1. Introduction
CRISPR (clustered regularly interspersed short palindromic repeats)-Cas (CRISPR-associated) systems evolved to defend microbes against bacteriophages and have been classified into different types based on their
cas genes [
1,
2]. Due to their RNA-based programmable DNA-targeting capability, the type-II Cas9 effector nucleases have been widely adapted as tools for gene editing, and beyond [
3]. More recently, the type V Cas12a effectors (formerly known as Cpf1) have also been shown to be active for gene editing [
4,
5]. The unique properties of Cas12a paralogues mean that, for many applications, they could become the gene editing enzyme of choice. Despite high-resolution crystal and electron microscopy (EM) structures, and further rapid progress, our knowledge of Cas12a is rudimentary [
6]. Here we sought to understand the nuclease mechanism of
Lachnospiraceae bacterium ND2006 Cas12a (LbCas12a) by examining the kinetics of DNA cleavage and the effect of DNA topology on the observed rates.
Activation of the Cas12a nuclease activity requires R-loop formation between the CRISPR RNA (crRNA) and the DNA protospacer sequences [
7,
8,
9,
10]. A Cas12a-crRNA binary complex first binds DNA though interaction between a T-rich Protospacer Adjacent Motif (PAM, 5′-TTTV-3′, where V = A/C/G) [
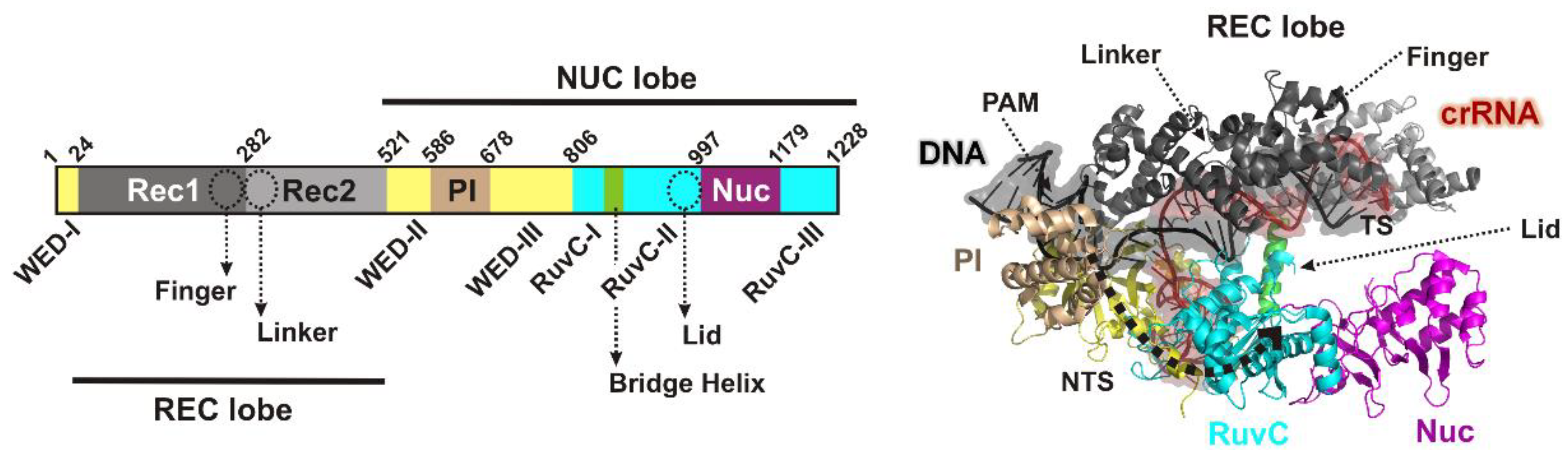
11], and a flexible pocket formed by the wedge (WED), REC1 and the PAM-interacting (PI) domains [
7,
11] (
Figure 1). PAM distortion leads to ATP-independent stand separation and the DNA target strand (TS) forms a heteroduplex with the pre-structured 3′ end of the crRNA spacer sequence (the “seed”) [
4,
7,
8,
12], displacing the non-target strand (NTS). A 20 bp R-loop then propagates by dsDNA unzipping and pseudo A-form RNA hybridization, triggering DNA cleavage with some variability in the precise cut sites [
4,
13]. How Cas12a generates a dsDNA break has been a matter of some debate, with recent breakthroughs that help to clarify our understanding of the mechanism.
For Cas9, there are separate, classifiable nuclease domains, RuvC and HNH, which target the NTS and TS, respectively [
14,
15,
16]. The HNH domain cleaves DNA faster than RuvC but it has been suggested that the conformational activation of the HNH domain controls the overall timing of DNA cleavage [
17,
18]. A classifiable RuvC domain is present in Cas12a, but a second nuclease domain was not identified from sequence/structure prediction alone [
4,
19]. An unclassified domain (Nuc,
Figure 1) was suggested as the second nuclease on the basis that mutations produced DNA nicking [
10]. Since RuvC mutations prevented any cleavage [
10], an ordered strand-cleavage mechanism was proposed where the RuvC must act first and only then can Nuc carry out the second strand cleavage. Other groups argued that Nuc lacks identifiable catalytic residues and demonstrated that equivalent mutants still generated dsDNA cleavage [
7]. The alternative suggestion is that Nuc regulates access to the RuvC active site which cuts both strands [
6,
8]. Structures of the related type V enzyme Cas12b [
20,
21], were also more consistent with Nuc acting in a noncatalytic role.
Closure of the Cas12a lobes moves the PI, REC1 and REC2 domains, exposing the RuvC nuclease and first guiding the displaced NTS towards RuvC [
6,
8,
9,
22,
23], although none of the structures showed the DNA engaged with the active site. Stella et al. have recently identified a series of conformation checkpoints that couple R-loop propagation to nuclease activation [
8]: Firstly a loop connecting REC1 and REC2 lobes (the “linker”,
Figure 1) interacts with the 5th to 7th nucleotides of the crRNA as the R-loop forms; secondly, a loop (the “lid”,
Figure 1) changes conformation, breaks contacts with the catalytic side chains of the RuvC nuclease, and interacts with the 8th to 11th nucleotides of the crRNA; and thirdly, a helix in the REC1 lobe (the “finger”,
Figure 1) moves to interact with the 15th to 17th nucleotides of the crRNA. A requirement for more than 17 bp of hybrid to activate cleavage has been measured elsewhere [
5,
7,
24].
Once activated, the exposed nuclease can now bind ssDNA, with the NTS being the most closely located (
Figure 1). Accordingly, several studies have demonstrated a difference in the kinetics of NTS and TS cleavage. Jeon et al., (2018) used single-molecule fluorescence to identify distinct Fluorescence Resonance Energy Transfer (FRET) states that followed a sequential order during DNA cleavage by
Acidaminococcus sp. Cas12a (AsCas12a) [
25]. By using 51 bp oligoduplexes pre-nicked in either the NTS or TS, they assigned FRET states to NTS or TS cleavage events. The FRET transition profiles were then consistent with NTS cleavage before TS cleavage. But the lifetime of the states appeared similar (36 s and 58 s at 37 °C). In an extensive kinetic study, Strohkendl et al. followed NTS and TS cleavage by AsCas12a in separate reactions by individual radiolabeling of each strand of a 57 bp oligoduplex [
26]. They determined maximal cleavage rate constants of 0.05 s
−1 and 0.005 s
−1 for the NTS and TS, respectively (at 25 °C), consistent with NTS cleavage preceding TS cleavage. Stella et al. [
8] measured individual strand cleavage rates using labelled oligoduplexes. An NTS cleavage rate of ~0.007 s
−1 (at 37 °C) was measured using a partially double-strand duplex where the NTS spacer was 24 nt but the TS spacer was 20 nt. A TS cleavage rate of ~0.0009 s
−1 (at 37 °C) was measured using a partially double-strand duplex where the TS spacer was 20 nt but the TS spacer was only 14 nt. Again, the data is consistent with the NTS being cleaved before the TS. We note however that other studies used labelled DNA to follow NTS and TS cleavage but did not observe measurable differences in product appearance.
The activated open nuclease may accept the NTS more readily because of its orientation and location, with the TS only entering more slowly following a conformational transition (i.e., there isn’t a strict sequential order per se) [
8]. This idea is consistent with the observation of
in trans cleavage of bystander nonspecific ssDNA that is catalyzed by Cas12a upon recognition of a TS strand [
22,
23]. Alternatively, the single RuvC-Nuc nuclease might transition sequentially between the NTS and TS in a strictly ordered fashion, but other DNA could access the site at any point.
Many studies of CRISPR Cas effectors have utilized short oligoduplexes as convenient substrates to monitor nucleic acid interactions and nuclease activity. However, using a single-molecule magnetic tweezers (MT) assay, we showed with both type II Cas9 and type I Cascade that on topologically-constrained linear DNA, negative supercoiling accelerates R-loop formation rates [
27]. Due to the low/zero negative torque on unconstrained linear DNA, R-loop formation may become rate-limiting relative to other steps. Additionally, short oligoduplexes may become destabilized during R-loop formation, leading to fraying of DNA ends. This may favor R-loop formation in the absence of topology but may not mimic the situation in a genomic context where protospacers are away from free ends.
To address the torque-dependence of R-loop formation and DNA cleavage by LbCas12a, we first used the previously-described MT assay [
27] to follow R-loop formation by LbCas12a in real time. The data shows that LbCas12a is torque-dependent, as expected, but less torque-stable than Cas9 or Cascade. By comparing cleavage kinetics on plasmid, nicked and linear DNA, we show that the microscopic DNA cleavage rate constants for the first cleavage event is >15-fold faster than the second, and that the observed cleavage of nicked and linear DNA were slower by at least 50-fold compared to negatively-supercoiled DNA. This can be explained by R-loop formation being rate-limiting on unconstrained linear DNA relative to cleavage. By labelling the TS and NTS strands of the plasmid-length linear DNA and comparing the data to simulations using the measured first and second strand rate constants, we could show that the more rapid cleavage rate is due to NTS cleavage. However, we could not distinguish between strict sequential and random models from the kinetics alone. We suggest that future studies of Cas12a (and other CRISPR-Cas effector nucleases) should always consider the effect of R-loop formation kinetics on the observed rates of downstream events including domain motions and strand cleavage events.
2. Materials and Methods
2.1. Protein
Saccharomyces cerevisiae Ubiquitin-like-specific protease 1 (SUMO-protease ULP1) was purified using pFGET19_Ulp1 and the published protocol [
28]. A synthetic gene for LbCas12a, codon-optimized for
Escherichia coli expression, was supplied by IDT, and cloned into pE-SUMO(Kan) (LifeSensors Inc, Malvern, PA, USA) using NEBuilder
® HiFi DNA Assembly (New England Biolabs, Ipswich, MA, USA). To express the protein,
E. coli Tuner (DE3) were transformed with pE-SUMO(Kan)-Cas12a. Following overnight growth at 37 °C on LB-agar (kanamycin, 50 μg/mL), a single colony was used to inoculate 5 mL LB (kanamycin, 50 μg/mL). Following overnight growth at 37 °C, the 5 mL culture was used to inoculate 500 mL LB (kanamycin, 50 μg/mL), and the cells grown in a 2.5 L flask at 37 °C and 250 rpm shaking. At an OD
600 of ~0.6–0.7, isopropyl β-D-1-thiogalactopyranoside was added to 1 mM, and cell continued overnight at 16 °C and 250 rpm shaking.
Cells were harvested, resuspended in 25 mL of Buffer A [50 mM Tris-Cl (pH 8.0), 500 mM NaCl, 5 mM MgCl2, 0.5 mM EDTA, 1 mM β-mercaptoethanol and EDTA-free protease inhibitor tablets according to the manufacturer’s instructions (Roche, Welwyn Garden City, UK)] and lysed by sonication. The cell extract was clarified by centrifugation at 100,736 g for 40 min. The supernatant was dialyzed for >1 hr at 4 °C against Buffer B [50 mM Tris-Cl (pH 8.0), 500 mM NaCl, 1 mM β-mercaptoethanol, 30 mM imidazole] using 10,000 Da cut-off Snake Skin dialysis tubing. The dialyzed sample was 0.45 µm filtered and loaded onto a 5-mL HisTrap column (GE Healthcare UK Ltd, Little Chalfont, UK), equilibrated in Buffer B. Bound proteins were eluted with a linear gradient of Imidazole in Buffer B (30–500 mM, 100 mL). Fractions containing Cas12a were pooled and dialyzed for 1 hr at 4 °C against two changes of Buffer C [50 mM Tris-Cl (pH 8.0), 200 mM NaCl, 1 mM β-mercaptoethanol] using 10,000 Da cut-off Snake Skin dialysis tubing. Ulp1 was added to the dialysis tubing and incubated overnight at 4 °C in fresh Buffer C.
NaCl and imidazole were added to 500 mM and 30 mM, respectively, and the dialyzed sample 0.45 μm filtered and loaded onto a 5-mL HisTrap column (GE Healthcare UK Ltd), equilibrated in Buffer B. The flow-through containing Cas12a was collected and the Sumo-tag and ULP1 protease eluted separately using Buffer B supplemented to 500 mM imidazole. Cas12a was concentrated and equilibrated into Buffer C using 50 kDa cut-off centrifugal filter units (Millipore, MA, USA). For storage at −20 °C, glycerol was added to 50% (v/v).
2.2. crRNA and Ribonucleoprotein Complex Assembly
The crRNA was produced by in vitro transcription. The oligodeoxyribonucleotides 5′-GAAATTAATACGACTCACTATCGGG-3′ and 5′-AGCTCGAATTGAAATTCTAAACGCATCTACACTTAGTAGAAATTCCCTATAGTGAGTCGTATTAATTTC-3′ (Eurofins genomics) were annealed at 40 μM in 50 mM NaCl by heating for 5 min at 95 °C followed by slow-cooling to room temperature (RT). This oligoduplex was then used in the HiScribe T7 high yield RNA synthesis kit (New England Biolabs) according to the manufacturer’s protocol. The samples were subjected to the optional DNase I treatment, and the crRNA purified using an RNA Clean & Concentrator (Zymo Research, Irvine, CA, USA). RNA concentrations were determined from UV absorbance at 260 nm. To assemble the ribonucleoprotein complex, 500 nM Cas12a and 500 nM crRNA were incubated in buffer SB [10 mM Tris (pH 7.5), 100 mM NaCl, 1 EDTA, 0.1 mM DTT, 5 μg/mL BSA] with RNase inhibitor (1U/ μL SUPERase in, Invitrogen, Carlsbad, CA, USA) for 1 hr at 37 °C, and the assembled complex used immediately.
2.3. DNA Substrates
For the MT assay, the plasmid pSP1 was used to prepare the biotin- and digoxigenin-labelled linear DNA substrate (see main text), as described previously [
27,
29]. For the supercoiled DNA used in the cleavage assays,
E. coli Top10 (Invitrogen) or XL10-Gold (Stratagene, San Diego, CA, USA) were transformed with pSP1 [
27], grown in M9 minimal medium supplemented with 37 MBq/l [
3H-methyl] thymidine, and the DNA extracted using commercial protocols (Qiagen, Hilden, Germany) or by density gradient centrifugation in CsCl-ethidium bromide [
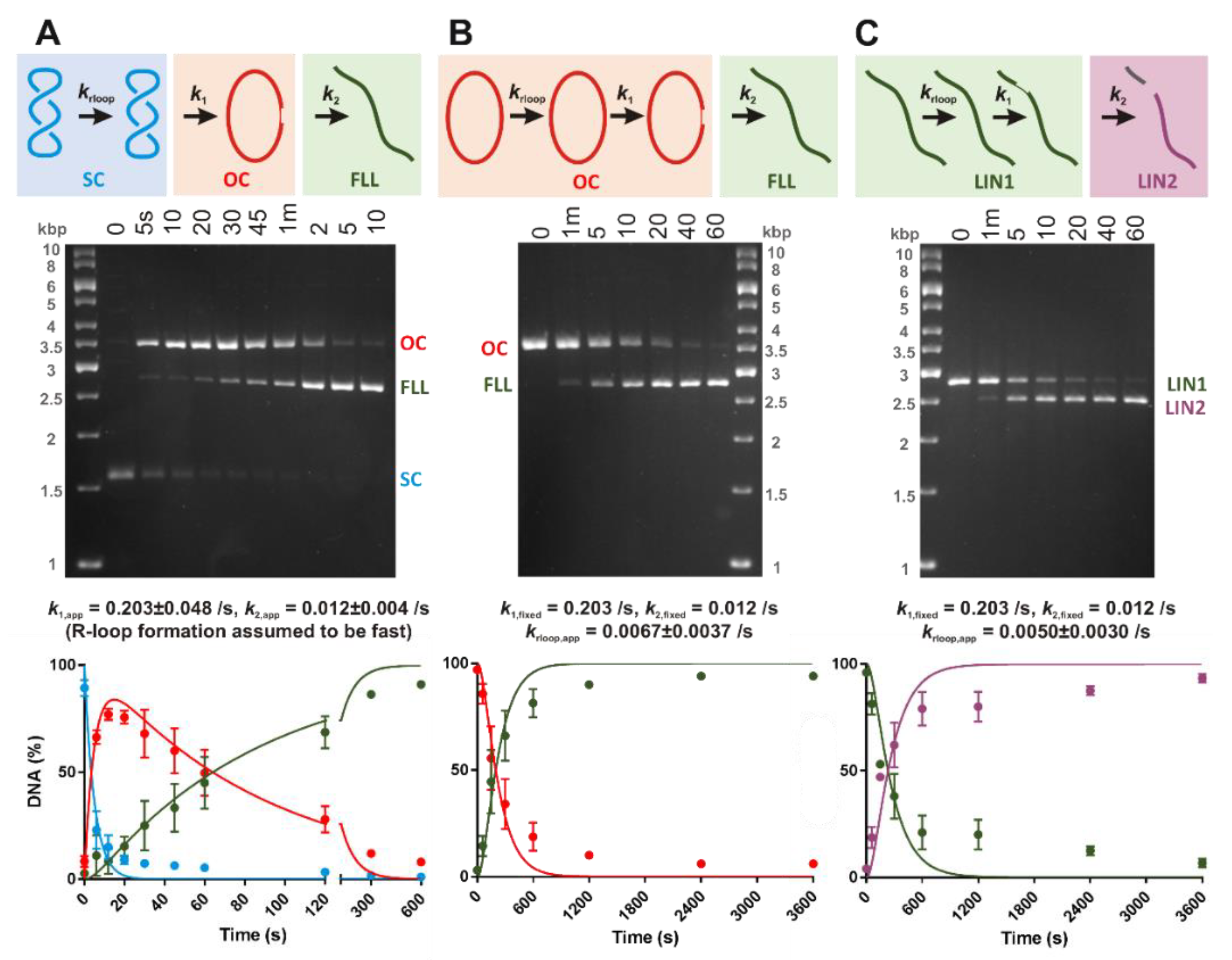
30]. Open Circle or LIN1 DNA substrates were generated by incubating
3H-labelled pSP1 with 0.5 U/μl of BspQI or Nt.BspQI in NEBuffer 3.1 (New England Biolabs) for 1 hr at 50 °C. The DNA was purified by phenol/chloroform and chloroform extraction followed by ethanol precipitation. DNA concentrations were determined from UV absorbance at 260 nm, assuming that an optical density of 1 corresponds to 50 μg/mL DNA and a molecular weight of 6.6 × 10
5 Da/kbp. On the linear substrate, the PAM sequence is 258 and 1853 bp from the free DNA ends.
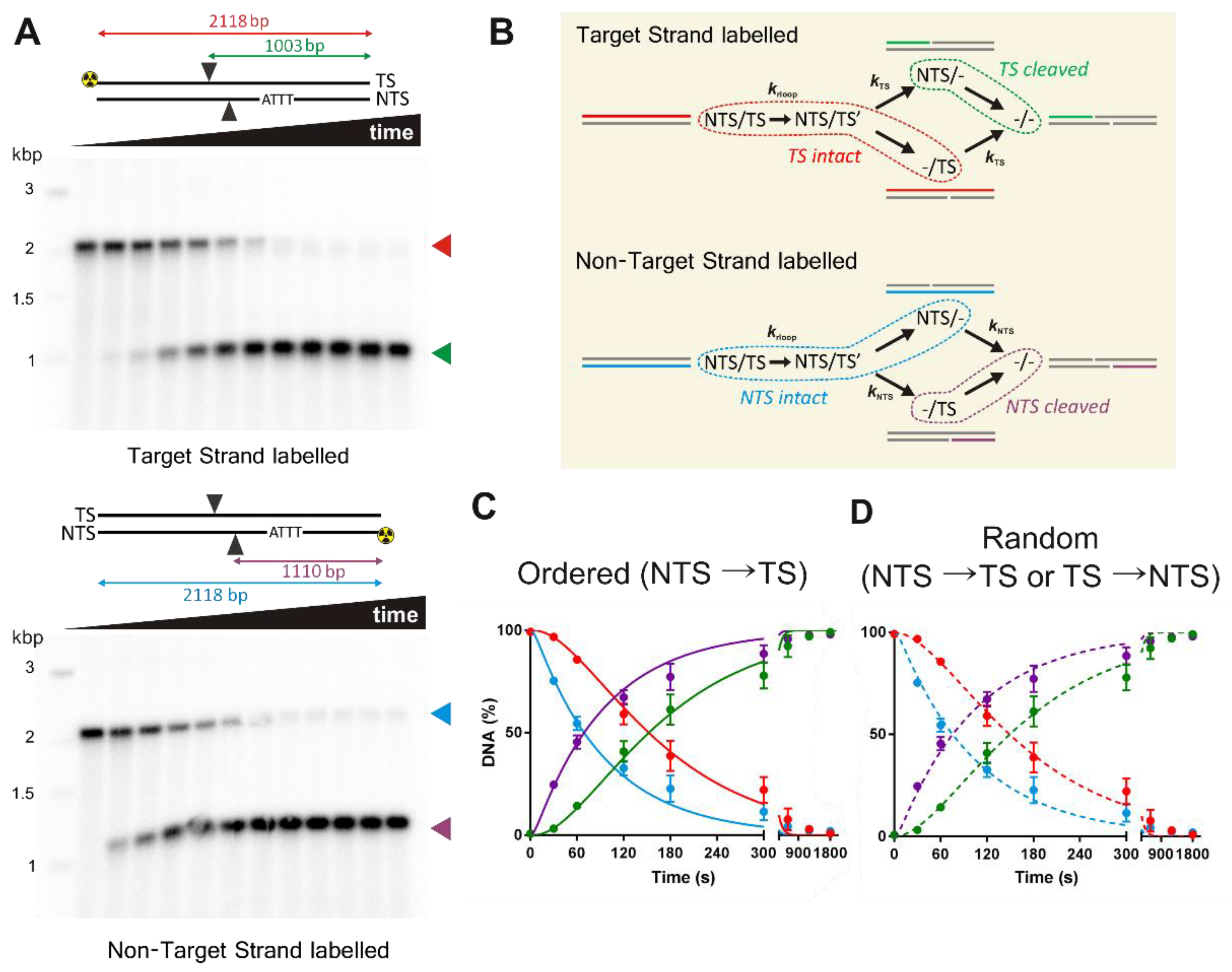
For the strand-specificity experiments, PCR was used to generate a 2118 bp linear pSP1 fragment with the forward primer 5′-GCGTAAGTCTCGAGAACTAGTTCCGTAAGATGCTTTTCTGTGACT-3′ and the reverse primer 5′-GCGTAAGTGCGGCCGCTTCGTTCCACTGAGCGTCAGA-3′. To label the TS or NTS, the forward or reverse primer, respectively, was first 5′-labelled with 32P using T4-polynucleotide kinase. The PCR reactions were purified using a QIAquick PCR purification kit (Qiagen) and the DNA concentrations determined as above. As a marker, a commercial 1 kb DNA Ladder (New England Biolabs) was labelled with 32P using T4-polynucleotide kinase.
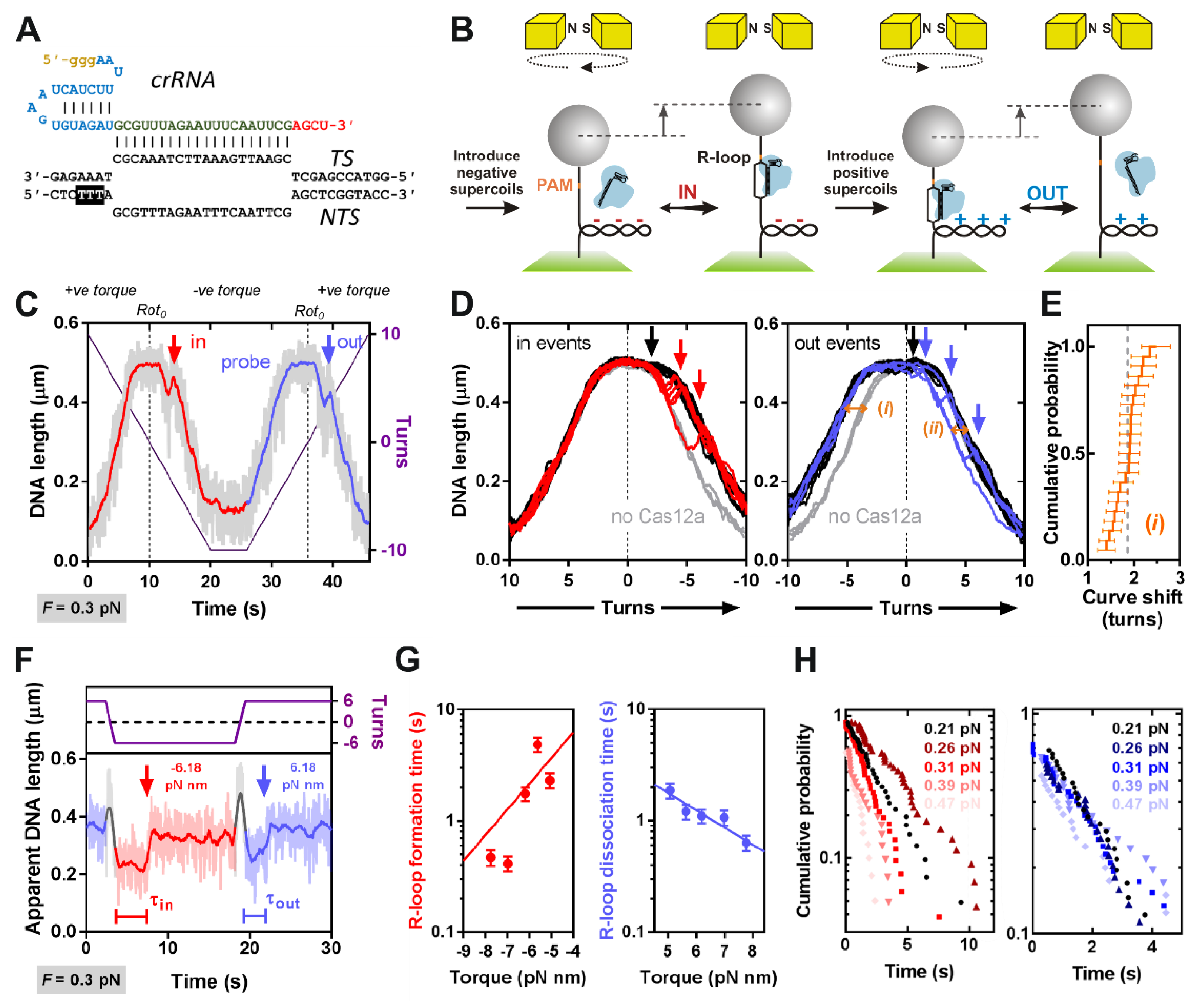
2.4. Magnetic Tweezers Assay
Magnetic tweezers measurements were performed using a commercial PicoTwist microscope (Fleurieux sur L’Arbresle, France) equipped with a Jai CV-A10 GE camera (image acquisition at 60 Hz) [
31]. DNA molecules were tethered to 1-μm MyOne paramagnetic beads (Invitrogen) and anchored in flow cells as previously described [
27]. Suitable topologically-constrained DNA were identified from rotation curves and the rotational zero reference (
Rot0) set from a rotation curve at 0.3 pN. R-loop reactions were analyzed in Buffer SB at 25 °C, using 5-nM LbCas12a-crRNA. The shift in
Rot0 upon R-loop formation was estimated by comparing the slope of the rotation curves when an R-loop was trapped in negative torque with the equivalent slopes in the absence of enzyme. Each trace in a reference set of 10 rotation curves collected in the absence of enzyme was compared to each rotation curve when an R-loop was trapped in the presence of enzyme (a total of 22) by using a least squares method to iteratively fit the matched data sets at −4.5 to –6.0 turns to parallel lines. The 220 parametrized linear fits where used to estimate the shift in the rotation curves and a local average was obtained for each R-loop event. A global average of the shifts in
Rot0 was subsequently obtained by parametrizing the distribution of the local averages to a Gaussian function by maximum likelihood estimation. To analyze the shift due to the R-loop that remains trapped at positive torque, the same principle was applied. In this case, each trace in a reference set of 18 rotation curves collected in the presence of enzyme where the R-loop had already dissociated spontaneously at lower positive torque was compared to each of 2 curves in which the R-loop remained bound at +3.0 to +5.0 turns, and the global average of the shift in
Rot0 was obtained from the 36 fits without assumption about the statistical distribution of the data. Torque values were calculated using software described in Ref [
27]. Errors in the mean R-loop formation and dissociation times were calculated as the standard error of an exponential distribution, by dividing the mean reaction time by the square root of the number of events [
29].
2.5. DNA Cleavage Assays
In vitro DNA cleavage reactions contained 3 nM DNA substrate and 50 nM LbCas12a-crRNA in Buffer RB [10 mM Tris (pH 7.5), 100 mM NaCl, 10 mM MgCl
2, 0.1 mM DTT, 5 μg/mL BSA] at 25 °C. Reactions were initiated by addition of Cas12a-crRNA. For the native 1.5% (
w/
v) agarose gel electrophoresis in 1× TAE buffer, reactions were quenched at the time points indicated by 0.5 volumes of STEB [0.1 M Tris (pH 7.5), 0.2 M EDTA, 40% (
w/
v) sucrose, 0.4 mg/mL bromophenol blue] and heated for 10 min at 80 °C. The percentage of DNA in each band per lane was ascertained by scintillation counting [
30]. Note that the smaller product of LIN1 cleavage was not clearly resolved on the gel. For alkaline denaturing 2% (
w/
v) agarose gel electrophoresis in 50 mM NaOH, 1 mM EDTA, reactions were quenched at the time points indicated by 0.5 volumes of Alkaline Buffer [300 mM NaOH, 6 mM EDTA, 18% (
w/
v) Ficoll 400, 0.1% (
w/
v) Bromocresol green and 0.1% (
w/
v) Xylene Cyanol] and heated for 10 min at 80 °C. Following electrophoresis, the gels were neutralized for 1 h in 500 mM Tris-HCl (pH 8), compressed for one hour, dried under vacuum for one hour, and a 16-bit densitometric phosphor screen scan analyzed using the 1D gel analysis software of ImageQuant (GE Healthcare). DNA fitting and simulations used numerical integration in Berkeley Madonna (
www.berkeleymadonna.com). For the fits, averages and errors were calculated from the values returned from individual datasets.
4. Discussion
We measured R-loop formation for LbCas12a (
Figure 1) using the MT assays previously applied to Cas9 and Cascade (
Figure 2) [
27]. Our data are consistent with an ~20 bp R-loop as observed in the crystal structures (although the rotational shifts measured in
Figure 2D,E are an indirect measurement that can be altered by DNA wrapping/bending, [
27]). The data is most similar to that observed with type II Cas9, which was strikingly less torque stable than type I Cascade. Both the rotation curves and kinetic data (
Figure 2D,G) indicate that Cas12a is more torque sensitive than Cas9, with spontaneous R-loop formation and dissociation at low torque values. For
Streptococcus thermophilus DGCC7710 CRISPR3 Cas9, the estimated transition time for R-loop dissociation at zero torque was ~130 s [
27], suggesting it is >10-fold more stable than LbCas12a (
Table 1). Cas12a torque-instability may be a benefit in preventing off target effects, as mismatches may have a greater destabilization effect. A survey of mismatch effects has been undertaken for linear DNA targets in vitro by Strohkendl et al. [
26], as well as by others [
4,
7,
24]. It would be interesting to determine whether the effects of mismatches are impacted by DNA supercoiling.
We observed a striking difference in the apparent DNA cleavage rates on negatively supercoiled plasmid DNA compared to topologically-unconstrained substrates (
Figure 3). Since negative DNA supercoiling favors R-loop formation (
Figure 2), this can be readily explained by DNA cleavage being rate-limited by R-loop formation in the absence of negative torque; R-loops will form within a second on negatively supercoiled DNA but could take tens to hundreds of seconds on linear or nicked DNA. Many previous studies of type V systems have used linear DNA substrates. Caution is needed when analyzing downstream events (e.g., cleavage kinetics) to ensure that the effect of R-loop formation rates is considered. Rates may also be affected by the position of the protospacer relative to DNA ends; being close to an end may allow more rapid R-loop formation due to capture of free ssDNA produced by thermal fraying and/or because the thermodynamics of strand separation are altered.
Consistent with other studies [
8,
26], we observed a more than 15-fold difference in the rate of cleavage of the NTS compared to the TS (
Figure 3 and
Figure 4,
Table 2). Because of the differences in the first and second strand cleavage rates, it is difficult to distinguish between a strict ordered mechanism and a random mechanism based on our kinetics. A limitation of the approach in
Figure 4 is that the labelled ssDNA can represent multiple species on the reaction pathway (
Figure 4B). A potential solution is a substrate where the TS and NTS nicked intermediates can be resolved. One way to achieve this would be a self-complementary hairpin oligonucleotide substrate, with an analysis label (e.g., a fluorophore) within the ssDNA hairpin loop, and separation of the DNA species on a denaturing gel. For a staggered cut as produced by Cas12a, this would allow the resolution in a single reaction of labelled substrate, both intermediates, and product as fragments of different lengths. However, because of the 15-fold difference in strand cleavage rates observed here, and compounded by slow R-loop formation rate on relaxed DNA, one would need to be confident in distinguishing 0% TS nicked intermediate (for an NTS→TS ordered model) from ~0.2% maximum TS nicked intermediate (for a random model). Even if the R-loop formation rate could be accelerated, the difference in expected TS intermediate species would still only be 0% and 2%, respectively.
The slower observed rate constant for TS cleavage could be due to a slow conformational transition that is required to engage the TS in the RuvC active site [
7,
8]. The PAM-distal TS cleavage site is distant (>40 Å) from RuvC in the Cas12a structures (
Figure 1). Therefore, significant movement of RuvC-Nuc and/or the DNA-RNA must be required. One option is that the TS region must unwind following NTS cleavage to expose ssDNA for cleavage and this moves into the active site (“DNA reeling”). Stella et al. suggest that the R-loop may partially unwind to facilitate this [
8]. Alternatively, the RuvC-Nuc domains could move towards the distal TS cleavage site (i.e., closing the nuclease and Rec lobes). A conformation change step following NTS cleavage was not included in the consideration of our kinetic models. Without being able to measure the intermediate species, adding further parameters to the model would not have returned sensible fitted values. Alternative approaches, such as FRET or structural determination (e.g., [
8,
24]), will be required to reveal what movements in the protein and DNA/RNA are required.







{kind=link}
{kind=link}
{kind=link}
{kind=link}