Comprehensive Expression Profiling Analysis of Pituitary Indicates that circRNA Participates in the Regulation of Sheep Estrus


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animals
2.3. RNA Extraction and Quality Test
2.4. Circular RNAs Identification
2.5. Analysis of Differential Expression Circular RNAs
2.6. Gene Ontology, Kyoto Encyclopedia of Genes and Genomes Enrichment Analysis and Binding Sites Prediction
2.7. Reverse Transcription PCR Analysis, RNase-R Treatment and Quantitative PCR Analysis
3. Results
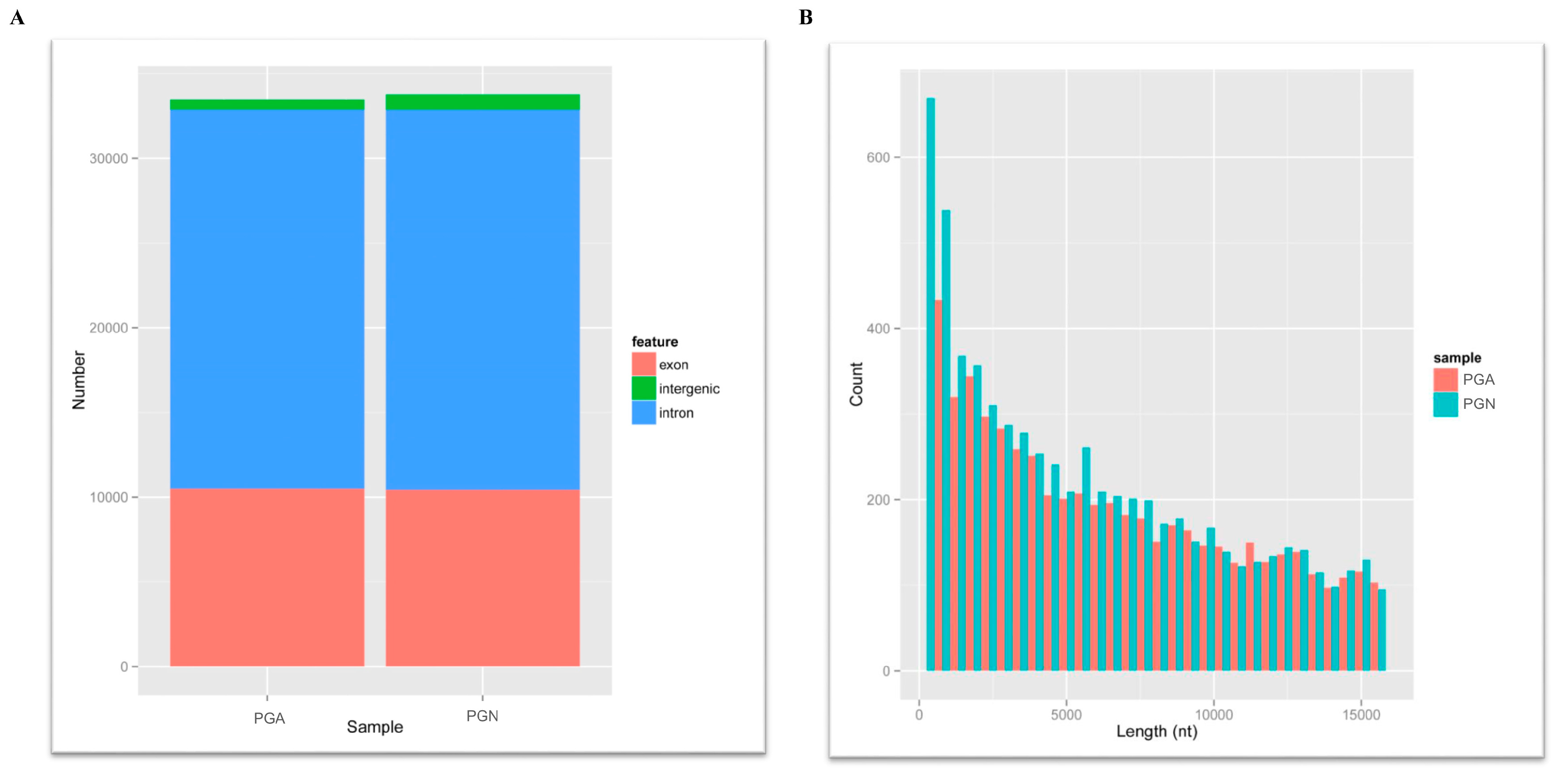
3.1. Sequencing Quality Description
3.2. Screening and Identification of Circular RNAs
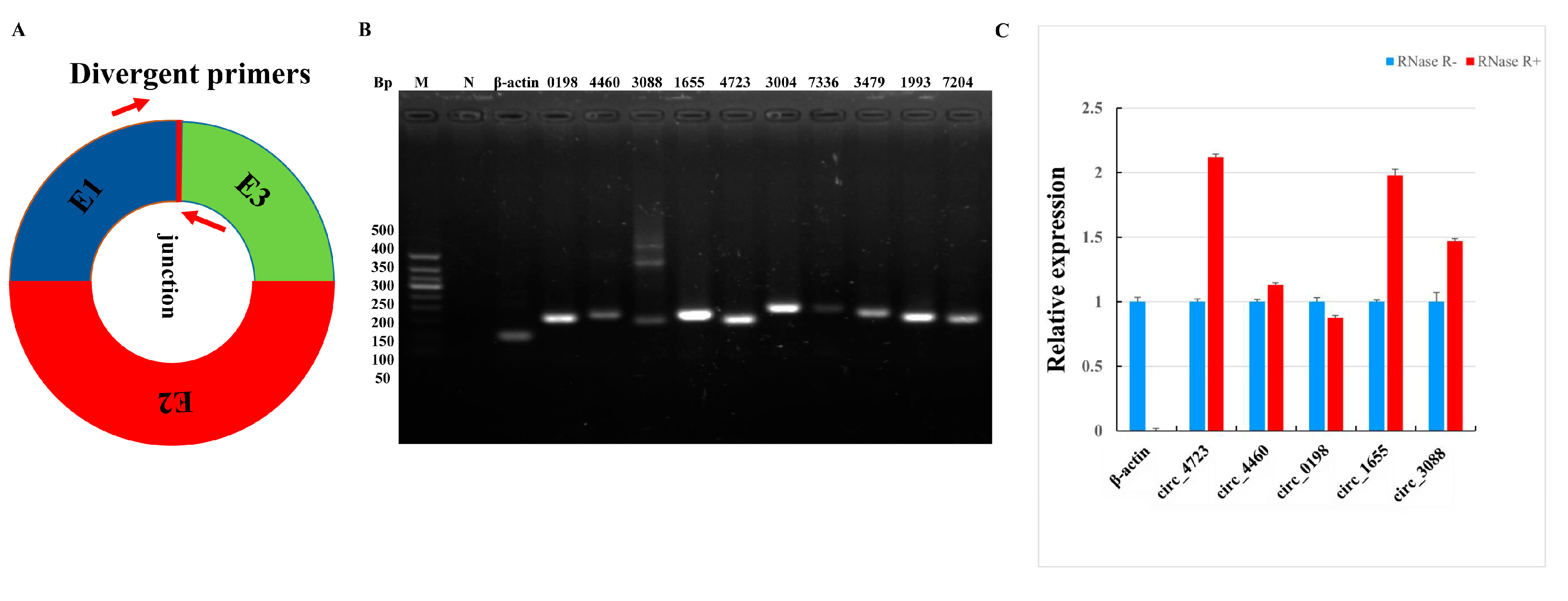
3.3. Validation of Sheep Circular RNAs
3.4. Analysis and Validation of Differentially Expressed Circular RNAs between Anestrus and Estrus
3.5. Enrichment Analysis of Circular RNAs
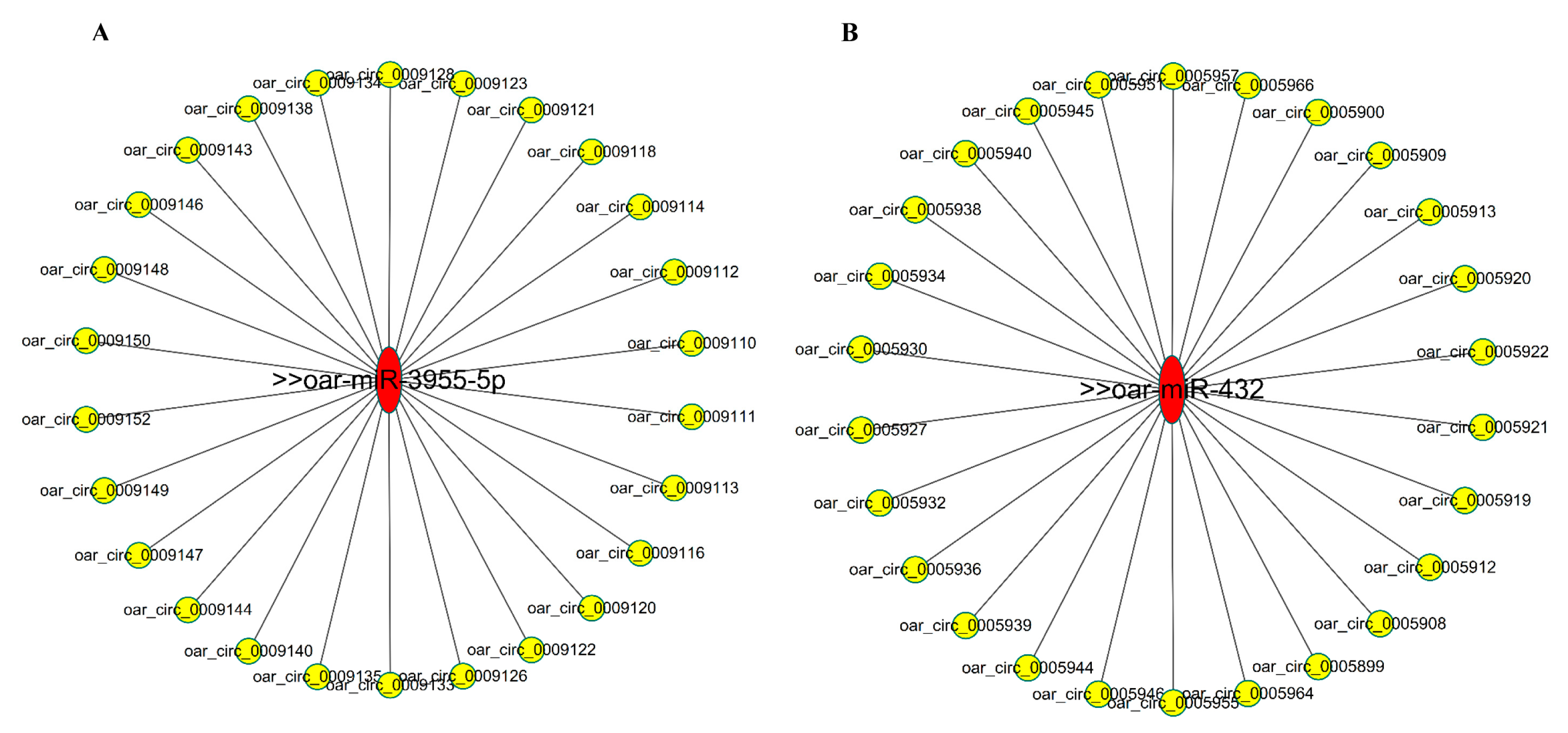
3.6. Bioinformatics Analysis of CircRNA-miRNA Networks
4. Discussion
Data Availability
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rybak-Wolf, A.; Stottmeister, C.; Glažar, P.; Jens, M.; Pino, N.; Giusti, S.; Hanan, M.; Behm, M.; Bartok, O.; Ashwal-Fluss, R.; et al. Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol. Cell 2015, 58, 870–885. [Google Scholar] [CrossRef] [PubMed]
- McBride, D.; Carre, W.; Sontakke, S.; Hogg, C.O.; Law, A.S.; Donadeu, F.X.; Clinton, M. Identification of miRNAs associated with the follicular-luteal transition in the ruminant ovary. Reproduction 2012, 144, 221–233. [Google Scholar] [CrossRef]
- Anderson, L.L.; Jeftinija, S.; Scanes, C.G. Growth hormone secretion: Molecular and cellular mechanisms and in vivo approaches. Exp. Biol. Med. 2004, 229, 291–302. [Google Scholar] [CrossRef]
- Ahmed, S.; Rekha, R.S.; Ahsan, K.B.; Doi, M.; Grandér, M.; Roy, A.K.; Ekström, E.C.; Wagatsuma, Y.; Vahter, M.; Raqib, R. Arsenic exposure affects plasma insulin-like growth factor 1 (IGF-1) in children in rural Bangladesh. PLoS ONE 2013, 8, e81530. [Google Scholar] [CrossRef]
- Zhang, X.D.; Zhang, Y.H.; Ling, Y.H.; Liu, Y.; Cao, H.G.; Yin, Z.J.; Ding, J.P.; Zhang, X.R. Characterization and differential expression of microRNAs in the ovaries of pregnant and non-pregnant goats (Capra hircus). BMC Genom. 2013, 14, 157. [Google Scholar] [CrossRef] [PubMed]
- D’angelo, D.; Palmieri, D.; Mussnich, P.; Roche, M.; Wierinckx, A.; Raverot, G.; Fedele, M.; Croce, C.M.; Trouillas, J.; Fusco, A. Altered microRNA expression profile in human pituitary GH adenomas: Down-regulation of miRNA targeting HMGA1, HMGA2, and E2F1. J. Clin. Endocrinol. Metab. 2012, 97, E1128–E1138. [Google Scholar] [CrossRef] [PubMed]
- Bahn, J.H.; Zhang, Q.; Li, F.; Chan, T.M.; Lin, X.; Kim, Y.; Wong, D.T.W.; Xiao, X. The landscape of microRNA, piwi-interacting RNA, and circular RNA in human saliva. Clin. Chem. 2015, 61, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Jiang, Y.; Ko, W.K.; Li, W.; Wong, A.O. Paracrine regulation of growth hormone gene expression by gonadotrophin release in grass carp pituitary cells: Functional implications, molecular mechanisms and signal transduction. J. Mol. Endocrinol. 2005, 34, 415–432. [Google Scholar] [CrossRef]
- Bloomston, M.; Frankel, W.L.; Petrocca, F.; Volinia, S.; Alder, H.; Hagan, J.P.; Liu, C.G.; Bhatt, D.; Taccioli, C.; Croce, C.M. MicroRNA expression patterns to differentiate pancreatic adenocarcinoma from normal pancreas and chronic pancreatitis. JAMA 2007, 297, 1901–1908. [Google Scholar] [CrossRef]
- Ghosal, S.; Das, S.; Sen, R.; Basak, P.; Chakrabarti, J. Circ2Traits: A comprehensive database for circular RNA potentially associated with disease and traits. Front. Genet. 2013, 4, 283. [Google Scholar] [CrossRef]
- Boeckel, J.N.; Jaé, N.; Heumüller, A.W.; Chen, W.; Boon, R.A.; Stellos, K.; Zeiher, A.M.; John, D.; Uchida, S.; Dimmeler, S. Identification and characterization of hypoxia-regulated endothelial circular RNA. Circ. Res. 2015, 117, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Burd, C.E.; Jeck, W.R.; Liu, Y.; Sanoff, H.K.; Wang, Z.; Sharpless, N.E. Expression of linear and novel circular forms of an INK4/ARF-associated non-coding RNA correlates with atherosclerosis risk. PLoS Genet. 2010, 6, e1001233. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.L.; Yang, L. Regulation of circRNA biogenesis. RNA Biol. 2015, 12, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Grivna, S.T.; Beyret, E.; Wang, Z.; Lin, H. A novel class of small RNAs in mouse spermatogenic cells. Genes Dev. 2006, 20, 1709–1714. [Google Scholar] [CrossRef]
- Salzman, J.; Chen, R.E.; Olsen, M.N.; Wang, P.L.; Brown, P.O. Cell-type specific features of circular RNA expression. PLoS Genet. 2013, 9, e1003777. [Google Scholar] [CrossRef]
- Lupu, F.; Terwilliger, J.D.; Lee, K.; Segre, G.V.; Efstratiadis, A. Roles of growth hormone and insulin-like growth factor 1 in mouse postnatal growth. Dev. Biol. 2001, 229, 141–162. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Robertson, H.A. The endogenous control of estrus and ovulation in sheep, cattle, and swine. In Vitamins & Hormones; Academic Press: Cambridge, MA, USA, 1970. [Google Scholar]
- Zhai, M.; Xie, Y.; Liang, H.; Lei, X.; Zhao, Z. Comparative profiling of differentially expressed microRNAs in estrous ovaries of Kazakh sheep in different seasons. Gene 2018, 664, 181–191. [Google Scholar] [CrossRef]
- Di, R.; He, J.; Song, S.; Tian, D.; Liu, Q.; Liang, X.; Cao, G. Characterization and comparative profiling of ovarian microRNAs during ovine anestrus and the breeding season. BMC Genom. 2014, 15, 899. [Google Scholar] [CrossRef]
- Chu, Q.; Zhou, B.; Xu, F.; Chen, R.; Shen, C.; Liang, T.; Schinckel, A.P. Genome-wide differential mRNA expression profiles in follicles of two breeds and at two stages of estrus cycle of gilts. Sci. Rep. 2017, 7, 5052. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Y.; Shen, H.; Jia, B.; Zhang, Y.S.; Wang, X.H.; Zeng, X.C. Differential gene expression in ovaries of Qira black sheep and Hetian sheep using RNA-Seq technique. PLoS ONE 2015, 10, e0120170. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liu, K.; Zhao, Z.; Blair, H.T.; Zhang, P.; Li, D.; Ma, R.Z. Identification of sheep ovary genes potentially associated with off-season reproduction. J. Genet. Genom. 2012, 39, 181–190. [Google Scholar] [CrossRef]
- Zhou, L.; Chen, J.; Li, Z.; Li, X.; Hu, X.; Huang, Y.; Zhao, X.; Liang, C.; Wang, Y.; Sun, L.; et al. Integrated profiling of microRNAs and mRNAs: MicroRNAs located on Xq27.3 associate with clear cell renal cell carcinoma. PLoS ONE 2010, 5, e15224. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2009, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Tuggle, C.K.; Trenkle, A. Control of growth hormone synthesis. Domest. Anim. Endocrinol. 1996, 13, 1–33. [Google Scholar] [CrossRef]
- Shan, L.; Wu, Q.; Li, Y.; Shang, H.; Guo, K.; Wu, J.; Wei, H.; Zhao, J.; Yu, J.; Li, M.H. Transcriptome profiling identifies differentially expressed genes in postnatal developing pituitary gland of miniature pig. DNA Res. 2013, 21, 207–216. [Google Scholar] [CrossRef]
- Zhang, Z.; Florez, S.; Gutierrez-Hartmann, A.; Martin, J.F.; Amendt, B.A. MicroRNAs regulate pituitary development and microRNA 26b specifically targets lymphoid enhancer factor 1 (Lef-1), which modulates pituitary transcription factor 1 (Pit-1) expression. J. Biol. Chem. 2010, 285, 34718–34728. [Google Scholar] [CrossRef]
- You, X.; Vlatkovic, I.; Babic, A.; Will, T.; Epstein, I.; Tushev, G.; Akbalik, G.; Wang, M.; Glock, C.; Quedenau, C.; et al. Neural circular RNAs are derived from synaptic genes and regulated by development and plasticity. Nat. Neurosci. 2015, 18, 603–610. [Google Scholar] [CrossRef]
- Li, P.; Chen, S.; Chen, H.; Mo, X.; Li, T.; Shao, Y.; Xiao, B.; Guo, J. Using circular RNA as a novel type of biomarker in the screening of gastric cancer. Clin. Chim. Acta 2015, 444, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Du, W.W.; Li, X.; Yee, A.J.; Yang, B.B. Foxo3 activity promoted by non-coding effects of circular RNA and Foxo3 pseudogene in the inhibition of tumor growth and angiogenesis. Oncogene 2016, 35, 3919–3931. [Google Scholar] [CrossRef] [PubMed]
- Qu, S.; Song, W.; Yang, X.; Wang, J.; Zhang, R.; Zhang, Z.; Zhang, H.; Li, H. Microarray expression profile of circular RNAs in human pancreatic ductal adenocarcinoma. Genom. Data 2015, 5, 385–387. [Google Scholar] [CrossRef] [PubMed]
- Ortavant, R.; Bocquier, F.; Pelletier, J.; Ravault, J.P.; Thimonier, J.; Volland-Nail, P. Seasonality of reproduction in sheep and its control by photoperiod. Aust. J. Biol. Sci. 1988, 41, 69–86. [Google Scholar] [CrossRef] [PubMed]
- Glidewell-Kenney, C.; Hurley, L.A.; Pfaff, L.; Weiss, J.; Levine, J.E.; Jameson, J.L. Nonclassical estrogen receptor α signaling mediates negative feedback in the female mouse reproductive axis. Proc. Natl. Acad. Sci. USA 2007, 104, 8173–8177. [Google Scholar] [CrossRef] [PubMed]
- Ott, T.L.; Yin, J.; Wiley, A.A.; Kim, H.T.; Gerami-Naini, B.; Spencer, T.E.; Bartol, F.F.; Burghardt, R.C.; Bazer, F.W. Effects of the estrous cycle and early pregnancy on uterine expression of Mx protein in sheep (Ovis aries). Biol. Reprod. 1998, 59, 784–794. [Google Scholar] [CrossRef] [PubMed]
- Colledge, W.H. Kisspeptins and GnRH neuronal signalling. Trends Endocrinol. Metab. 2009, 20, 115–121. [Google Scholar] [CrossRef]
- Yao, G.; Yin, M.; Lian, J.; Tian, H.; Liu, L.; Li, X.; Sun, F. MicroRNA-224 is involved in transforming growth factor-β-mediated mouse granulosa cell proliferation and granulosa cell function by targeting Smad4. Mol. Endocrinol. 2010, 24, 540–551. [Google Scholar] [CrossRef]
- Wang, K.; Long, B.; Liu, F.; Wang, J.X.; Liu, C.Y.; Zhao, B.; Zhou, L.Y.; Sun, T.; Wang, M.; Yu, T.; et al. A circular RNA protects the heart from pathological hypertrophy and heart failure by targeting miR-223. Eur. Heart J. 2016, 37, 2602–2611. [Google Scholar] [CrossRef]
- Westholm, J.O.; Miura, P.; Olson, S.; Shenker, S.; Joseph, B.; Sanfilippo, P.; Celniker, S.E.; Graveley, B.R.; Lai, E.C. Genome-wide analysis of drosophila circular RNAs reveals their structural and sequence properties and age-dependent neural accumulation. Cell Rep. 2014, 9, 1966–1980. [Google Scholar] [CrossRef]
- Li, Z.; Huang, C.; Bao, C.; Chen, L.; Lin, M.; Wang, X.; Zhong, G.; Yu, B.; Hu, W.; Dai, L.; et al. Exon-intron circular RNAs regulate transcription in the nucleus. Nat. Struct. Mol. Biol. 2015, 22, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Bao, C.; Guo, W.; Li, S.; Chen, J.; Chen, B.; Luo, Y.; Lyu, D.; Li, Y.; Shi, G.; et al. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat. Commun. 2016, 7, 11215. [Google Scholar] [CrossRef] [PubMed]
- Muller, E.E.; Locatelli, V.; Cocchi, D. Neuroendocrine control of growth hormone secretion. Physiol. Rev. 1999, 79, 511–607. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Sun, M.; Liu, H.; Yao, Y.; Song, Y. Long non-coding RNAs: A new frontier in the study of human diseases. Cancer Lett. 2013, 339, 159–166. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Li, C.; Wei, J.; Ni, W.; Xu, Y.; Yao, R.; Zhang, M.; Li, H.; Liu, L.; Dang, H.; et al. Comprehensive Expression Profiling Analysis of Pituitary Indicates that circRNA Participates in the Regulation of Sheep Estrus. Genes 2019, 10, 90. https://doi.org/10.3390/genes10020090
Li X, Li C, Wei J, Ni W, Xu Y, Yao R, Zhang M, Li H, Liu L, Dang H, et al. Comprehensive Expression Profiling Analysis of Pituitary Indicates that circRNA Participates in the Regulation of Sheep Estrus. Genes. 2019; 10(2):90. https://doi.org/10.3390/genes10020090
Chicago/Turabian StyleLi, Xiaoyue, Cunyuan Li, Junchang Wei, Wei Ni, Yueren Xu, Rui Yao, Mengdan Zhang, Huixiang Li, Li Liu, Hanli Dang, and et al. 2019. "Comprehensive Expression Profiling Analysis of Pituitary Indicates that circRNA Participates in the Regulation of Sheep Estrus" Genes 10, no. 2: 90. https://doi.org/10.3390/genes10020090
APA StyleLi, X., Li, C., Wei, J., Ni, W., Xu, Y., Yao, R., Zhang, M., Li, H., Liu, L., Dang, H., Hazi, W., & Hu, S. (2019). Comprehensive Expression Profiling Analysis of Pituitary Indicates that circRNA Participates in the Regulation of Sheep Estrus. Genes, 10(2), 90. https://doi.org/10.3390/genes10020090