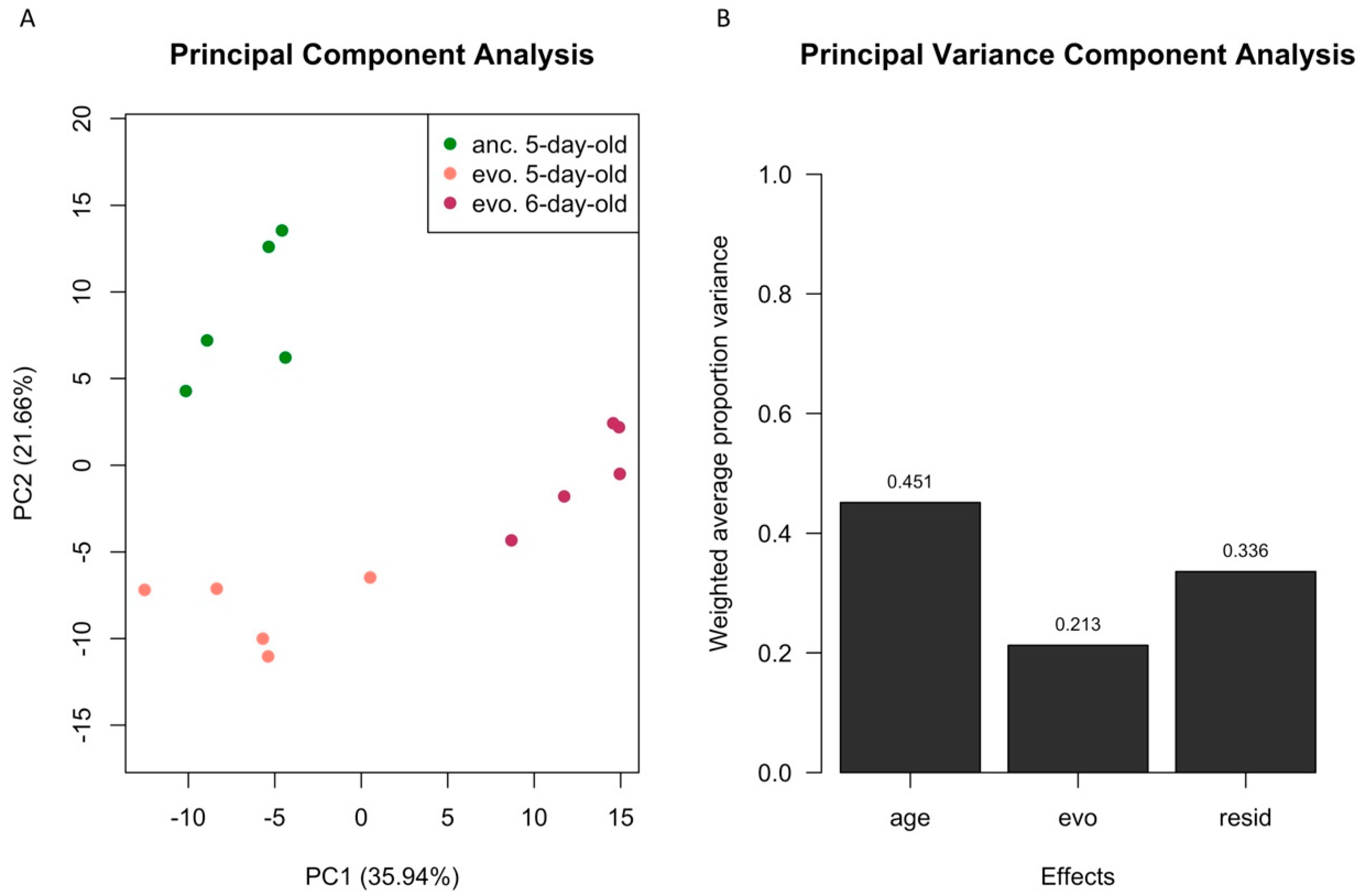
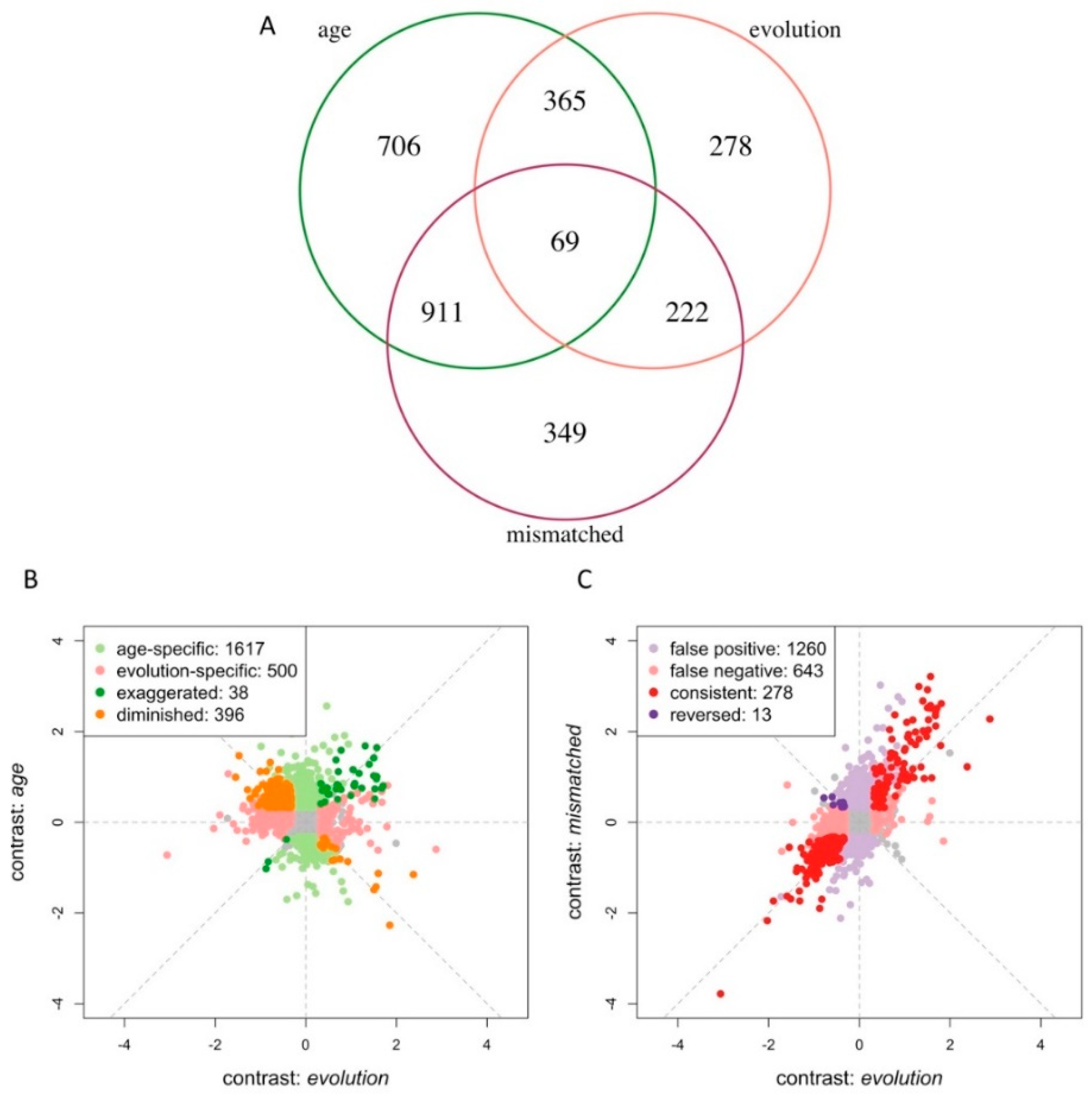
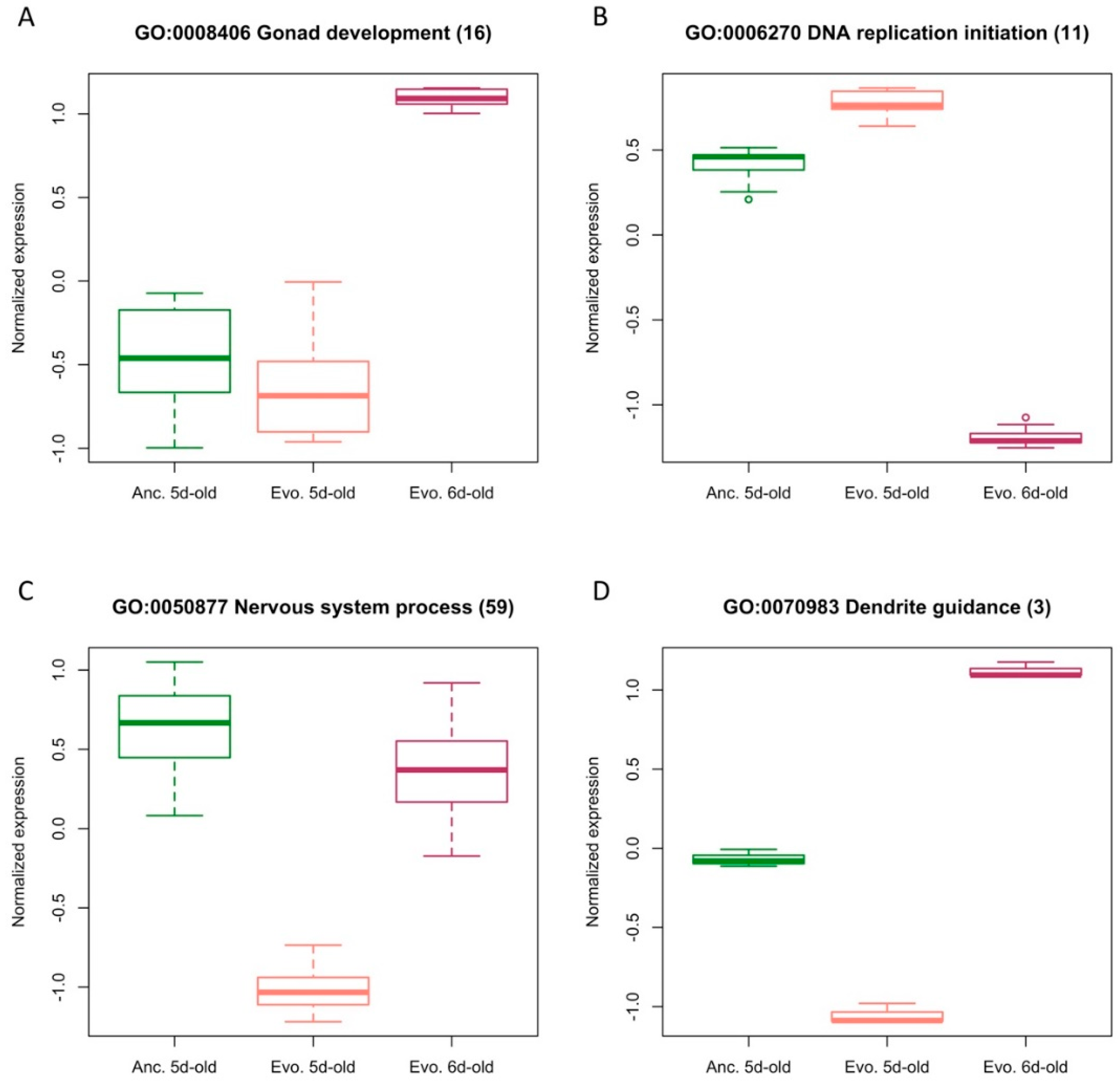
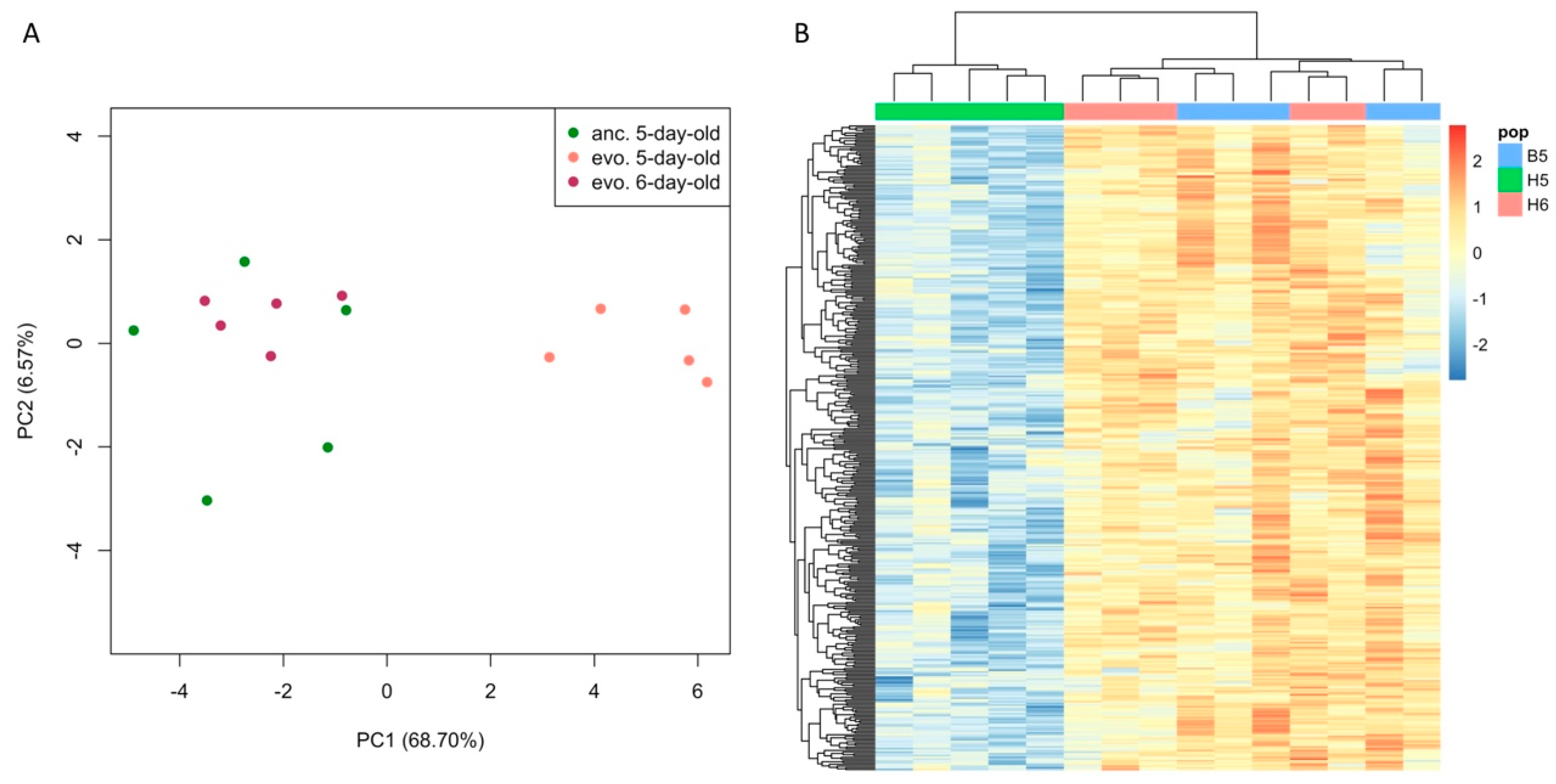
3. Results
3.1. Age Difference Explains more Variance than Evolutionary Responses
3.2. Functional Implications of a 24 h Age Difference or Temperature Adaptation
3.3. Age Difference Confounds Signals of Adaptive Responses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Graveley, B.R.; Brooks, A.N.; Carlson, J.W.; Duff, M.O.; Landolin, J.M.; Yang, L.; Artieri, C.G.; van Baren, M.J.; Boley, N.; Booth, B.W.; et al. The developmental transcriptome of Drosophila melanogaster. Nature 2011, 471, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Gosselin, D.; Skola, D.; Coufal, N.G.; Holtman, I.R.; Schlachetzki, J.C.M.; Sajti, E.; Jaeger, B.N.; O’Connor, C.; Fitzpatrick, C.; Pasillas, M.P.; et al. An environment-dependent transcriptional network specifies human microglia identity. Science 2017, 356, eaal3222. [Google Scholar] [CrossRef] [PubMed]
- Ohama, N.; Sato, H.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Transcriptional regulatory network of plant heat stress response. Trends Plant Sci. 2017, 22, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Nolte, V.; Schlötterer, C. Temperature-related reaction norms of gene expression: Regulatory architecture and functional implications. Mol. Biol. Evol. 2015, 32, 2393–2402. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Nolte, V.; Schlötterer, C. Temperature stress mediates decanalization and dominance of gene expression in Drosophila melanogaster. PLoS Genet. 2015, 11, 1–19. [Google Scholar] [CrossRef]
- Gibbons, T.C.; Metzger, D.C.H.; Healy, T.M.; Schulte, P.M. Gene expression plasticity in response to salinity acclimation in threespine stickleback ecotypes from different salinity habitats. Mol. Ecol. 2017, 26, 2711–2725. [Google Scholar] [CrossRef] [PubMed]
- Levis, N.A.; Serrato-Capuchina, A.; Pfennig, D.W. Genetic accommodation in the wild: Evolution of gene expression plasticity during character displacement. J. Evol. Biol. 2017, 30, 1712–1723. [Google Scholar] [CrossRef]
- Kenkel, C.D.; Matz, M.V. Gene expression plasticity as a mechanism of coral adaptation to a variable environment. Nat. Ecol. Evol. 2016, 1, 0014. [Google Scholar] [CrossRef]
- Hwang, B.; Lee, J.H.; Bang, D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med. 2018, 50, 96. [Google Scholar] [CrossRef]
- Huang, Y.; Agrawal, A.F. Experimental evolution of gene expression and plasticity in alternative selective regimes. PLoS Genet. 2016, 12, e1006336. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Carbone, M.A.; Magwire, M.M.; Peiffer, J.A.; Lyman, R.F.; Stone, E.A.; Anholt, R.R.H.; Mackay, T.F.C. Genetic basis of transcriptome diversity in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 2015, 112, E6010–E6019. [Google Scholar] [CrossRef] [PubMed]
- West, M.A.L.; Kim, K.; Kliebenstein, D.J.; van Leeuwen, H.; Michelmore, R.W.; Doerge, R.W.; Clair, D.A.S. Global eQTL mapping reveals the complex genetic architecture of transcript-level variation in Arabidopsis. Genetics 2007, 175, 1441–1450. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.L.; Bonduriansky, R.; Sgro, C.M.; Chenoweth, S.F. Sex-biased transcriptome divergence along a latitudinal gradient. Mol. Ecol. 2017, 26, 1256–1272. [Google Scholar] [CrossRef] [PubMed]
- Hutter, S.; Saminadin-Peter, S.S.; Stephan, W.; Parsch, J. Gene expression variation in African and European populations of Drosophila melanogaster. Genome Biol. 2008, 9, R12. [Google Scholar] [CrossRef]
- Zhao, L.; Wit, J.; Svetec, N.; Begun, D.J. Parallel gene expression differences between low and high latitude populations of Drosophila melanogaster and D. simulans. PLoS Genet. 2015, 11, e1005184. [Google Scholar] [CrossRef] [PubMed]
- Müller, L.; Hutter, S.; Stamboliyska, R.; Saminadin-Peter, S.S.; Stephan, W.; Parsch, J. Population transcriptomics of Drosophila melanogaster females. BMC Genom. 2011, 12, 81. [Google Scholar] [CrossRef] [PubMed]
- Mallard, F.; Nolte, V.; Tobler, R.; Kapun, M.; Schlötterer, C. A simple genetic basis of adaptation to a novel thermal environment results in complex metabolic rewiring in Drosophila. Genome Biol. 2018, 19, 119. [Google Scholar] [CrossRef] [PubMed]
- Garlapow, M.E.; Everett, L.J.; Zhou, S.; Gearhart, A.W.; Fay, K.A.; Huang, W.; Morozova, T.V.; Arya, G.H.; Turlapati, L.; St. Armour, G.; et al. Genetic and genomic response to selection for food consumption in Drosophila melanogaster. Behav. Genet. 2017, 47, 227–243. [Google Scholar] [CrossRef]
- Yampolsky, L.Y.; Glazko, G.V.; Fry, J.D. Evolution of gene expression and expression plasticity in long-term experimental populations of Drosophila melanogaster maintained under constant and variable ethanol stress. Mol. Ecol. 2012, 21, 4287–4299. [Google Scholar] [CrossRef] [PubMed]
- Arbeitman, M.N.; Furlong, E.E.M.; Imam, F.; Johnson, E.; Null, B.H.; Baker, B.S.; Krasnow, M.A.; Scott, M.P.; Davis, R.W.; White, K.P. Gene expression during the life cycle of Drosophila melanogaster. Science 2002, 297, 2270–2275. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, S.H.; Mank, J.E. Inferring regulatory change from gene expression: The confounding effects of tissue scaling. Mol. Ecol. 2016, 25, 5114–5128. [Google Scholar] [CrossRef] [PubMed]
- Marinkovic, D.; Ayala, E.J. Genetic variation for rate of development in natural populations of Drosophila melanogaster. Genetica 1986, 71, 123–132. [Google Scholar] [CrossRef]
- Mensch, J.; Lavagnino, N.; Carreira, V.P.; Massaldi, A.; Hasson, E.; Fanara, J.J. Identifying candidate genes affecting developmental time in Drosophila melanogaster: Pervasive pleiotropy and gene-by-environment interaction. BMC Dev. Biol. 2008, 8, 78. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Riley, R.M.; Wolfinger, R.D.; White, K.P.; Passador-Gurgel, G.; Gibson, G. The contributions of sex, genotype and age to transcriptional variance in Drosophila melanogaster. Nat. Genet. 2001, 29, 389–395. [Google Scholar] [CrossRef]
- Coolon, J.D.; McManus, C.J.; Stevenson, K.R.; Graveley, B.R.; Wittkopp, P.J. Tempo and mode of regulatory evolution in Drosophila. Genome Res. 2014, 24, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Takada, Y.; Miyagi, R.; Takahashi, A.; Endo, T.; Osada, N. A generalized linear model for decomposing cis-regulatory, parent-of-origin, and maternal effects on allele-specific gene expression. G3 (Bethesda) 2017, 7, 2227–2234. [Google Scholar] [CrossRef] [PubMed]
- Qu, W.; Gurdziel, K.; Pique-Regi, R.; Ruden, D.M. Lead modulates trans- and cis-expression quantitative trait loci (eQTLs) in Drosophila melanogaster heads. Front. Genet. 2018, 9, 395. [Google Scholar] [CrossRef]
- Symonenko, A.V.; Roshina, N.V.; Krementsova, A.V.; Pasyukova, E.G. Reduced neuronal transcription of Escargot, the Drosophila gene encoding a Snail-type transcription factor, promotes longevity. Front. Genet. 2018, 9, 151. [Google Scholar] [CrossRef]
- Grotewiel, M.S.; Martin, I.; Bhandari, P.; Cook-Wiens, E. Functional senescence in Drosophila melanogaster. Ageing Res. Rev. 2005, 4, 372–397. [Google Scholar] [CrossRef]
- Helfand, S.L.; Rogina, B. Genetics of aging in the fruit fly, Drosophila melanogaster. Annu. Rev. Genet. 2003, 37, 329–348. [Google Scholar] [CrossRef]
- Kim, S.N.; Rhee, J.-H.; Song, Y.-H.; Park, D.Y.; Hwang, M.; Lee, S.; Kim, J.E.; Gim, B.S.; Yoon, J.H.; Kim, Y.-J.; et al. Age-dependent changes of gene expression in the Drosophila head. Neurobiol. Aging 2005, 26, 1083–1091. [Google Scholar] [CrossRef]
- Zhan, M.; Yamaza, H.; Sun, Y.; Sinclair, J.; Li, H.; Zou, S. Temporal and spatial transcriptional profiles of aging in Drosophila melanogaster. Genome Res. 2007, 17, 1236–1243. [Google Scholar] [CrossRef] [PubMed]
- Carlson, K.A.; Gardner, K.; Pashaj, A.; Carlson, D.J.; Yu, F.; Eudy, J.D.; Zhang, C.; Harshman, L.G. Genome-wide gene expression in relation to age in large laboratory cohorts of Drosophila melanogaster. Genet. Res. Int. 2015, 2015, 835624. [Google Scholar] [CrossRef] [PubMed]
- Nouhaud, P.; Tobler, R.; Nolte, V.; Schlötterer, C. Ancestral population reconstitution from isofemale lines as a tool for experimental evolution. Ecol. Evol. 2016, 6, 7169–7175. [Google Scholar] [CrossRef] [PubMed]
- Barghi, N.; Tobler, R.; Nolte, V.; Jaksic, A.M.; Mallard, F.; Otte, K.; Dolezal, M.; Taus, T.; Kofler, R.; Schlötterer, C. Polygenic adaptation fuels genetic redundancy in Drosophila. bioRxiv 2018, 332122. [Google Scholar] [CrossRef]
- Gómez-Sánchez, D.; Schlötterer, C. ReadTools: A universal toolkit for handling sequence data from different sequencing platforms. Mol. Ecol. Resour. 2018, 18, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.D.; Nacu, S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 2010, 26, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, N.; Nolte, V.; Chen, J.; Schlötterer, C. Genome assembly and annotation of a Drosophila simulans strain from Madagascar. Mol. Ecol. Resour. 2015, 15, 372–381. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. The subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013, 41, e108. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Boedigheimer, M.J.; Wolfinger, R.D.; Bass, M.B.; Bushel, P.R.; Chou, J.W.; Cooper, M.; Corton, J.C.; Fostel, J.; Hester, S.; Lee, J.S.; et al. Sources of variation in baseline gene expression levels from toxicogenomics study control animals across multiple laboratories. BMC Genom. 2008, 9, 285. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y.; Society, R.S. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Alexa, A.; Rahnenfuhrer, J.; Lengauer, T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 2006, 22, 1600–1607. [Google Scholar] [CrossRef] [PubMed]
- Alexa, A.; Rahnenführer, J. Gene Set Enrichment Analysis with topGO; Roswell Park Cancer Institute: Buffalo, NY, USA, 2018. [Google Scholar]
- Technau, G.M. Fiber number in the mushroom bodies of adult Drosophila melanogaster depends on age, sex and experience. J. Neurogenet. 1984, 1, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Wallenfang, M.R.; Nayak, R.; DiNardo, S. Dynamics of the male germline stem cell population during aging of Drosophila melanogaster. Aging Cell 2006, 5, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Gorbunova, V.; Seluanov, A.; Mao, Z.; Hine, C. Changes in DNA repair during aging. Nucleic Acids Res. 2007, 35, 7466–7474. [Google Scholar] [CrossRef]
- Lombard, D.B.; Chua, K.F.; Mostoslavsky, R.; Franco, S.; Gostissa, M.; Alt, F.W. DNA repair, genome stability, and aging. Cell 2005, 120, 497–512. [Google Scholar] [CrossRef]
- Zander, G.; Hackmann, A.; Bender, L.; Becker, D.; Lingner, T.; Salinas, G.; Krebber, H. mRNA quality control is bypassed for immediate export of stress-responsive transcripts. Nature 2016, 540, 593–596. [Google Scholar] [CrossRef]
- Bond, U. Stressed out! Effects of environmental stress on mRNA metabolism. FEMS Yeast Res. 2006, 6, 160–170. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Short-Term 1 | Long-Term 2 [33] | Consistency 3 | |
|---|---|---|---|
| Genes | 2051 | 966 | 366 (37.9%) *** |
| GO terms | 312 | 89 | 24 (27.0%) *** |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, S.-K.; Jakšić, A.M.; Nolte, V.; Barghi, N.; Mallard, F.; Otte, K.A.; Schlötterer, C. A 24 h Age Difference Causes Twice as Much Gene Expression Divergence as 100 Generations of Adaptation to a Novel Environment. Genes 2019, 10, 89. https://doi.org/10.3390/genes10020089
Hsu S-K, Jakšić AM, Nolte V, Barghi N, Mallard F, Otte KA, Schlötterer C. A 24 h Age Difference Causes Twice as Much Gene Expression Divergence as 100 Generations of Adaptation to a Novel Environment. Genes. 2019; 10(2):89. https://doi.org/10.3390/genes10020089
Chicago/Turabian StyleHsu, Sheng-Kai, Ana Marija Jakšić, Viola Nolte, Neda Barghi, François Mallard, Kathrin A. Otte, and Christian Schlötterer. 2019. "A 24 h Age Difference Causes Twice as Much Gene Expression Divergence as 100 Generations of Adaptation to a Novel Environment" Genes 10, no. 2: 89. https://doi.org/10.3390/genes10020089
APA StyleHsu, S.-K., Jakšić, A. M., Nolte, V., Barghi, N., Mallard, F., Otte, K. A., & Schlötterer, C. (2019). A 24 h Age Difference Causes Twice as Much Gene Expression Divergence as 100 Generations of Adaptation to a Novel Environment. Genes, 10(2), 89. https://doi.org/10.3390/genes10020089