KDM4B: A Nail for Every Hammer?


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
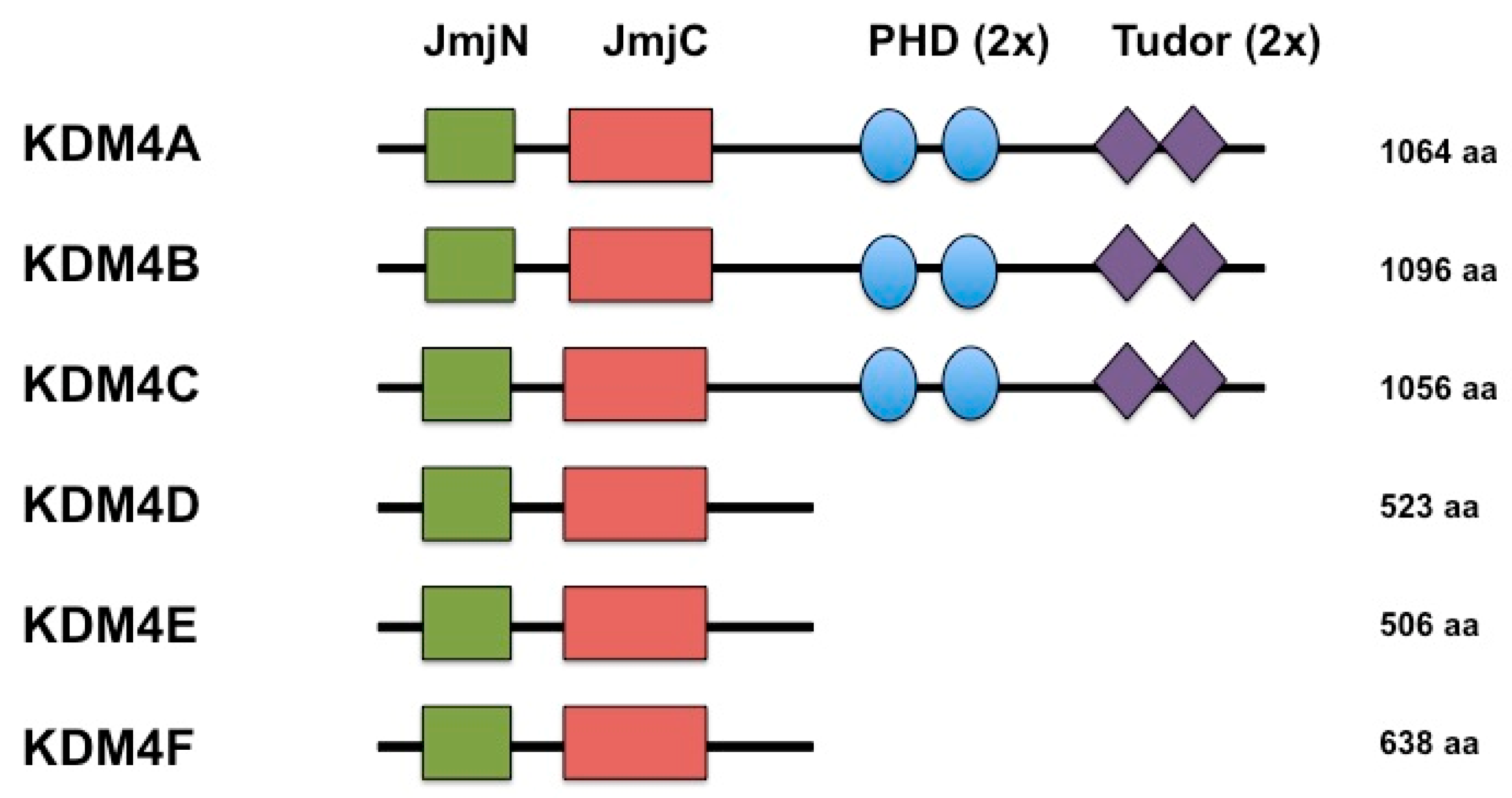
2. KDMB is a Histone Demethylase that Reverses Repressive Histone Modifications
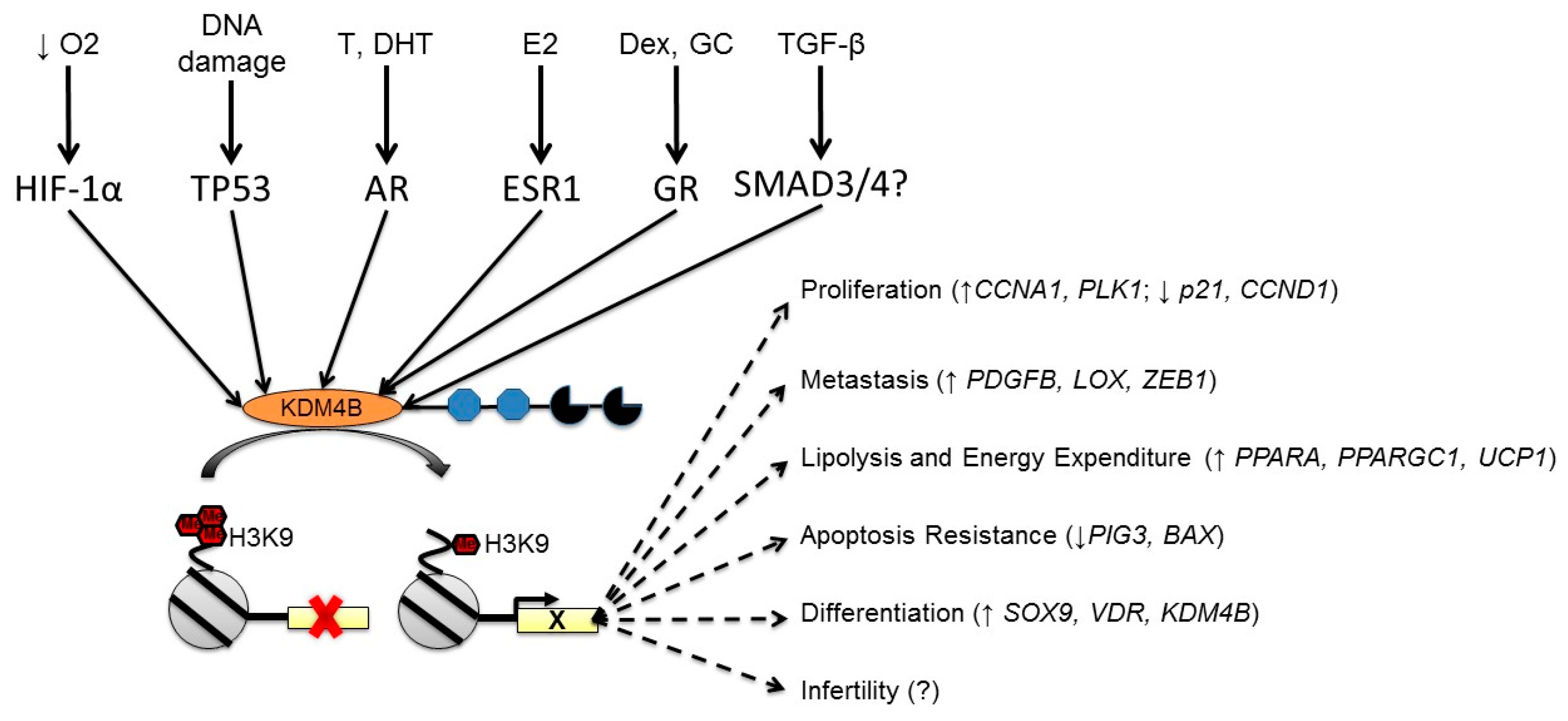
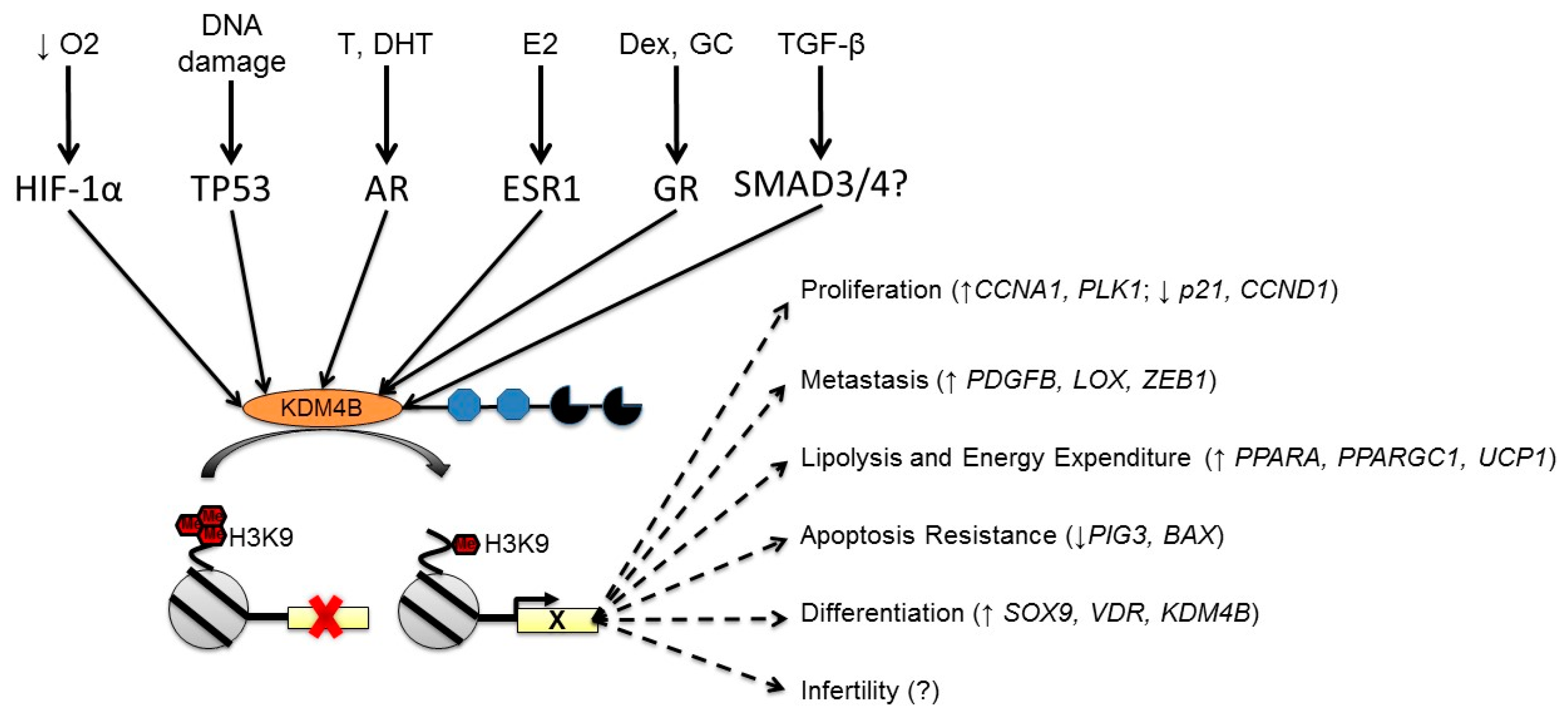
3. KDM4B is Regulated by Multiple Cellular Stimuli
3.1. Regulation of KDM4B by Hypoxia
3.2. Regulation of KDM4B by Nuclear Hormone Receptors
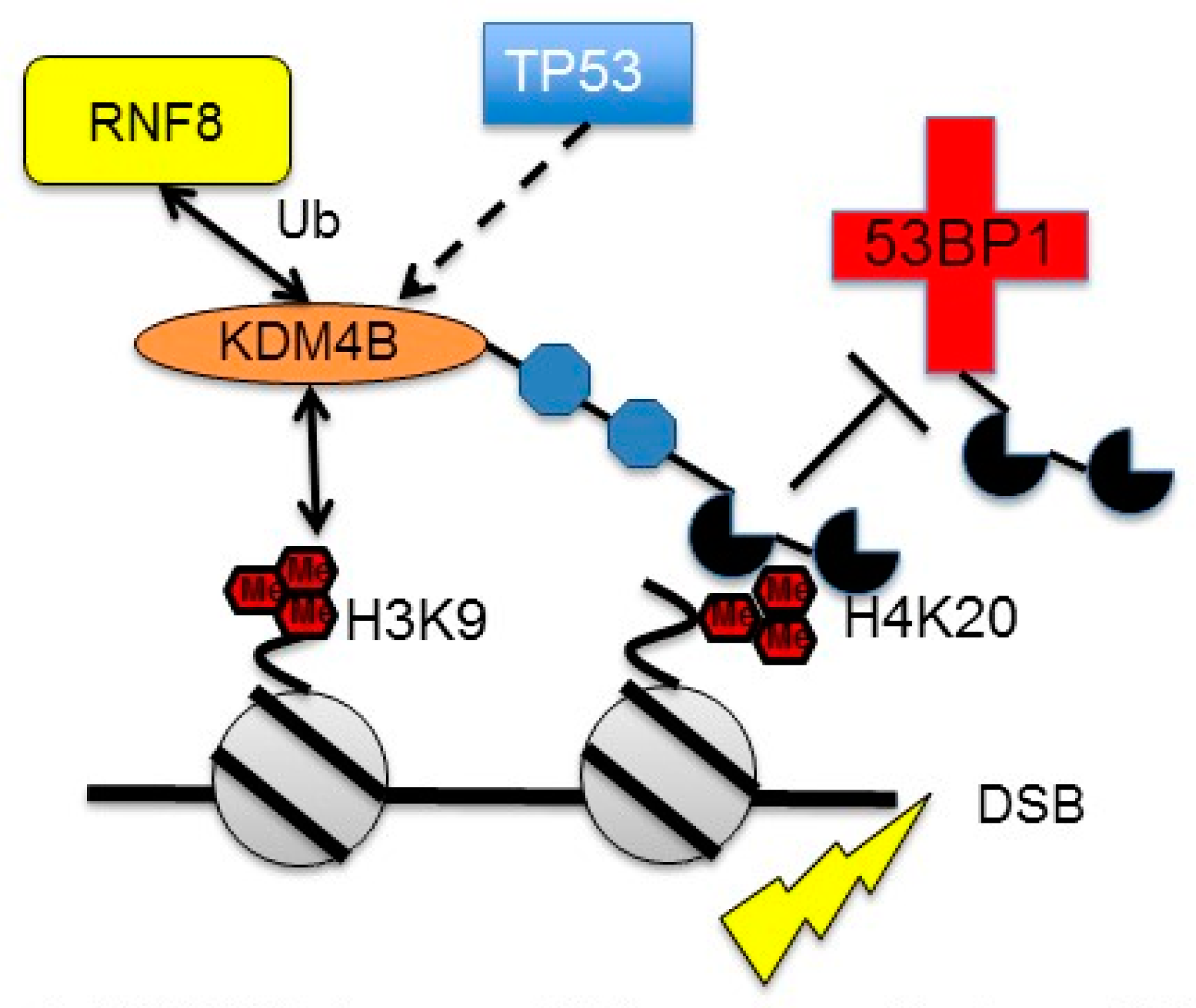
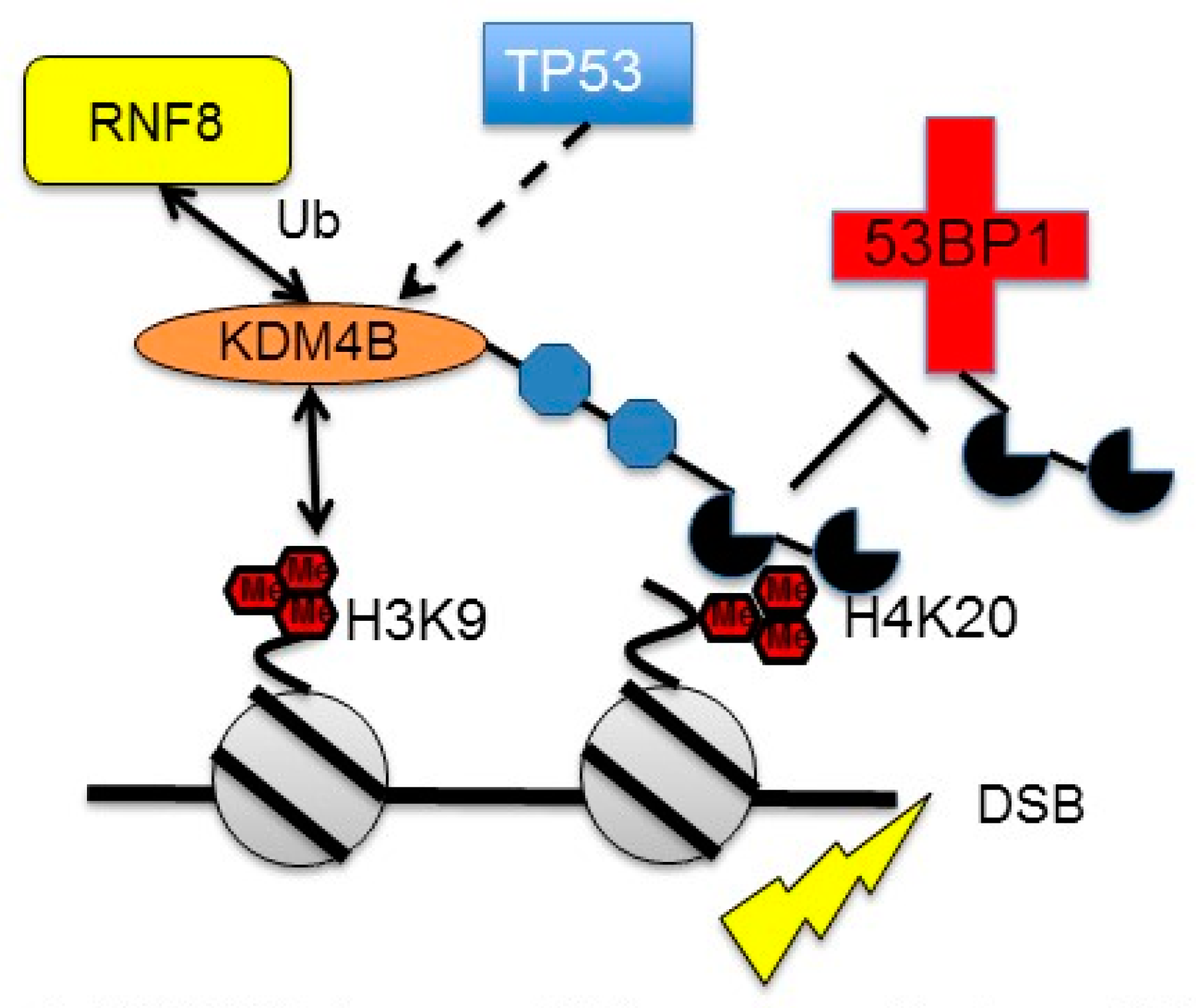
3.3. Regulation of KDM4B by DNA Damage
4. Integrating KDM4B Expression Phenomena with Biological Processes
4.1. KDM4B and Stem Cell Biology
4.2. KDM4B and Mesenchymal Tissues
4.3. KDM4B in the Central Nervous System
4.4. KDM4B in Ear Development
4.5. KDM4B in Reproductive Tissues
4.6. KDM4B in Non-Mammalian Systems
5. KDM4B in Cancer
5.1. KDM4B in Breast Cancer
5.2. KDM4B in Prostate Cancer
5.3. KDM4B in Colorectal Cancer
5.4. KDM4B in Gastric Cancer
5.5. KDM4B in Osteosarcoma
5.6. KDM4B in Hematological Tumors
5.7. KDM4B in Gynecological Cancers
5.8. KDM4B in Other Cancers
6. KDM4B as a Therapeutic Target (i.e., a Nail for Every Hammer)?
Funding
Acknowledgments
Conflicts of Interest
References
- Black, J.C.; Van Rechem, C.; Whetstine, J.R. Histone lysine methylation dynamics: Establishment, regulation, and biological impact. Mol. Cell 2012, 48, 491–507. [Google Scholar] [CrossRef] [PubMed]
- Trewick, S.C.; McLaughlin, P.J.; Allshire, R.C. Methylation: Lost in hydroxylation? EMBO Rep. 2005, 6, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, Y.; Fang, J.; Erdjument-Bromage, H.; Warren, M.E.; Borchers, C.H.; Tempst, P.; Zhang, Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature 2006, 439, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Whetstine, J.R.; Nottke, A.; Lan, F.; Huarte, M.; Smolikov, S.; Chen, Z.; Spooner, E.; Li, E.; Zhang, G.; Colaiacovo, M.; et al. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell 2006, 125, 467–481. [Google Scholar] [CrossRef]
- Cloos, P.A.; Christensen, J.; Agger, K.; Maiolica, A.; Rappsilber, J.; Antal, T.; Hansen, K.H.; Helin, K. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature 2006, 442, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, T.; Yamazaki, Y.; Katoh-Fukui, Y.; Tsuchiya, R.; Kondo, S.; Motoyama, J.; Higashinakagawa, T. Gene trap capture of a novel mouse gene, jumonji, required for neural tube formation. Genes Dev. 1995, 9, 1211–1222. [Google Scholar] [CrossRef]
- Klose, R.J.; Kallin, E.M.; Zhang, Y. JmjC-domain-containing proteins and histone demethylation. Nat. Rev. Genet. 2006, 7, 715–727. [Google Scholar] [CrossRef]
- Pedersen, M.T.; Helin, K. Histone demethylases in development and disease. Trends Cell Biol. 2010, 20, 662–671. [Google Scholar] [CrossRef]
- Hancock, R.L.; Dunne, K.; Walport, L.J.; Flashman, E.; Kawamura, A. Epigenetic regulation by histone demethylases in hypoxia. Epigenomics 2015, 7, 791–811. [Google Scholar] [CrossRef]
- Huang, F.; Chandrasekharan, M.B.; Chen, Y.C.; Bhaskara, S.; Hiebert, S.W.; Sun, Z.W. The JmjN domain of Jhd2 is important for its protein stability, and the plant homeodomain (PHD) finger mediates its chromatin association independent of H3K4 methylation. J. Biol. Chem. 2010, 285, 24548–24561. [Google Scholar] [CrossRef] [PubMed]
- Quan, Z.; Oliver, S.G.; Zhang, N. JmjN interacts with JmjC to ensure selective proteolysis of Gis1 by the proteasome. Microbiology 2011, 157, 2694–2701. [Google Scholar] [CrossRef] [PubMed]
- Pasini, D.; Cloos, P.A.; Walfridsson, J.; Olsson, L.; Bukowski, J.P.; Johansen, J.V.; Bak, M.; Tommerup, N.; Rappsilber, J.; Helin, K. JARID2 regulates binding of the Polycomb repressive complex 2 to target genes in ES cells. Nature 2010, 464, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.C.; Valouev, A.; Swigut, T.; Zhang, J.; Zhao, Y.; Sidow, A.; Wysocka, J. Jarid2/Jumonji coordinates control of PRC2 enzymatic activity and target gene occupancy in pluripotent cells. Cell 2009, 139, 1290–1302. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Kim, W.; Fujiwara, Y.; Simon, M.D.; Liu, Y.; Mysliwiec, M.R.; Yuan, G.C.; Lee, Y.; Orkin, S.H. Jumonji modulates polycomb activity and self-renewal versus differentiation of stem cells. Cell 2009, 139, 1303–1314. [Google Scholar] [CrossRef] [PubMed]
- Berry, W.L.; Janknecht, R. KDM4/JMJD2 histone demethylases: Epigenetic regulators in cancer cells. Cancer Res. 2013, 73, 2936–2942. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M.; Katoh, M. Identification and characterization of JMJD2 family genes in silico. Int. J. Oncol. 2004, 24, 1623–1628. [Google Scholar] [PubMed]
- Levin, M.; Stark, M.; Assaraf, Y.G. The JmjN domain as a dimerization interface and a targeted inhibitor of KDM4 demethylase activity. Oncotarget 2018, 9, 16861–16882. [Google Scholar] [CrossRef]
- Hillringhaus, L.; Yue, W.W.; Rose, N.R.; Ng, S.S.; Gileadi, C.; Loenarz, C.; Bello, S.H.; Bray, J.E.; Schofield, C.J.; Oppermann, U. Structural and evolutionary basis for the dual substrate selectivity of human KDM4 histone demethylase family. J. Biol. Chem. 2011, 286, 41616–41625. [Google Scholar] [CrossRef]
- Fodor, B.D.; Kubicek, S.; Yonezawa, M.; O’Sullivan, R.J.; Sengupta, R.; Perez-Burgos, L.; Opravil, S.; Mechtler, K.; Schotta, G.; Jenuwein, T. Jmjd2b antagonizes H3K9 trimethylation at pericentric heterochromatin in mammalian cells. Genes Dev. 2006, 20, 1557–1562. [Google Scholar] [CrossRef]
- Rotili, D.; Mai, A. Targeting Histone Demethylases: A New Avenue for the Fight against Cancer. Genes Cancer 2011, 2, 663–679. [Google Scholar] [CrossRef]
- Rose, N.R.; McDonough, M.A.; King, O.N.; Kawamura, A.; Schofield, C.J. Inhibition of 2-oxoglutarate dependent oxygenases. Chem. Soc. Rev. 2011, 40, 4364–4397. [Google Scholar] [CrossRef]
- Pedersen, M.T.; Kooistra, S.M.; Radzisheuskaya, A.; Laugesen, A.; Johansen, J.V.; Hayward, D.G.; Nilsson, J.; Agger, K.; Helin, K. Continual removal of H3K9 promoter methylation by Jmjd2 demethylases is vital for ESC self-renewal and early development. EMBO J. 2016, 35, 1550–1564. [Google Scholar] [CrossRef]
- Roadmap Epigenomics, C.; Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar]
- Mozzetta, C.; Boyarchuk, E.; Pontis, J.; Ait-Si-Ali, S. Sound of silence: The properties and functions of repressive Lys methyltransferases. Nat. Rev. Mol. Cell Biol. 2015, 16, 499–513. [Google Scholar] [CrossRef]
- Mikkelsen, T.S.; Ku, M.; Jaffe, D.B.; Issac, B.; Lieberman, E.; Giannoukos, G.; Alvarez, P.; Brockman, W.; Kim, T.K.; Koche, R.P.; et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 2007, 448, 553–560. [Google Scholar] [CrossRef]
- Wang, Z.; Zang, C.; Rosenfeld, J.A.; Schones, D.E.; Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Peng, W.; Zhang, M.Q.; et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008, 40, 897–903. [Google Scholar] [CrossRef]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef]
- Vakoc, C.R.; Sachdeva, M.M.; Wang, H.; Blobel, G.A. Profile of histone lysine methylation across transcribed mammalian chromatin. Mol. Cell. Biol. 2006, 26, 9185–9195. [Google Scholar] [CrossRef]
- Vakoc, C.R.; Mandat, S.A.; Olenchock, B.A.; Blobel, G.A. Histone H3 lysine 9 methylation and HP1γ are associated with transcription elongation through mammalian chromatin. Mol. Cell 2005, 19, 381–391. [Google Scholar] [CrossRef]
- Yokochi, T.; Poduch, K.; Ryba, T.; Lu, J.; Hiratani, I.; Tachibana, M.; Shinkai, Y.; Gilbert, D.M. G9a selectively represses a class of late-replicating genes at the nuclear periphery. Proc. Natl. Acad. Sci. USA 2009, 106, 19363–19368. [Google Scholar] [CrossRef]
- Kind, J.; Pagie, L.; Ortabozkoyun, H.; Boyle, S.; de Vries, S.S.; Janssen, H.; Amendola, M.; Nolen, L.D.; Bickmore, W.A.; van Steensel, B. Single-cell dynamics of genome-nuclear lamina interactions. Cell 2013, 153, 178–192. [Google Scholar] [CrossRef]
- Wu, R.; Terry, A.V.; Singh, P.B.; Gilbert, D.M. Differential subnuclear localization and replication timing of histone H3 lysine 9 methylation states. Mol. Biol. Cell 2005, 16, 2872–2881. [Google Scholar] [CrossRef]
- Das, P.P.; Shao, Z.; Beyaz, S.; Apostolou, E.; Pinello, L.; De Los Angeles, A.; O’Brien, K.; Atsma, J.M.; Fujiwara, Y.; Nguyen, M.; et al. Distinct and combinatorial functions of Jmjd2b/Kdm4b and Jmjd2c/Kdm4c in mouse embryonic stem cell identity. Mol. Cell 2014, 53, 32–48. [Google Scholar] [CrossRef]
- Wilson, C.; Qiu, L.; Hong, Y.; Karnik, T.; Tadros, G.; Mau, B.; Ma, T.; Mu, Y.; New, J.; Louie, R.J.; et al. The histone demethylase KDM4B regulates peritoneal seeding of ovarian cancer. Oncogene 2017, 36, 2565–2576. [Google Scholar] [CrossRef]
- Yang, J.; Jubb, A.M.; Pike, L.; Buffa, F.M.; Turley, H.; Baban, D.; Leek, R.; Gatter, K.C.; Ragoussis, J.; Harris, A.L. The histone demethylase JMJD2B is regulated by estrogen receptor α and hypoxia, and is a key mediator of estrogen induced growth. Cancer Res. 2010, 70, 6456–6466. [Google Scholar] [CrossRef]
- Loh, Y.H.; Zhang, W.; Chen, X.; George, J.; Ng, H.H. Jmjd1a and Jmjd2c histone H3 Lys 9 demethylases regulate self-renewal in embryonic stem cells. Genes Dev. 2007, 21, 2545–2557. [Google Scholar] [CrossRef]
- Castellini, L.; Moon, E.J.; Razorenova, O.V.; Krieg, A.J.; von Eyben, R.; Giaccia, A.J. KDM4B/JMJD2B is a p53 target gene that modulates the amplitude of p53 response after DNA damage. Nucleic Acids Res. 2017, 45, 3674–3692. [Google Scholar] [CrossRef]
- Ponnaluri, V.K.; Vavilala, D.T.; Putty, S.; Gutheil, W.G.; Mukherji, M. Identification of non-histone substrates for JMJD2A-C histone demethylases. Biochem. Biophys. Res. Commun. 2009, 390, 280–284. [Google Scholar] [CrossRef]
- Yang, J.; Harris, A.L.; Davidoff, A.M. Hypoxia and Hormone-Mediated Pathways Converge at the Histone Demethylase KDM4B in Cancer. Int. J. Mol. Sci. 2018, 19, 240. [Google Scholar] [CrossRef]
- Lee, J.; Thompson, J.R.; Botuyan, M.V.; Mer, G. Distinct binding modes specify the recognition of methylated histones H3K4 and H4K20 by JMJD2A-tudor. Nat. Struct. Mol. Biol. 2008, 15, 109–111. [Google Scholar] [CrossRef]
- Su, Z.; Wang, F.; Lee, J.H.; Stephens, K.E.; Papazyan, R.; Voronina, E.; Krautkramer, K.A.; Raman, A.; Thorpe, J.J.; Boersma, M.D.; et al. Reader domain specificity and lysine demethylase-4 family function. Nat. Commun. 2016, 7, 13387. [Google Scholar] [CrossRef]
- Mallette, F.A.; Mattiroli, F.; Cui, G.; Young, L.C.; Hendzel, M.J.; Mer, G.; Sixma, T.K.; Richard, S. RNF8- and RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1 recruitment to DNA damage sites. EMBO J. 2012, 31, 1865–1878. [Google Scholar] [CrossRef]
- Beyer, S.; Kristensen, M.M.; Jensen, K.S.; Johansen, J.V.; Staller, P. The histone demethylases JMJD1A and JMJD2B are transcriptional targets of hypoxia-inducible factor HIF. J. Biol. Chem. 2008, 283, 36542–36552. [Google Scholar] [CrossRef]
- Pollard, P.J.; Loenarz, C.; Mole, D.R.; McDonough, M.A.; Gleadle, J.M.; Schofield, C.J.; Ratcliffe, P.J. Regulation of Jumonji-domain-containing histone demethylases by hypoxia-inducible factor (HIF)-1α. Biochem. J. 2008, 416, 387–394. [Google Scholar] [CrossRef]
- Xia, X.; Lemieux, M.E.; Li, W.; Carroll, J.S.; Brown, M.; Liu, X.S.; Kung, A.L. Integrative analysis of HIF binding and transactivation reveals its role in maintaining histone methylation homeostasis. Proc. Natl. Acad. Sci. USA 2009, 106, 4260–4265. [Google Scholar] [CrossRef]
- Ramachandran, S.; Ient, J.; Gottgens, E.L.; Krieg, A.J.; Hammond, E.M. Epigenetic Therapy for Solid Tumors: Highlighting the Impact of Tumor Hypoxia. Genes 2015, 6, 935–956. [Google Scholar] [CrossRef]
- Hammond, E.M.; Giaccia, A.J. Hypoxia-inducible factor-1 and p53: Friends, acquaintances, or strangers? Clin. Cancer Res. 2006, 12, 5007–5009. [Google Scholar] [CrossRef]
- Thomlinson, R.H.; Gray, L.H. The histological structure of some human lung cancers and the possible implications for radiotherapy. Br. J. Cancer 1955, 9, 539–549. [Google Scholar] [CrossRef]
- Trotter, M.J.; Chaplin, D.J.; Durand, R.E.; Olive, P.L. The use of fluorescent probes to identify regions of transient perfusion in murine tumors. Int. J. Radiat. Oncol. Biol. Phys. 1989, 16, 931–934. [Google Scholar] [CrossRef]
- Brown, J.M.; Giaccia, A.J. The unique physiology of solid tumors: Opportunities (and problems) for cancer therapy. Cancer Res. 1998, 58, 1408–1416. [Google Scholar]
- Chan, D.A.; Giaccia, A.J. Hypoxia, gene expression, and metastasis. Cancer Metastasis Rev. 2007, 26, 333–339. [Google Scholar] [CrossRef]
- Rankin, E.B.; Giaccia, A.J. Hypoxic control of metastasis. Science 2016, 352, 175–180. [Google Scholar] [CrossRef]
- Mole, D.R.; Blancher, C.; Copley, R.R.; Pollard, P.J.; Gleadle, J.M.; Ragoussis, J.; Ratcliffe, P.J. Genome-wide association of hypoxia-inducible factor (HIF)-1α and HIF-2α DNA binding with expression profiling of hypoxia-inducible transcripts. J. Biol. Chem. 2009, 284, 16767–16775. [Google Scholar] [CrossRef]
- Raval, R.R.; Lau, K.W.; Tran, M.G.; Sowter, H.M.; Mandriota, S.J.; Li, J.L.; Pugh, C.W.; Maxwell, P.H.; Harris, A.L.; Ratcliffe, P.J. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol. Cell. Biol. 2005, 25, 5675–5686. [Google Scholar] [CrossRef]
- Manalo, D.J.; Rowan, A.; Lavoie, T.; Natarajan, L.; Kelly, B.D.; Ye, S.Q.; Garcia, J.G.; Semenza, G.L. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 2005, 105, 659–669. [Google Scholar] [CrossRef]
- Epstein, A.C.; Gleadle, J.M.; McNeill, L.A.; Hewitson, K.S.; O’Rourke, J.; Mole, D.R.; Mukherji, M.; Metzen, E.; Wilson, M.I.; Dhanda, A.; et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 2001, 107, 43–54. [Google Scholar] [CrossRef]
- Ivan, M.; Kaelin, W.G., Jr. The EGLN-HIF O2-Sensing System: Multiple Inputs and Feedbacks. Mol. Cell 2017, 66, 772–779. [Google Scholar] [CrossRef]
- Ivan, M.; Haberberger, T.; Gervasi, D.C.; Michelson, K.S.; Gunzler, V.; Kondo, K.; Yang, H.; Sorokina, I.; Conaway, R.C.; Conaway, J.W.; et al. Biochemical purification and pharmacological inhibition of a mammalian prolyl hydroxylase acting on hypoxia-inducible factor. Proc. Natl. Acad. Sci. USA 2002, 99, 13459–13464. [Google Scholar] [CrossRef]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef]
- Masson, N.; Willam, C.; Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J. Independent function of two destruction domains in hypoxia-inducible factor-α chains activated by prolyl hydroxylation. EMBO J. 2001, 20, 5197–5206. [Google Scholar] [CrossRef]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFα targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef]
- Min, J.H.; Yang, H.; Ivan, M.; Gertler, F.; Kaelin, W.G., Jr.; Pavletich, N.P. Structure of an HIF-1α -pVHL complex: Hydroxyproline recognition in signaling. Science 2002, 296, 1886–1889. [Google Scholar] [CrossRef]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef]
- Wang, G.L.; Semenza, G.L. Purification and characterization of hypoxia-inducible factor 1. J. Biol. Chem. 1995, 270, 1230–1237. [Google Scholar] [CrossRef]
- Krieg, A.J.; Rankin, E.B.; Chan, D.; Razorenova, O.; Fernandez, S.; Giaccia, A.J. Regulation of the histone demethylase JMJD1A by hypoxia-inducible factor 1 α enhances hypoxic gene expression and tumor growth. Mol. Cell. Biol. 2010, 30, 344–353. [Google Scholar] [CrossRef]
- Hancock, R.L.; Masson, N.; Dunne, K.; Flashman, E.; Kawamura, A. The Activity of JmjC Histone Lysine Demethylase KDM4A is Highly Sensitive to Oxygen Concentrations. ACS Chem. Biol. 2017, 12, 1011–1019. [Google Scholar] [CrossRef]
- Tarhonskaya, H.; Hardy, A.P.; Howe, E.A.; Loik, N.D.; Kramer, H.B.; McCullagh, J.S.; Schofield, C.J.; Flashman, E. Kinetic Investigations of the Role of Factor Inhibiting Hypoxia-inducible Factor (FIH) as an Oxygen Sensor. J. Biol. Chem. 2015, 290, 19726–19742. [Google Scholar] [CrossRef]
- Koivunen, P.; Hirsila, M.; Gunzler, V.; Kivirikko, K.I.; Myllyharju, J. Catalytic properties of the asparaginyl hydroxylase (FIH) in the oxygen sensing pathway are distinct from those of its prolyl 4-hydroxylases. J. Biol. Chem. 2004, 279, 9899–9904. [Google Scholar] [CrossRef]
- Kang, C.; Saso, K.; Ota, K.; Kawazu, M.; Ueda, T.; Okada, H. JMJD2B/KDM4B inactivation in adipose tissues accelerates obesity and systemic metabolic abnormalities. Genes Cells 2018, 23, 767–777. [Google Scholar] [CrossRef]
- Cheng, Y.; Yuan, Q.; Vergnes, L.; Rong, X.; Youn, J.Y.; Li, J.; Yu, Y.; Liu, W.; Cai, H.; Lin, J.D.; et al. KDM4B protects against obesity and metabolic dysfunction. Proc. Natl. Acad. Sci. USA 2018, 115, E5566–E5575. [Google Scholar] [CrossRef]
- Shi, L.; Sun, L.; Li, Q.; Liang, J.; Yu, W.; Yi, X.; Yang, X.; Li, Y.; Han, X.; Zhang, Y.; et al. Histone demethylase JMJD2B coordinates H3K4/H3K9 methylation and promotes hormonally responsive breast carcinogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 7541–7546. [Google Scholar] [CrossRef]
- Kawazu, M.; Saso, K.; Tong, K.I.; McQuire, T.; Goto, K.; Son, D.O.; Wakeham, A.; Miyagishi, M.; Mak, T.W.; Okada, H. Histone demethylase JMJD2B functions as a co-factor of estrogen receptor in breast cancer proliferation and mammary gland development. PLoS ONE 2011, 6, e17830. [Google Scholar] [CrossRef]
- Gaughan, L.; Stockley, J.; Coffey, K.; O’Neill, D.; Jones, D.L.; Wade, M.; Wright, J.; Moore, M.; Tse, S.; Rogerson, L.; et al. KDM4B is a master regulator of the estrogen receptor signalling cascade. Nucleic Acids Res. 2013, 41, 6892–6904. [Google Scholar] [CrossRef]
- West, D.C.; Pan, D.; Tonsing-Carter, E.Y.; Hernandez, K.M.; Pierce, C.F.; Styke, S.C.; Bowie, K.R.; Garcia, T.I.; Kocherginsky, M.; Conzen, S.D. GR and ER Coactivation Alters the Expression of Differentiation Genes and Associates with Improved ER+ Breast Cancer Outcome. Mol. Cancer Res. 2016, 14, 707–719. [Google Scholar] [CrossRef]
- Coffey, K.; Rogerson, L.; Ryan-Munden, C.; Alkharaif, D.; Stockley, J.; Heer, R.; Sahadevan, K.; O’Neill, D.; Jones, D.; Darby, S.; et al. The lysine demethylase, KDM4B, is a key molecule in androgen receptor signalling and turnover. Nucleic Acids Res. 2013, 41, 4433–4446. [Google Scholar] [CrossRef]
- Chu, C.H.; Wang, L.Y.; Hsu, K.C.; Chen, C.C.; Cheng, H.H.; Wang, S.M.; Wu, C.M.; Chen, T.J.; Li, L.T.; Liu, R.; et al. KDM4B as a target for prostate cancer: Structural analysis and selective inhibition by a novel inhibitor. J. Med. Chem. 2014, 57, 5975–5985. [Google Scholar] [CrossRef]
- Heinlein, C.A.; Chang, C. Androgen receptor in prostate cancer. Endocr. Rev. 2004, 25, 276–308. [Google Scholar] [CrossRef]
- Li, X.; Dong, S. Histone demethylase JMJD2B and JMJD2C induce fibroblast growth factor 2: Mediated tumorigenesis of osteosarcoma. Med. Oncol. 2015, 32, 53. [Google Scholar] [CrossRef]
- Zheng, H.; Chen, L.; Pledger, W.J.; Fang, J.; Chen, J. p53 promotes repair of heterochromatin DNA by regulating JMJD2b and SUV39H1 expression. Oncogene 2014, 33, 734–744. [Google Scholar] [CrossRef]
- Kim, J.G.; Yi, J.M.; Park, S.J.; Kim, J.S.; Son, T.G.; Yang, K.; Yoo, M.A.; Heo, K. Histone demethylase JMJD2B-mediated cell proliferation regulated by hypoxia and radiation in gastric cancer cell. Biochim. Biophys. Acta 2012, 1819, 1200–1207. [Google Scholar] [CrossRef]
- Zimmermann, M.; de Lange, T. 53BP1: Pro choice in DNA repair. Trends Cell Biol. 2014, 24, 108–117. [Google Scholar] [CrossRef]
- Mishra, S.; Van Rechem, C.; Pal, S.; Clarke, T.L.; Chakraborty, D.; Mahan, S.D.; Black, J.C.; Murphy, S.E.; Lawrence, M.S.; Daniels, D.L.; et al. Cross-talk between Lysine-Modifying Enzymes Controls Site-Specific DNA Amplifications. Cell 2018, 175, 1716. [Google Scholar] [CrossRef]
- Black, J.C.; Atabakhsh, E.; Kim, J.; Biette, K.M.; Van Rechem, C.; Ladd, B.; Burrowes, P.D.; Donado, C.; Mattoo, H.; Kleinstiver, B.P.; et al. Hypoxia drives transient site-specific copy gain and drug-resistant gene expression. Genes Dev. 2015, 29, 1018–1031. [Google Scholar] [CrossRef]
- Black, J.C.; Manning, A.L.; Van Rechem, C.; Kim, J.; Ladd, B.; Cho, J.; Pineda, C.M.; Murphy, N.; Daniels, D.L.; Montagna, C.; et al. KDM4A lysine demethylase induces site-specific copy gain and rereplication of regions amplified in tumors. Cell 2013, 154, 541–555. [Google Scholar] [CrossRef]
- Xiang, Y.; Yan, K.; Zheng, Q.; Ke, H.; Cheng, J.; Xiong, W.; Shi, X.; Wei, L.; Zhao, M.; Yang, F.; et al. Histone demethylase KDM4B promotes DNA damage by activating long interspersed nuclear element-1. Cancer Res. 2018. [Google Scholar] [CrossRef]
- Deng, W.W.; Hu, Q.; Liu, Z.R.; Chen, Q.H.; Wang, W.X.; Zhang, H.G.; Zhang, Q.; Huang, Y.L.; Zhang, X.K. KDM4B promotes DNA damage response via STAT3 signaling and is a target of CREB in colorectal cancer cells. Mol. Cell. Biochem. 2018, 449, 81–90. [Google Scholar] [CrossRef]
- Lee, H.L.; Yu, B.; Deng, P.; Wang, C.Y.; Hong, C. Transforming Growth Factor-beta-Induced KDM4B Promotes Chondrogenic Differentiation of Human Mesenchymal Stem Cells. Stem Cells 2016, 34, 711–719. [Google Scholar] [CrossRef]
- Dambacher, S.; Hahn, M.; Schotta, G. Epigenetic regulation of development by histone lysine methylation. Heredity 2010, 105, 24–37. [Google Scholar] [CrossRef]
- Katoh, Y.; Katoh, M. Comparative integromics on JMJD2A, JMJD2B and JMJD2C: Preferential expression of JMJD2C in undifferentiated ES cells. Int. J. Mol. Med. 2007, 20, 269–273. [Google Scholar] [CrossRef]
- Volarevic, V.; Bojic, S.; Nurkovic, J.; Volarevic, A.; Ljujic, B.; Arsenijevic, N.; Lako, M.; Stojkovic, M. Stem cells as new agents for the treatment of infertility: Current and future perspectives and challenges. Biomed. Res. Int. 2014, 2014, 507234. [Google Scholar] [CrossRef]
- Antony, J.; Oback, F.; Chamley, L.W.; Oback, B.; Laible, G. Transient JMJD2B-mediated reduction of H3K9me3 levels improves reprogramming of embryonic stem cells into cloned embryos. Mol. Cell. Biol. 2013, 33, 974–983. [Google Scholar] [CrossRef]
- Liu, Z.; Cai, Y.; Wang, Y.; Nie, Y.; Zhang, C.; Xu, Y.; Zhang, X.; Lu, Y.; Wang, Z.; Poo, M.; et al. Cloning of Macaque Monkeys by Somatic Cell Nuclear Transfer. Cell 2018, 174, 245. [Google Scholar] [CrossRef]
- Iwamori, N.; Zhao, M.; Meistrich, M.L.; Matzuk, M.M. The testis-enriched histone demethylase, KDM4D, regulates methylation of histone H3 lysine 9 during spermatogenesis in the mouse but is dispensable for fertility. Biol. Reprod. 2011, 84, 1225–1234. [Google Scholar] [CrossRef]
- Ye, L.; Fan, Z.; Yu, B.; Chang, J.; Al Hezaimi, K.; Zhou, X.; Park, N.H.; Wang, C.Y. Histone demethylases KDM4B and KDM6B promotes osteogenic differentiation of human MSCs. Cell Stem Cell 2012, 11, 50–61. [Google Scholar] [CrossRef]
- Qu, B.; Liu, O.; Fang, X.; Zhang, H.; Wang, Y.; Quan, H.; Zhang, J.; Zhou, J.; Zuo, J.; Tang, J.; et al. Distal-less homeobox 2 promotes the osteogenic differentiation potential of stem cells from apical papilla. Cell Tissue Res. 2014, 357, 133–143. [Google Scholar] [CrossRef]
- Yang, H.; Fan, J.; Cao, Y.; Gao, R.; Fan, Z. Distal-less homeobox 5 promotes the osteo-/dentinogenic differentiation potential of stem cells from apical papilla by activating histone demethylase KDM4B through a positive feedback mechanism. Exp. Cell. Res. 2019, 374, 221–230. [Google Scholar] [CrossRef]
- Guo, L.; Li, X.; Huang, J.X.; Huang, H.Y.; Zhang, Y.Y.; Qian, S.W.; Zhu, H.; Zhang, Y.D.; Liu, Y.; Liu, Y.; et al. Histone demethylase Kdm4b functions as a co-factor of C/EBPbeta to promote mitotic clonal expansion during differentiation of 3T3-L1 preadipocytes. Cell Death Differ. 2012, 19, 1917–1927. [Google Scholar] [CrossRef]
- Tang, Q.Q.; Otto, T.C.; Lane, M.D. Mitotic clonal expansion: A synchronous process required for adipogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 44–49. [Google Scholar] [CrossRef]
- Fujiwara, K.; Fujita, Y.; Kasai, A.; Onaka, Y.; Hashimoto, H.; Okada, H.; Yamashita, T. Deletion of JMJD2B in neurons leads to defective spine maturation, hyperactive behavior and memory deficits in mouse. Transl. Psychiatry 2016, 6, e766. [Google Scholar] [CrossRef]
- Uribe, R.A.; Buzzi, A.L.; Bronner, M.E.; Strobl-Mazzulla, P.H. Histone demethylase KDM4B regulates otic vesicle invagination via epigenetic control of Dlx3 expression. J. Cell Biol. 2015, 211, 815–827. [Google Scholar] [CrossRef]
- Yoshioka, H.; McCarrey, J.R.; Yamazaki, Y. Dynamic nuclear organization of constitutive heterochromatin during fetal male germ cell development in mice. Biol. Reprod. 2009, 80, 804–812. [Google Scholar] [CrossRef]
- Krieg, A.J.; Mullinax, S.R.; Grimstad, F.; Marquis, K.; Constance, E.; Hong, Y.; Krieg, S.A.; Roby, K.F. Histone demethylase KDM4A and KDM4B expression in granulosa cells from women undergoing in vitro fertilization. J. Assist. Reprod. Genet. 2018, 35, 993–1003. [Google Scholar] [CrossRef]
- Speroff, L.; Fritz, M.A. Clinical Gynaecologic Endocrinology and Infertility, 7th ed.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2005. [Google Scholar]
- Krieg, S.A.; Fan, X.; Hong, Y.; Sang, Q.X.; Giaccia, A.; Westphal, L.M.; Lathi, R.B.; Krieg, A.J.; Nayak, N.R. Global alteration in gene expression profiles of deciduas from women with idiopathic recurrent pregnancy loss. Mol. Hum. Reprod. 2012, 18, 442–450. [Google Scholar] [CrossRef]
- Qiu, M.T.; Fan, Q.; Zhu, Z.; Kwan, S.Y.; Chen, L.; Chen, J.H.; Ying, Z.L.; Zhou, Y.; Gu, W.; Wang, L.H.; et al. KDM4B and KDM4A promote endometrial cancer progression by regulating androgen receptor, c-myc, and p27kip1. Oncotarget 2015, 6, 31702–31720. [Google Scholar] [CrossRef]
- Tsurumi, A.; Dutta, P.; Shang, R.; Yan, S.J.; Li, W.X. Drosophila Kdm4 demethylases in histone H3 lysine 9 demethylation and ecdysteroid signaling. Sci. Rep. 2013, 3, 2894. [Google Scholar] [CrossRef]
- Palomera-Sanchez, Z.; Bucio-Mendez, A.; Valadez-Graham, V.; Reynaud, E.; Zurita, M. Drosophila p53 is required to increase the levels of the dKDM4B demethylase after UV-induced DNA damage to demethylate histone H3 lysine 9. J. Biol. Chem. 2010, 285, 31370–31379. [Google Scholar] [CrossRef]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef]
- Varier, R.A.; Timmers, H.T. Histone lysine methylation and demethylation pathways in cancer. Biochim. Biophys. Acta 2011, 1815, 75–89. [Google Scholar] [CrossRef]
- Hoffmann, I.; Roatsch, M.; Schmitt, M.L.; Carlino, L.; Pippel, M.; Sippl, W.; Jung, M. The role of histone demethylases in cancer therapy. Mol. Oncol. 2012, 6, 683–703. [Google Scholar] [CrossRef]
- Thinnes, C.C.; England, K.S.; Kawamura, A.; Chowdhury, R.; Schofield, C.J.; Hopkinson, R.J. Targeting histone lysine demethylases—Progress, challenges, and the future. Biochim. Biophys. Acta 2014, 1839, 1416–1432. [Google Scholar] [CrossRef]
- Chizuka, A.; Suda, M.; Shibata, T.; Kusumi, E.; Hori, A.; Hamaki, T.; Kodama, Y.; Horigome, K.; Kishi, Y.; Kobayashi, K.; et al. Difference between hematological malignancy and solid tumor research articles published in four major medical journals. Leukemia 2006, 20, 1655–1657. [Google Scholar] [CrossRef]
- Hui, Z.; Yiling, C.; Wenting, Y.; XuQun, H.; ChuanYi, Z.; Hui, L. miR-491-5p functions as a tumor suppressor by targeting JMJD2B in ERα-positive breast cancer. FEBS Lett. 2015, 589, 812–821. [Google Scholar] [CrossRef]
- Johmura, Y.; Maeda, I.; Suzuki, N.; Wu, W.; Goda, A.; Morita, M.; Yamaguchi, K.; Yamamoto, M.; Nagasawa, S.; Kojima, Y.; et al. Fbxo22-mediated KDM4B degradation determines selective estrogen receptor modulator activity in breast cancer. J. Clin. Investig. 2018, 128, 5603–5619. [Google Scholar] [CrossRef]
- Wang, W.; Oguz, G.; Lee, P.L.; Bao, Y.; Wang, P.; Terp, M.G.; Ditzel, H.J.; Yu, Q. KDM4B-regulated unfolded protein response as a therapeutic vulnerability in PTEN-deficient breast cancer. J. Exp. Med. 2018, 215, 2833–2849. [Google Scholar] [CrossRef]
- Hwang, S.Y.; Heo, K.; Kim, J.S.; Im, J.W.; Lee, S.M.; Cho, M.; Kang, D.H.; Heo, J.; Lee, J.W.; Choi, C.W.; et al. Emodin attenuates radioresistance induced by hypoxia in HepG2 cells via the enhancement of PARP1 cleavage and inhibition of JMJD2B. Oncol. Rep. 2015, 33, 1691–1698. [Google Scholar] [CrossRef]
- Toyokawa, G.; Cho, H.S.; Iwai, Y.; Yoshimatsu, M.; Takawa, M.; Hayami, S.; Maejima, K.; Shimizu, N.; Tanaka, H.; Tsunoda, T.; et al. The histone demethylase JMJD2B plays an essential role in human carcinogenesis through positive regulation of cyclin-dependent kinase 6. Cancer Prev. Res. 2011, 4, 2051–2061. [Google Scholar] [CrossRef]
- Berry, W.L.; Kim, T.D.; Janknecht, R. Stimulation of beta-catenin and colon cancer cell growth by the KDM4B histone demethylase. Int. J. Oncol. 2014, 44, 1341–1348. [Google Scholar] [CrossRef]
- Han, F.; Ren, J.; Zhang, J.; Sun, Y.; Ma, F.; Liu, Z.; Yu, H.; Jia, J.; Li, W. JMJD2B is required for Helicobacter pylori-induced gastric carcinogenesis via regulating COX-2 expression. Oncotarget 2016. [Google Scholar] [CrossRef]
- Li, W.; Zhao, L.; Zang, W.; Liu, Z.; Chen, L.; Liu, T.; Xu, D.; Jia, J. Histone demethylase JMJD2B is required for tumor cell proliferation and survival and is overexpressed in gastric cancer. Biochem. Biophys. Res. Commun. 2011, 416, 372–378. [Google Scholar] [CrossRef]
- Agger, K.; Miyagi, S.; Pedersen, M.T.; Kooistra, S.M.; Johansen, J.V.; Helin, K. Jmjd2/Kdm4 demethylases are required for expression of Il3ra and survival of acute myeloid leukemia cells. Genes Dev. 2016, 30, 1278–1288. [Google Scholar] [CrossRef]
- Zhao, L.; Li, W.; Zang, W.; Liu, Z.; Xu, X.; Yu, H.; Yang, Q.; Jia, J. JMJD2B promotes epithelial-mesenchymal transition by cooperating with beta-catenin and enhances gastric cancer metastasis. Clin. Cancer Res. 2013, 19, 6419–6429. [Google Scholar] [CrossRef]
- Jing, J.C.; Feng, Z.; Chen, Z.H.; Ji, B.N.; Hong, J.; Tang, N.; Yu, J.L.; Wang, S.Y. KDM4B promotes gastric cancer metastasis by regulating miR-125b-mediated activation of Wnt signaling. J. Cell. Biochem. 2018. [Google Scholar] [CrossRef]
- Abarrategi, A.; Tornin, J.; Martinez-Cruzado, L.; Hamilton, A.; Martinez-Campos, E.; Rodrigo, J.P.; Gonzalez, M.V.; Baldini, N.; Garcia-Castro, J.; Rodriguez, R. Osteosarcoma: Cells-of-Origin, Cancer Stem Cells, and Targeted Therapies. Stem Cells Int. 2016, 2016, 3631764. [Google Scholar] [CrossRef]
- Ipenberg, I.; Guttmann-Raviv, N.; Khoury, H.P.; Kupershmit, I.; Ayoub, N. Heat shock protein 90 (Hsp90) selectively regulates the stability of KDM4B/JMJD2B histone demethylase. J. Biol. Chem. 2013, 288, 14681–14687. [Google Scholar] [CrossRef]
- Young, L.C.; McDonald, D.W.; Hendzel, M.J. Kdm4b histone demethylase is a DNA damage response protein and confers a survival advantage following γ-irradiation. J. Biol. Chem. 2013, 288, 21376–21388. [Google Scholar] [CrossRef]
- Wen, L.; Chen, Y.; Zeng, L.L.; Zhao, F.; Li, R.; Liu, Y.; Zhang, C. Triptolide induces cell-cycle arrest and apoptosis of human multiple myeloma cells in vitro via altering expression of histone demethylase LSD1 and JMJD2B. Acta Pharmacol. Sin. 2012, 33, 109–119. [Google Scholar] [CrossRef]
- Filiu-Braga, L.D.C.; Serejo, T.R.T.; Lucena-Araujo, A.R.; Neves, F.A.R.; de Carvalho, J.L.; Rego, E.M.; Saldanha-Araujo, F. Unraveling KDM4 histone demethylase expression and its association with adverse cytogenetic findings in chronic lymphocytic leukemia. Med. Oncol. 2018, 36, 3. [Google Scholar] [CrossRef]
- Cho, K.R.; Shih Ie, M. Ovarian cancer. Annu. Rev. Pathol. 2009, 4, 287–313. [Google Scholar] [CrossRef]
- Armstrong, D.K. Relapsed ovarian cancer: Challenges and management strategies for a chronic disease. Oncologist 2002, 7 (Suppl. 5), 20–28. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar]
- Lindemann, K.; Gibbs, E.; Avall-Lundqvist, E.; dePont Christensen, R.; Woie, K.; Kalling, M.; Auranen, A.; Grenman, S.; Hoegberg, T.; Rosenberg, P.; et al. Chemotherapy vs tamoxifen in platinum-resistant ovarian cancer: A phase III, randomised, multicentre trial (Ovaresist). Br. J. Cancer 2017, 116, 455–463. [Google Scholar] [CrossRef]
- George, A.; McLachlan, J.; Tunariu, N.; Della Pepa, C.; Migali, C.; Gore, M.; Kaye, S.; Banerjee, S. The role of hormonal therapy in patients with relapsed high-grade ovarian carcinoma: A retrospective series of tamoxifen and letrozole. BMC Cancer 2017, 17, 456. [Google Scholar] [CrossRef]
- Lu, J.W.; Ho, Y.J.; Lin, L.I.; Huang, Y.C.; Yeh, K.T.; Lin, Y.H.; Lin, Y.M.; Tzeng, T.Y. JMJD2B as a potential diagnostic immunohistochemical marker for hepatocellular carcinoma: A tissue microarray-based study. Acta Histochem. 2015, 117, 14–19. [Google Scholar] [CrossRef]
- Yang, J.; AlTahan, A.M.; Hu, D.; Wang, Y.; Cheng, P.H.; Morton, C.L.; Qu, C.; Nathwani, A.C.; Shohet, J.M.; Fotsis, T.; et al. The role of histone demethylase KDM4B in Myc signaling in neuroblastoma. J. Natl. Cancer Inst. 2015, 107, djv080. [Google Scholar] [CrossRef]
- Herlihy, N.; Dogrusoz, M.; van Essen, T.H.; Harbour, J.W.; van der Velden, P.A.; van Eggermond, M.C.; Haasnoot, G.W.; van den Elsen, P.J.; Jager, M.J. Skewed expression of the genes encoding epigenetic modifiers in high-risk uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2015, 56, 1447–1458. [Google Scholar] [CrossRef]
- Sisley, K.; Cottam, D.W.; Rennie, I.G.; Parsons, M.A.; Potter, A.M.; Potter, C.W.; Rees, R.C. Non-random abnormalities of chromosomes 3, 6, and 8 associated with posterior uveal melanoma. Genes Chromos. Cancer 1992, 5, 197–200. [Google Scholar] [CrossRef]
- Hatch, S.B.; Yapp, C.; Montenegro, R.C.; Savitsky, P.; Gamble, V.; Tumber, A.; Ruda, G.F.; Bavetsias, V.; Fedorov, O.; Atrash, B.; et al. Assessing histone demethylase inhibitors in cells: Lessons learned. Epigenet. Chromat. 2017, 10, 9. [Google Scholar] [CrossRef]
- Kruidenier, L.; Chung, C.W.; Cheng, Z.; Liddle, J.; Che, K.; Joberty, G.; Bantscheff, M.; Bountra, C.; Bridges, A.; Diallo, H.; et al. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature 2012, 488, 404–408. [Google Scholar] [CrossRef]
- Rai, G.; Kawamura, A.; Tumber, A.; Liang, Y.; Vogel, J.L.; Arbuckle, J.H.; Rose, N.R.; Dexheimer, T.S.; Foley, T.L.; King, O.N.; et al. Discovery of ML324, a JMJD2 demethylase inhibitor with demonstrated antiviral activity. In Probe Reports from the NIH Molecular Libraries Program; National Center for Biotechnology Information: Bethesda, MD, USA, 2010. [Google Scholar]
- Rose, N.R.; Woon, E.C.; Kingham, G.L.; King, O.N.; Mecinovic, J.; Clifton, I.J.; Ng, S.S.; Talib-Hardy, J.; Oppermann, U.; McDonough, M.A.; et al. Selective inhibitors of the JMJD2 histone demethylases: Combined nondenaturing mass spectrometric screening and crystallographic approaches. J. Med. Chem. 2010, 53, 1810–1818. [Google Scholar] [CrossRef]
- Tumber, A.; Nuzzi, A.; Hookway, E.S.; Hatch, S.B.; Velupillai, S.; Johansson, C.; Kawamura, A.; Savitsky, P.; Yapp, C.; Szykowska, A.; et al. Potent and Selective KDM5 Inhibitor Stops Cellular Demethylation of H3K4me3 at Transcription Start Sites and Proliferation of MM1S Myeloma Cells. Cell. Chem. Biol. 2017, 24, 371–380. [Google Scholar] [CrossRef]
- Cascella, B.; Lee, S.G.; Singh, S.; Jez, J.M.; Mirica, L.M. The small molecule JIB-04 disrupts O2 binding in the Fe-dependent histone demethylase KDM4A/JMJD2A. Chem. Commun. 2017, 53, 2174–2177. [Google Scholar] [CrossRef]
- Gerken, P.A.; Wolstenhulme, J.R.; Tumber, A.; Hatch, S.B.; Zhang, Y.; Muller, S.; Chandler, S.A.; Mair, B.; Li, F.; Nijman, S.M.B.; et al. Discovery of a Highly Selective Cell-Active Inhibitor of the Histone Lysine Demethylases KDM2/7. Angew. Chem. Int. Ed. Engl. 2017, 56, 15555–15559. [Google Scholar] [CrossRef]
- Kawamura, A.; Munzel, M.; Kojima, T.; Yapp, C.; Bhushan, B.; Goto, Y.; Tumber, A.; Katoh, T.; King, O.N.; Passioura, T.; et al. Highly selective inhibition of histone demethylases by de novo macrocyclic peptides. Nat. Commun. 2017, 8, 14773. [Google Scholar] [CrossRef]
- Jambhekar, A.; Anastas, J.N.; Shi, Y. Histone Lysine Demethylase Inhibitors. Cold Spring Harb. Perspect. Med. 2017, 7, a026484. [Google Scholar] [CrossRef]
- Chowdhury, R.; Yeoh, K.K.; Tian, Y.M.; Hillringhaus, L.; Bagg, E.A.; Rose, N.R.; Leung, I.K.; Li, X.S.; Woon, E.C.; Yang, M.; et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011, 12, 463–469. [Google Scholar] [CrossRef]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef]
- Waitkus, M.S.; Diplas, B.H.; Yan, H. Biological Role and Therapeutic Potential of IDH Mutations in Cancer. Cancer Cell 2018, 34, 186–195. [Google Scholar] [CrossRef]
- Ward, P.S.; Cross, J.R.; Lu, C.; Weigert, O.; Abel-Wahab, O.; Levine, R.L.; Weinstock, D.M.; Sharp, K.A.; Thompson, C.B. Identification of additional IDH mutations associated with oncometabolite R(-)-2-hydroxyglutarate production. Oncogene 2012, 31, 2491–2498. [Google Scholar] [CrossRef]
- Laukka, T.; Myllykoski, M.; Looper, R.E.; Koivunen, P. Cancer-associated 2-oxoglutarate analogues modify histone methylation by inhibiting histone lysine demethylases. J. Mol. Biol. 2018, 430, 3081–3092. [Google Scholar] [CrossRef]
- Sulkowski, P.L.; Corso, C.D.; Robinson, N.D.; Scanlon, S.E.; Purshouse, K.R.; Bai, H.; Liu, Y.; Sundaram, R.K.; Hegan, D.C.; Fons, N.R.; et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci. Transl. Med. 2017, 9, eaal2463. [Google Scholar] [CrossRef]
- Sulkowski, P.L.; Sundaram, R.K.; Oeck, S.; Corso, C.D.; Liu, Y.; Noorbakhsh, S.; Niger, M.; Boeke, M.; Ueno, D.; Kalathil, A.N.; et al. Krebs-cycle-deficient hereditary cancer syndromes are defined by defects in homologous-recombination DNA repair. Nat. Genet. 2018, 50, 1086–1092. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilson, C.; Krieg, A.J. KDM4B: A Nail for Every Hammer? Genes 2019, 10, 134. https://doi.org/10.3390/genes10020134
Wilson C, Krieg AJ. KDM4B: A Nail for Every Hammer? Genes. 2019; 10(2):134. https://doi.org/10.3390/genes10020134
Chicago/Turabian StyleWilson, Cailin, and Adam J. Krieg. 2019. "KDM4B: A Nail for Every Hammer?" Genes 10, no. 2: 134. https://doi.org/10.3390/genes10020134
APA StyleWilson, C., & Krieg, A. J. (2019). KDM4B: A Nail for Every Hammer? Genes, 10(2), 134. https://doi.org/10.3390/genes10020134