Genome-Wide Identification and Analysis of High-Copy-Number LTR Retrotransposons in Asian Pears

Abstract
1. Introduction
2. Methods and Materials
2.1. Plant Materials and DNA Extraction
2.2. The Genome Data
2.3. Identification and Annotation of LTR-RTs
2.4. Whole-Genome Resequencing of Pyrus Species
2.5. Evaluation of the Relative Copy Numbers of LTR-RTs
2.6. The Insertion Times and Classifications of High-Copy-Number LTR-RT Subfamilies
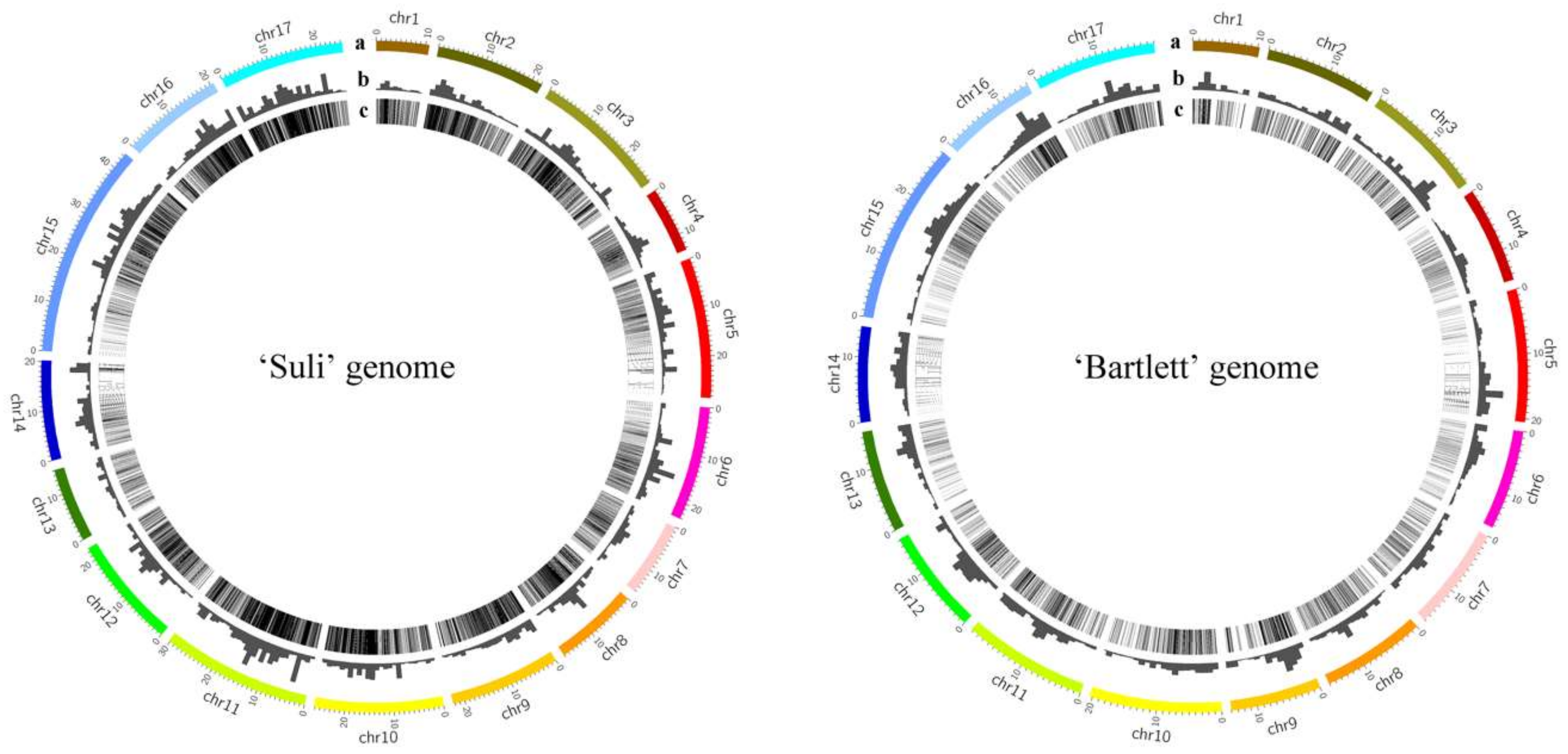
2.7. Genomic Distribution of High-Copy-Number LTR-RT Subfamilies
2.8. The Homologous and Specific Insertion Sites in the ‘Suli’ and ‘Bartlett’ Genomes
3. Results
3.1. Prediction and Classification of Retrotransposons in the Pyrus Genome
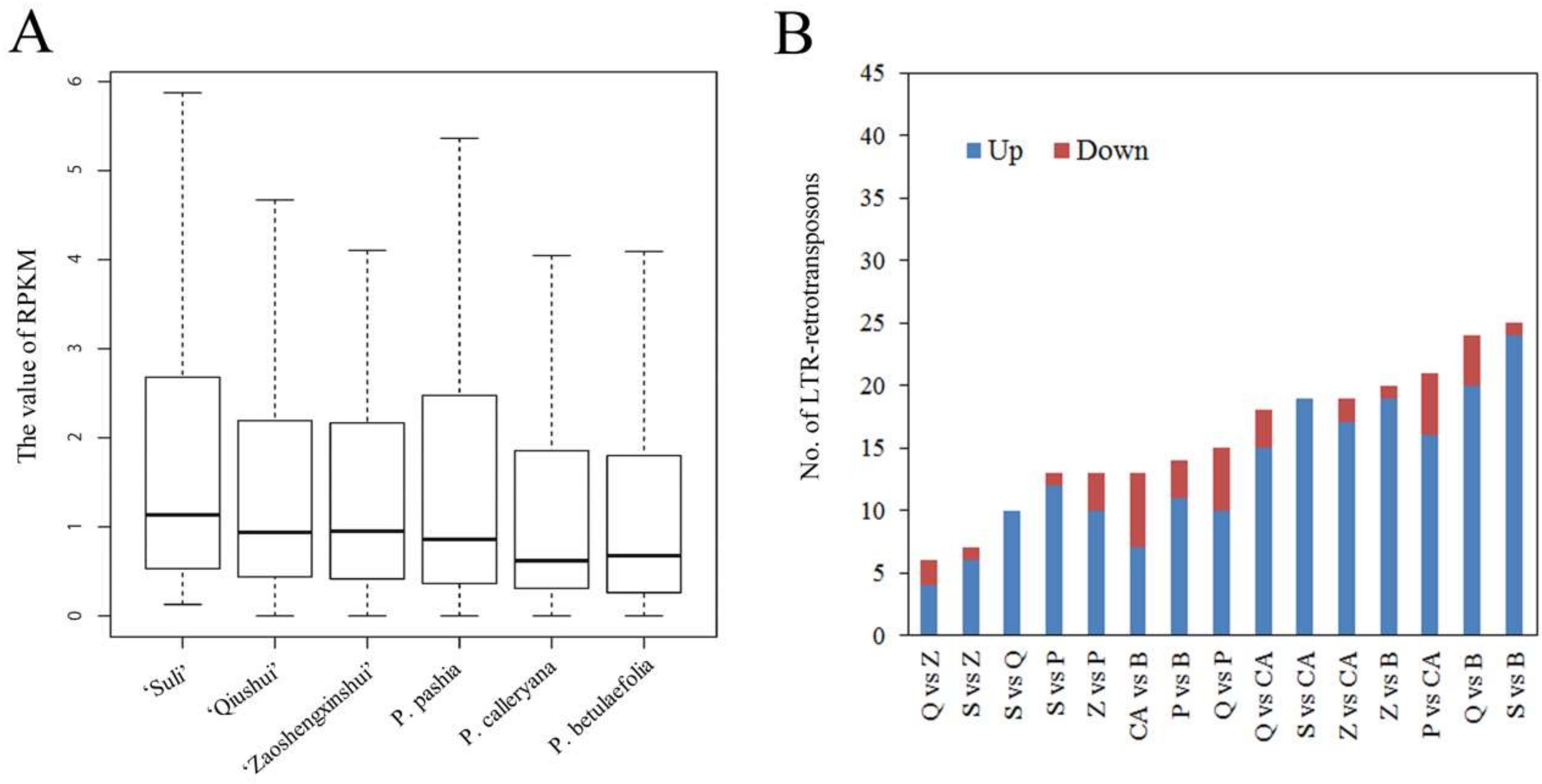
3.2. Whole-Genome Resequencing in Pyrus Species
3.3. Variable Relative Copy Numbers of LTR-RT Subfamilies in Pyrus Species
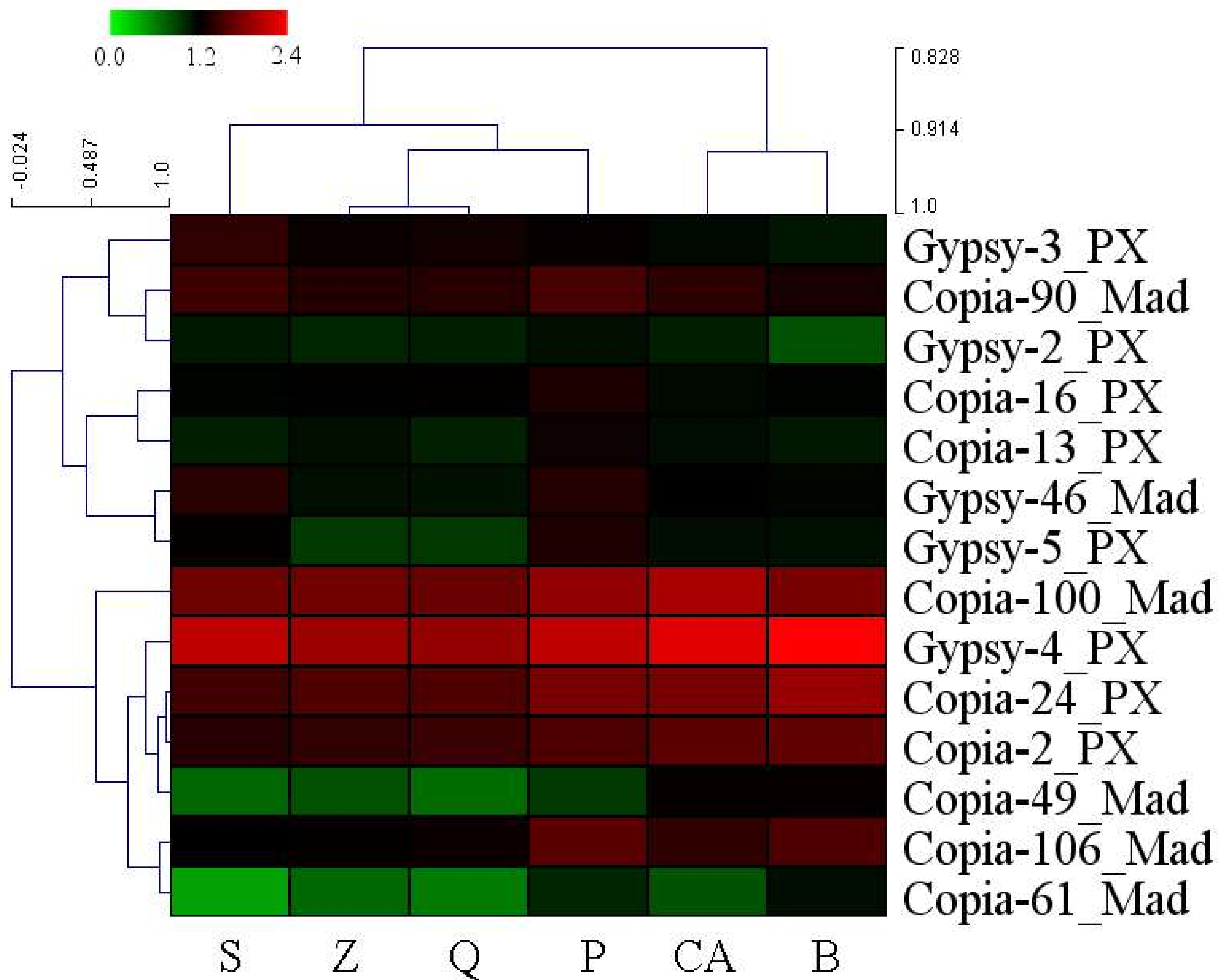
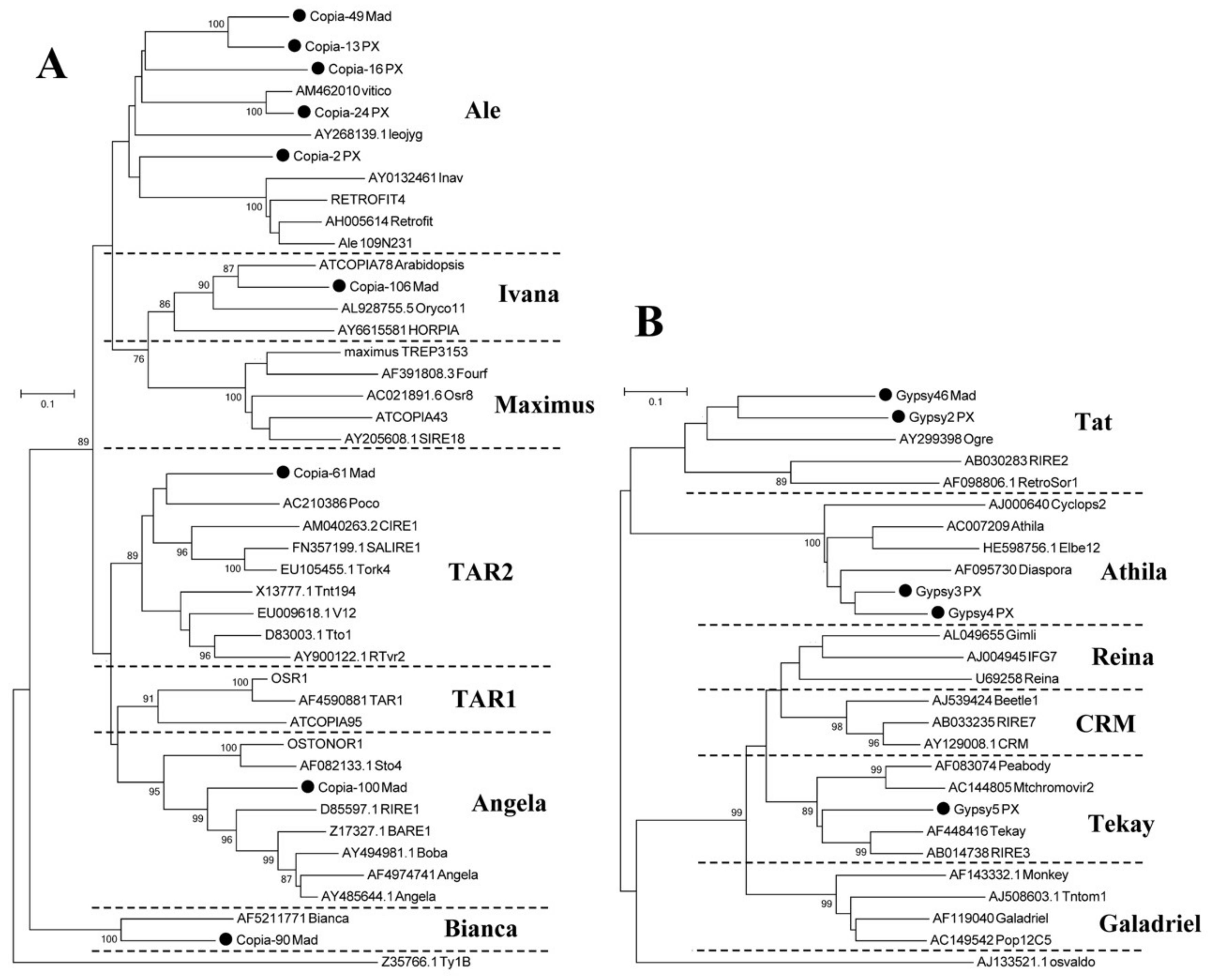
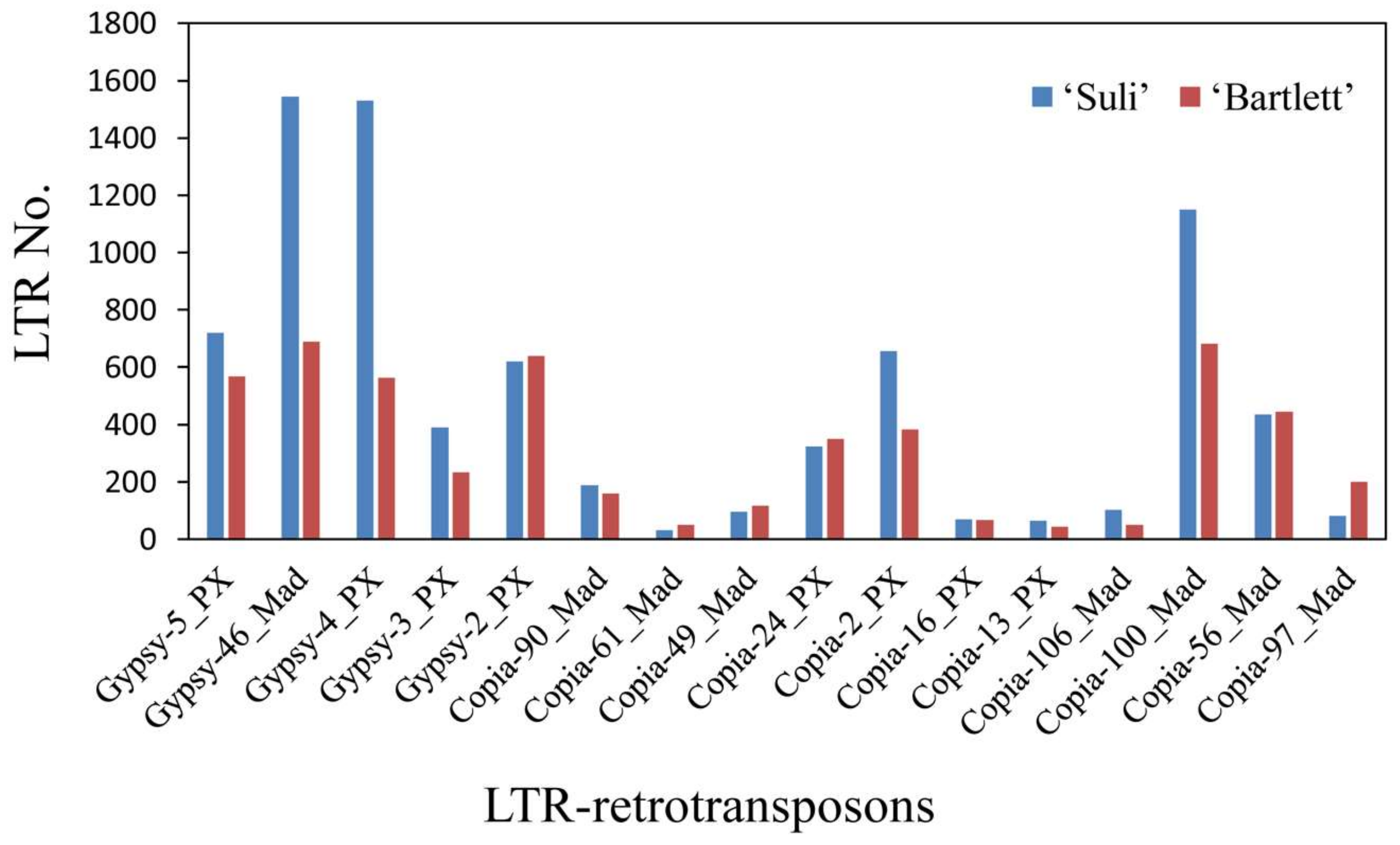
3.4. High-Copy-Number LTR-RT Subfamilies in Pyrus Species
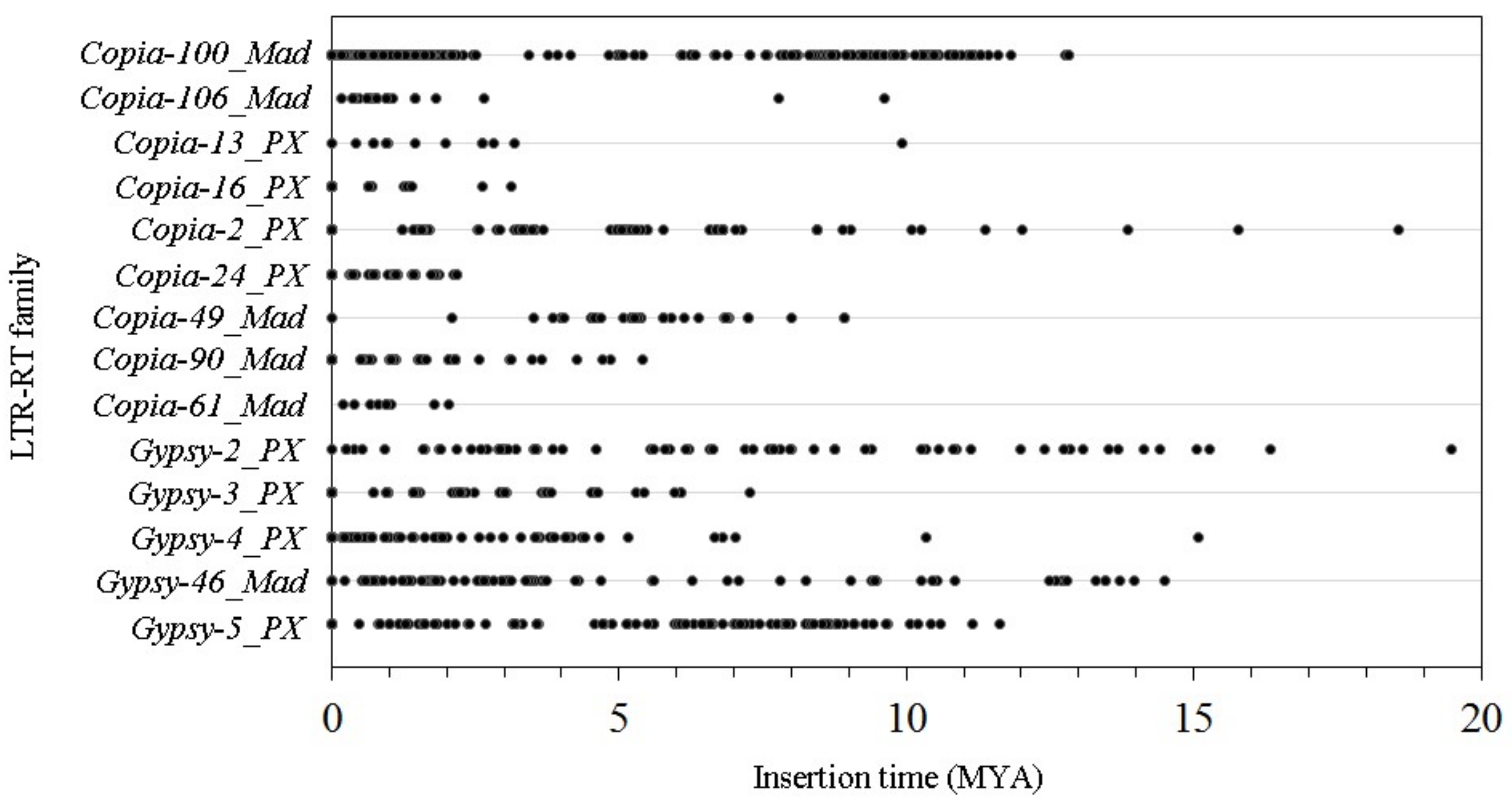
3.5. The Insertion Times of the 14 LTR-RT Subfamilies in the ‘Suli’ Genome
3.6. The Insertion Sites of the 14 LTR-RT Subfamilies in the ‘Suli’ and ‘Bartlett’ Genomes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgements
Conflicts of Interest
Abbreviations
| LTR-RT | long terminal repeat retrotransposon |
| RPKM | reads per kilobase per hundred thousand mapped pear reference genome (‘Suli’ genome) reads |
References
- Kumar, A.; Bennetzen, J.L. Plant retrotransposons. Annu. Rev. Genet. 1999, 33, 479–532. [Google Scholar] [CrossRef] [PubMed]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Sabot, F.; Schulman, A.H. Parasitism and the retrotransposon life cycle in plants: A hitchhiker’s guide to the genome. Heredity 2006, 97, 381–388. [Google Scholar] [CrossRef] [PubMed]
- SanMiguel, P.; Tikhonov, A.; Jin, Y.K.; Motchoulskaia, N.; Zakharov, D.; Melake-Berhan, A.; Springer, P.S.; Edwards, K.J.; Lee, M.; Avramova, Z.; et al. Nested retrotransposons in the intergenic regions of the maize genome. Science 1996, 274, 765–768. [Google Scholar] [CrossRef] [PubMed]
- Piegu, B.; Guyot, R.; Picault, N.; Roulin, A.; Saniyal, A.; Kim, H.; Collura, K.; Brar, D.S.; Jackson, S.; Wing, R.A.; et al. Doubling genome size without POLyploidization: Dynamics of retrotransposition-driven genomic expansions in Oryza australiensis, a wild relative of rice. Genome Res. 2006, 16, 1262–1269. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Devos, K.M.; Bennetzen, J.L. Analyses of LTR-retrotransposon structures reveal recent and rapid genomic DNA loss in rice. Genome Res. 2004, 14, 860–869. [Google Scholar] [CrossRef] [PubMed]
- Steinbiss, S.; Willhoeft, U.; Gremme, G.; Kurtz, S. Fine-grained annotation and classification of de novo predicted LTR retrotransposons. Nucleic Acids Res. 2009, 37, 7002–7013. [Google Scholar] [CrossRef]
- Marquet, R.; Isel, C.; Ehresmann, C.; Ehresmann, B. tRNAs as primer of reverse transcriptases. Biochimie 1995, 77, 113–124. [Google Scholar] [CrossRef]
- Mak, J.; Kleiman, L. Primer tRNAs for reverse transcription. J. Virol. 1997, 71, 8087–8095. [Google Scholar]
- Havecker, E.R.; Gao, X.; Voytas, D.F. The diversity of LTR retrotransposons. Genome Biol. 2004, 5, 225. [Google Scholar] [CrossRef][Green Version]
- Feschotte, C.; Jiang, N.; Wessler, S.R. Plant transposable elements: Where genetics meets genomics. Nat. Rev. Genet. 2002, 3, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, J.A. Retrotransposons and regulatory suites. BioEssays 2005, 27, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Goto-Yamamoto, N.; Hirochika, H. Retrotransposon-induced mutations in grape skin color. Science 2004, 304, 982. [Google Scholar] [CrossRef] [PubMed]
- Butelli, E.; Licciardello, C.; Zhang, Y.; Liu, J.; Mackay, S.; Bailey, P.; Reforgiato-Recupero, G.; Martin, C. Retrotransposons control fruit-specific, cold-dependent accumulation of anthocyanins in blood oranges. Plant Cell 2012, 24, 1242–1255. [Google Scholar] [CrossRef] [PubMed]
- Ni, F.; Qi, J.; Hao, Q.; Lyu, B.; Luo, M.; Wang, Y.; Chen, F.; Wang, S.; Zhang, C.; Epstein, L. Wheat Ms2 encodes for an orphan protein that confers male sterility in grass species. Nat. Commun. 2017, 8, 15121. [Google Scholar] [CrossRef] [PubMed]
- Turland, N.J.; Wiersema, J.H.; Barrie, F.R.; Greuter, W.; Hawksworth, D.L.; Herendeen, P.S.; Knapp, S.; Kusber, W.-H.; Li, D.-Z.; Marhold, K.; et al. International Code of Nomenclature for Algae, Fungi, and Plants (Shenzhen Code) Adopted by the Nineteenth International Botanical Congress Shenzhen, China, July 2017; Regnum Vegetabile 159; Koeltz Botanical Books: Glashütten, Germany, 2018. [Google Scholar]
- Rubstov, G.A. Geographical distribution of the genus Pyrus and trends and factors in its evolution. Am. Nat. 1944, 78, 358–366. [Google Scholar]
- Bailey, L.H. The Standard Cyclopedia of Horticulture; Macmillan Press: New York, NY, USA, USA, 1917; Volume 4, pp. 2865–2878. [Google Scholar]
- Teng, Y.; Tanabe, K. Reconsideration on the origin of cultivated pears native to East Asia. Acta Hortic. 2004, 634, 175–182. [Google Scholar] [CrossRef]
- Zheng, X.; Cai, D.; Potter, D.; Postmand, J.; Liu, J.; Teng, Y. Phylogeny and evolutionary histories of Pyrus L. revealed by phylogenetic trees and networks based on data from multiple DNA sequences. Mol. Phylogenet. Evolut. 2014, 80, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Chagne, D.; Crowhurst, R.N.; Pindo, M.; Thrimawithana, A.; Deng, C.; Ireland, H.; Fiers, M.; Dzierzon, H.; Cestaro, A.; Fontana, P.; et al. The draft genome sequence of European pear (Pyrus communis L. ‘Bartlett’). PLoS ONE 2014, 9, e92644. [Google Scholar] [CrossRef]
- Wu, J.; Wang, Z.; Shi, Z.; Zhang, S.; Ming, R.; Zhu, S.; Khan, M.A.; Tao, S.; Korban, S.S.; Wang, H.; et al. The genome of the pear (Pyrus bretschneideri Rehd.). Genome Res. 2013, 23, 396–408. [Google Scholar] [CrossRef]
- Velasco, R.; Zharkikh, A.; Affourtit, J.; Dhingra, A.; Cestaro, A.; Kalyanaraman, A.; Fontana, P.; Bhatnagar, S.K.; Troggio, M.; Pruss, D.; et al. The genome of the domesticated apple (Malus x domestica Borkh.). Nat. Genet. 2010, 42, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Verde, I.; Abbott, A.G.; Scalabrin, S.; Jung, S.; Shu, S.Q.; Marroni, F.; Zhebentyayeva, T.; Dettori, M.T.; Grimwood, J.; Cattonaro, F.; et al. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat. Genet. 2013, 45, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Du, J.C.; Li, L.T.; Jin, C.; Fan, L.; Li, M.; Wu, J.; Zhang, S.L. Comparative genomic analysis reveals multiple long terminal repeats, lineage-specific amplification, and frequent interelement recombination for Cassandra retrotransposon in pear (Pyrus bretschneideri Rehd.). Genome Biol. Evol. 2014, 6, 1423–1436. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Zong, Y.; Yue, X.; Postman, J.; Teng, Y.; Cai, D. Prediction of retrotransposons and assessment of genetic variability based on developed retrotransposon-based insertion POLymorphism (RBIP) markers in Pyrus L. Mol. Genet. Genomics 2015, 290, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Ellinghaus, D.; Kurtz, S.; Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformat. 2008, 9, 18. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 2015, 6, 11. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Thompson, J.D.; Gibson, T.J.; Higgins, D.G. Multiple sequence alignment using ClustalW and ClustalX. Curr. Protoc. Bioinformat. 2002. [Google Scholar] [CrossRef]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular Evolutionary Genetics Analysis Using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Cossu, R.M.; Buti, M.; Giordani, T.; Natali, L.; Cavallini, A. A computational study of the dynamics of LTR retrotransposons in the Populus trichocarpa genome. Tree Genet. Genomes 2012, 8, 61–75. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Baucom, R.S.; Estill, J.C.; Leebens-Mack, J.; Bennetzen, J.L. Natural selection on gene function drives the evolution of LTR retrotransposon families in the rice genome. Genome Res. 2009, 19, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Sun, H.; Zhao, G.; Dai, H.; Gao, X.; Li, H.; Zhang, Z. Isolation and characterization of genomic retrotransposon sequences from octoploid strawberry (Fragaria x ananassa Duch.). Plant Cell Rep. 2008, 27, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.H.; Wen, X.P.; Ding, G.J.; Cui, B.W. Isolation, identification, and characterization of genomic LTR retrotransposon sequences from masson pine (Pinus massoniana). Tree Genet. Genomes 2013, 9, 1237–1246. [Google Scholar] [CrossRef]
- Jiang, S.; Cai, D.; Sun, Y.; Teng, Y. Isolation and characterization of putative functional long terminal repeat retrotransposons in the Pyrus genome. Mob. DNA 2016, 7, 1. [Google Scholar] [CrossRef]
- Jedrzejczyk, I.; Sliwinska, E. Leaves and seeds as materials for flow cytometric estimation of the genome size of 11 rosaceae woody species containing DNA-Staining inhibitors. J. Bot. 2010, 2010, 930895. [Google Scholar] [CrossRef]
- Dickson, E.E.; Arumuganathan, K.; Kresovich, S.; Doyle, J.J. Nuclear-DNA Content Variation within the Rosaceae. Am. J. Bot. 1992, 79, 1081–1086. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ‘Suli’ * | ‘Qiushui’ | ‘Zaoshengxinshui’ | P. pashia | P. calleryana | P. betulaefolia | |
|---|---|---|---|---|---|---|
| Reads processed (M) | 296.7 | 45.5 | 50.1 | 57.4 | 49.5 | 51.8 |
| Mapped reference genome reads (M) | 207.7 (71.4%) | 23.2 (50.9%) | 24.4 (48.9%) | 24.1 (41.9%) | 22.4 (45.3%) | 22.5 (43.5%) |
| Mapped 4,912 LTR-RTs reads (M) | 49.5 | 5.2 | 5.5 | 6.7 | 6.7 | 7.9 |
| Mapped LTR-RTs/mapped reference genome | 23.8% | 22.4% | 22.5% | 27.8% | 29.9% | 35.1% |
| ‘Suli’ | ‘Qiushui’ | ‘Zaoshengxinshui’ | P. pashia | P. calleryana | P. betulaefolia | |
|---|---|---|---|---|---|---|
| RPKM value of 14 LTR-RT subfamilies | 396.5 | 337.1 | 326.2 | 508.8 | 545.2 | 625.0 |
| RPKM value of 198 LTR-RT subfamilies | 700.6 | 589.8 | 596.2 | 800.0 | 790.0 | 863.7 |
| Percent (%) | 56.6 | 57.1 | 54.7 | 63.6 | 69.0 | 72.4 |
| Mapped Sequences in ‘Suli’ | Specific Insertion Sites in ‘Suli’ | Mapped Sequences in ‘Bartlett’ | Specific Insertion Sites in ‘Bartlett’ | |
|---|---|---|---|---|
| Gypsy-2_PX | 412 | 241 | 396 | 232 |
| Gypsy-3_PX | 234 | 97 | 86 | 58 |
| Gypsy-4_PX | 1254 | 404 | 468 | 330 |
| Copia-24_PX | 302 | 145 | 247 | 160 |
| Copia-56_MAD | 302 | 84 | 167 | 37 |
| Total | 2504 | 971 | 1364 | 817 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, S.; Wang, X.; Shi, C.; Luo, J. Genome-Wide Identification and Analysis of High-Copy-Number LTR Retrotransposons in Asian Pears. Genes 2019, 10, 156. https://doi.org/10.3390/genes10020156
Jiang S, Wang X, Shi C, Luo J. Genome-Wide Identification and Analysis of High-Copy-Number LTR Retrotransposons in Asian Pears. Genes. 2019; 10(2):156. https://doi.org/10.3390/genes10020156
Chicago/Turabian StyleJiang, Shuang, Xiaoqing Wang, Chunhui Shi, and Jun Luo. 2019. "Genome-Wide Identification and Analysis of High-Copy-Number LTR Retrotransposons in Asian Pears" Genes 10, no. 2: 156. https://doi.org/10.3390/genes10020156
APA StyleJiang, S., Wang, X., Shi, C., & Luo, J. (2019). Genome-Wide Identification and Analysis of High-Copy-Number LTR Retrotransposons in Asian Pears. Genes, 10(2), 156. https://doi.org/10.3390/genes10020156