Selecting Near-Native Protein Structures from Predicted Decoy Sets Using Ordered Graphlet Degree Similarity



Abstract
:1. Introduction
2. Materials and Methods
2.1. Maximum Contact Map Overlap (CMO)
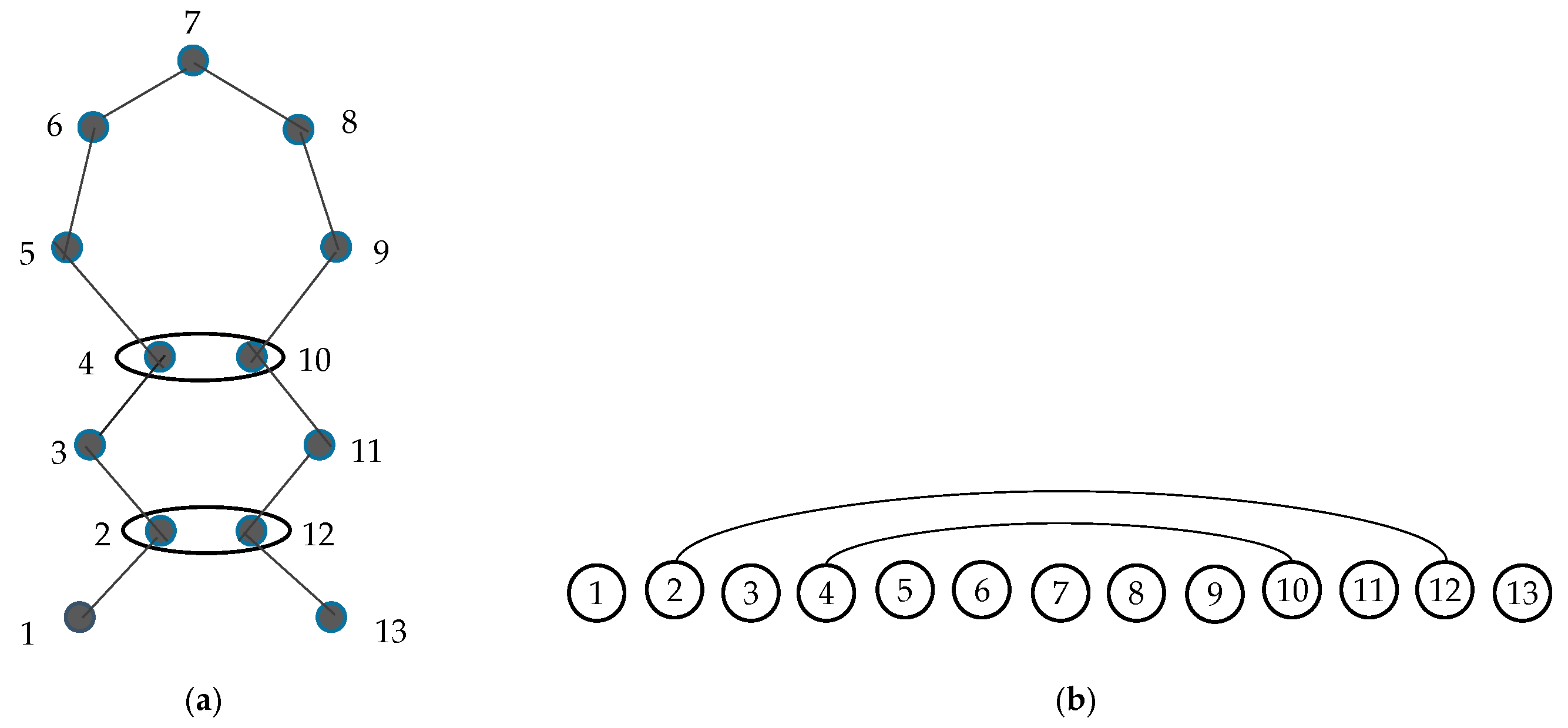
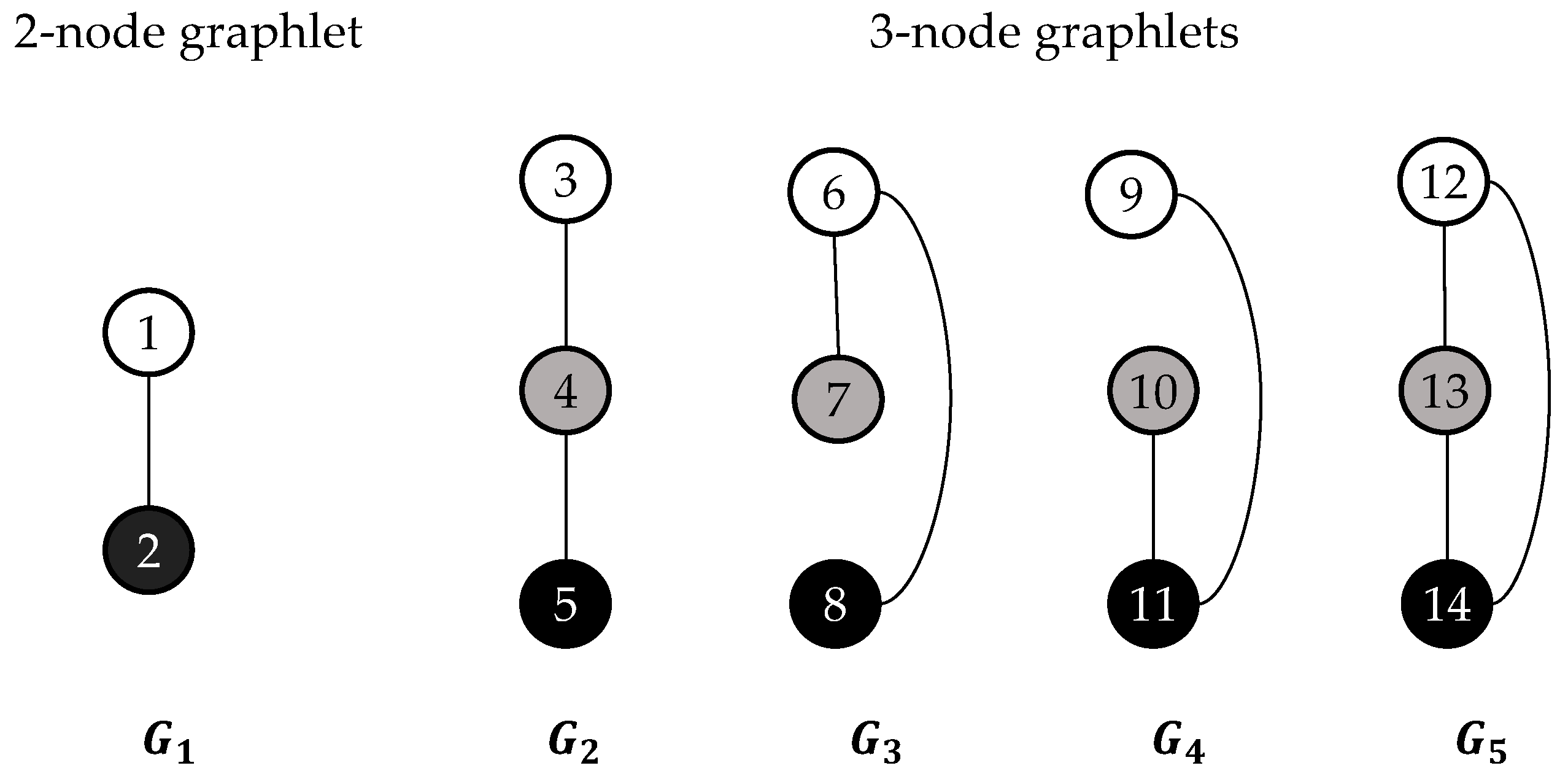
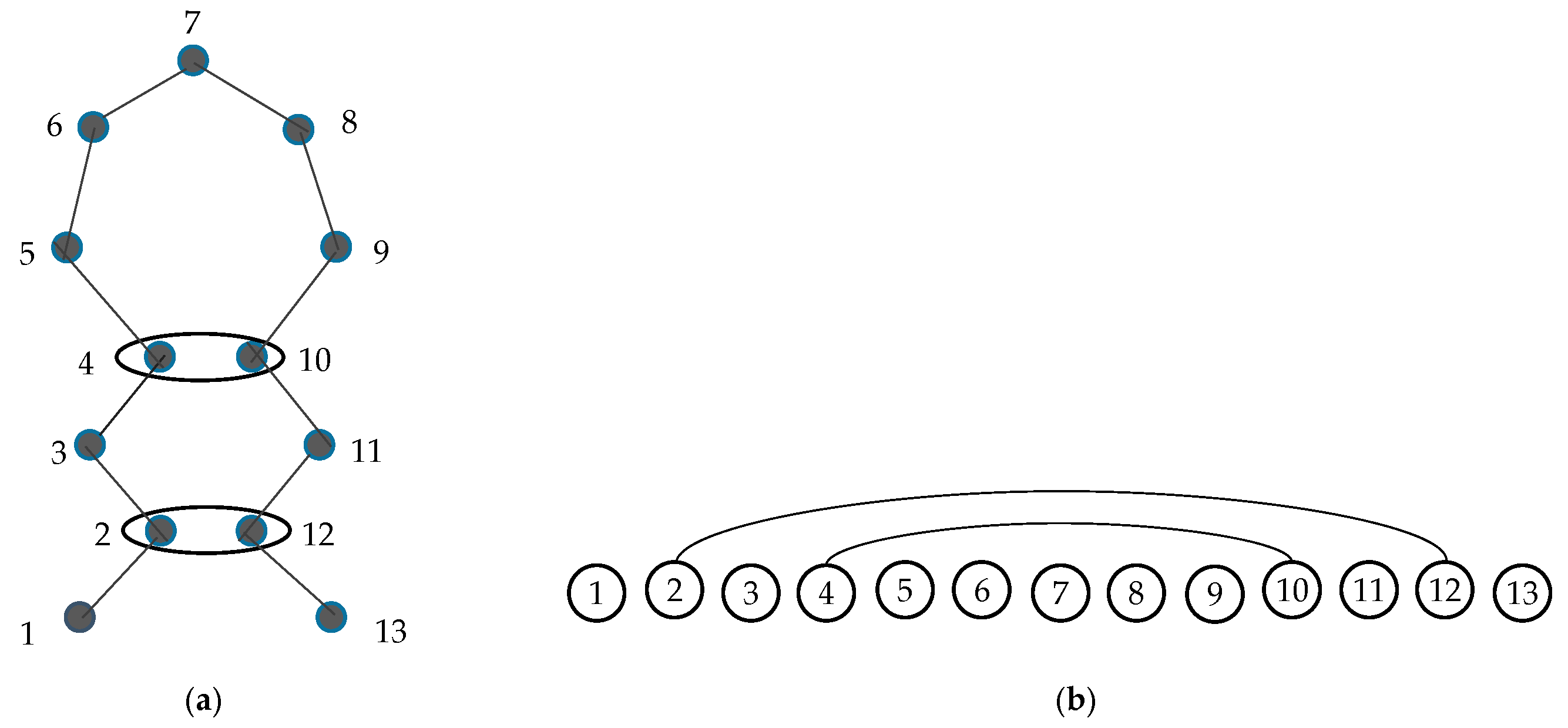
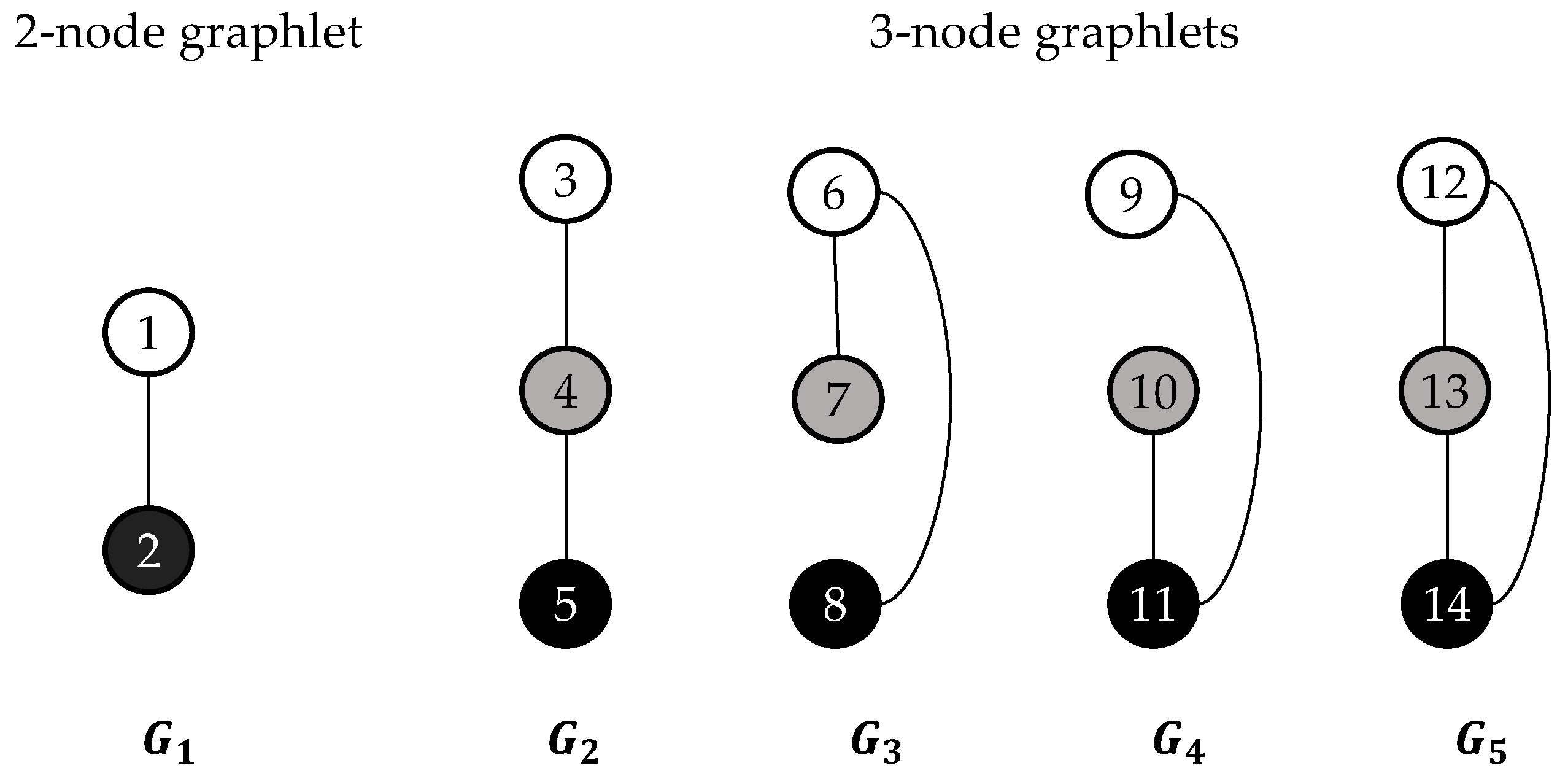
2.2. Graphlets and Graphlet Degrees
2.3. TM-Score
2.4. SPICKER
2.5. Ensemble Clustering
2.6. GR_score
2.6.1. Ordered Graphlet Degree Similarity.
2.6.2. Structure Alignment Algorithm.
2.6.3. Definition of the GR_score.
2.7. Constructing the Distance Matrix
2.8. Select the Near-native Structure using Ensemble Clustering
3. Results and Discussion
3.1. Dataset
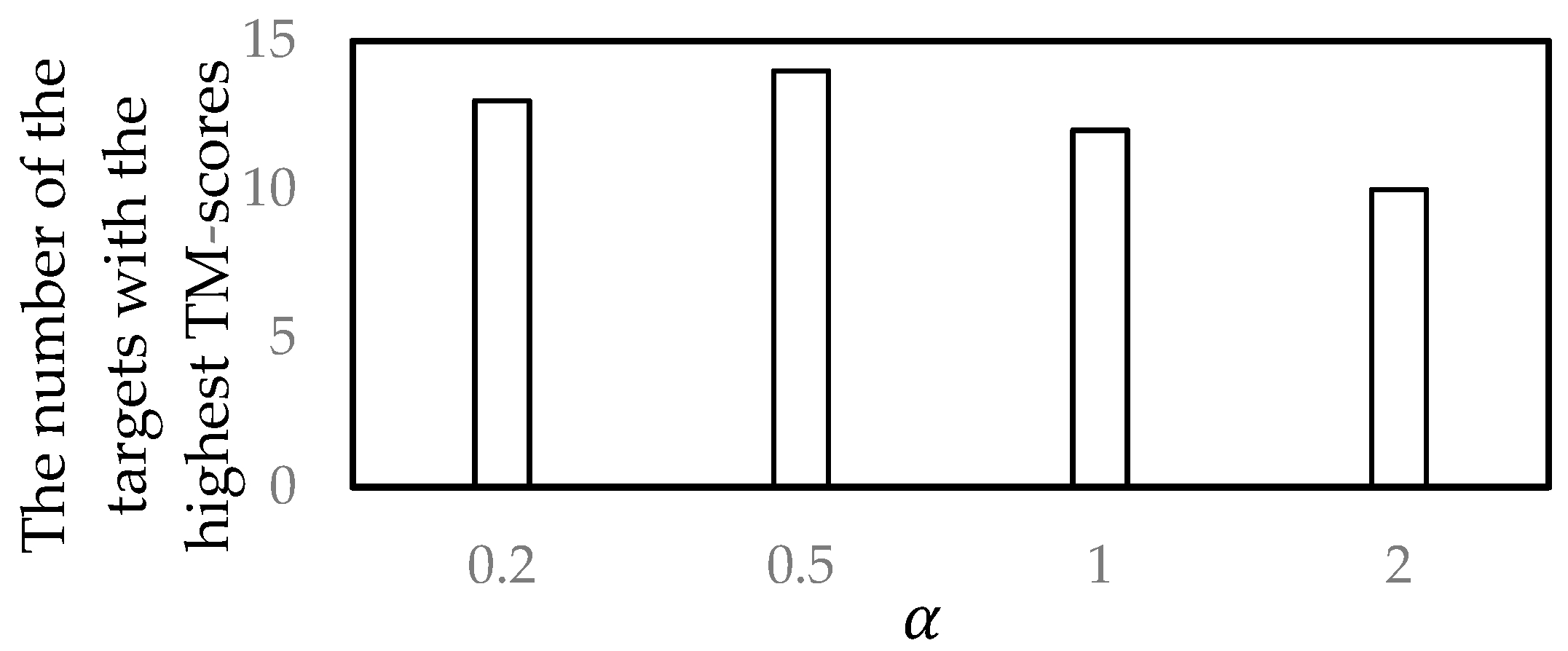
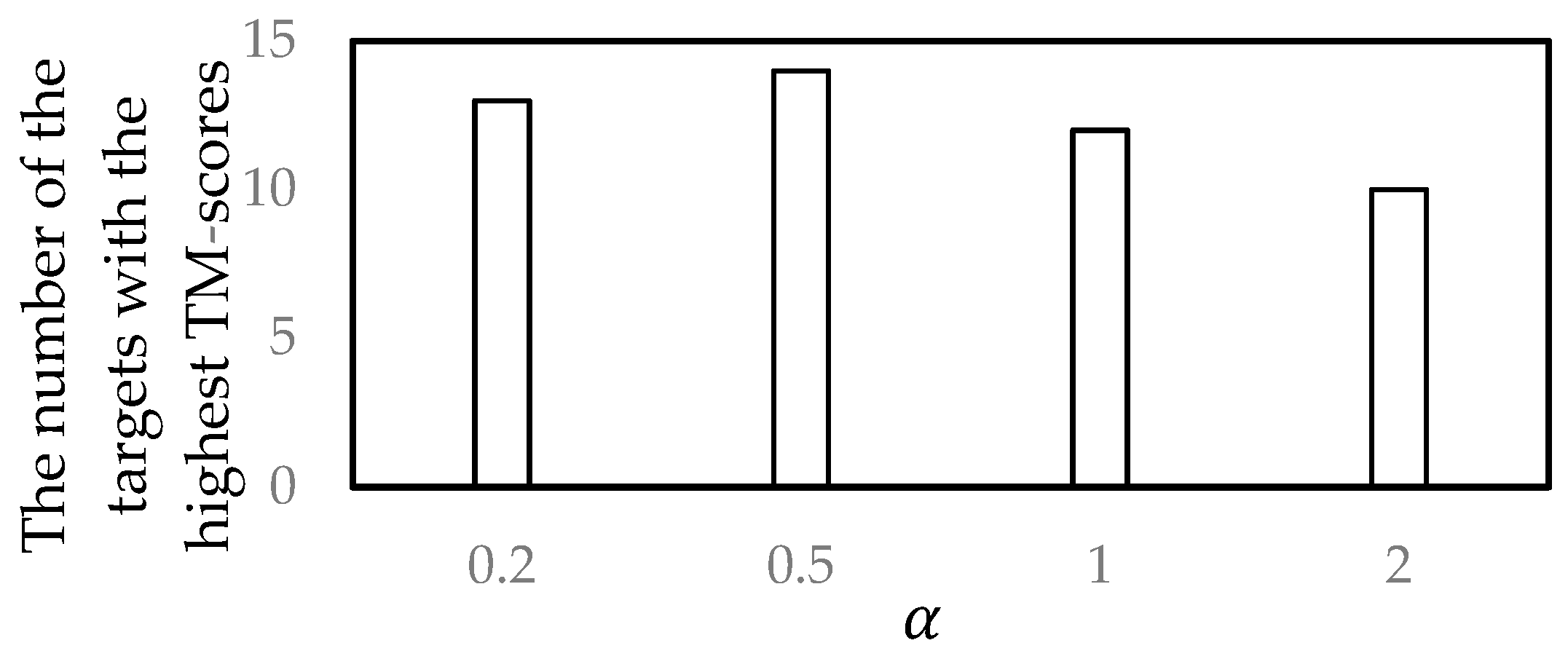
3.2. Parameter Selection
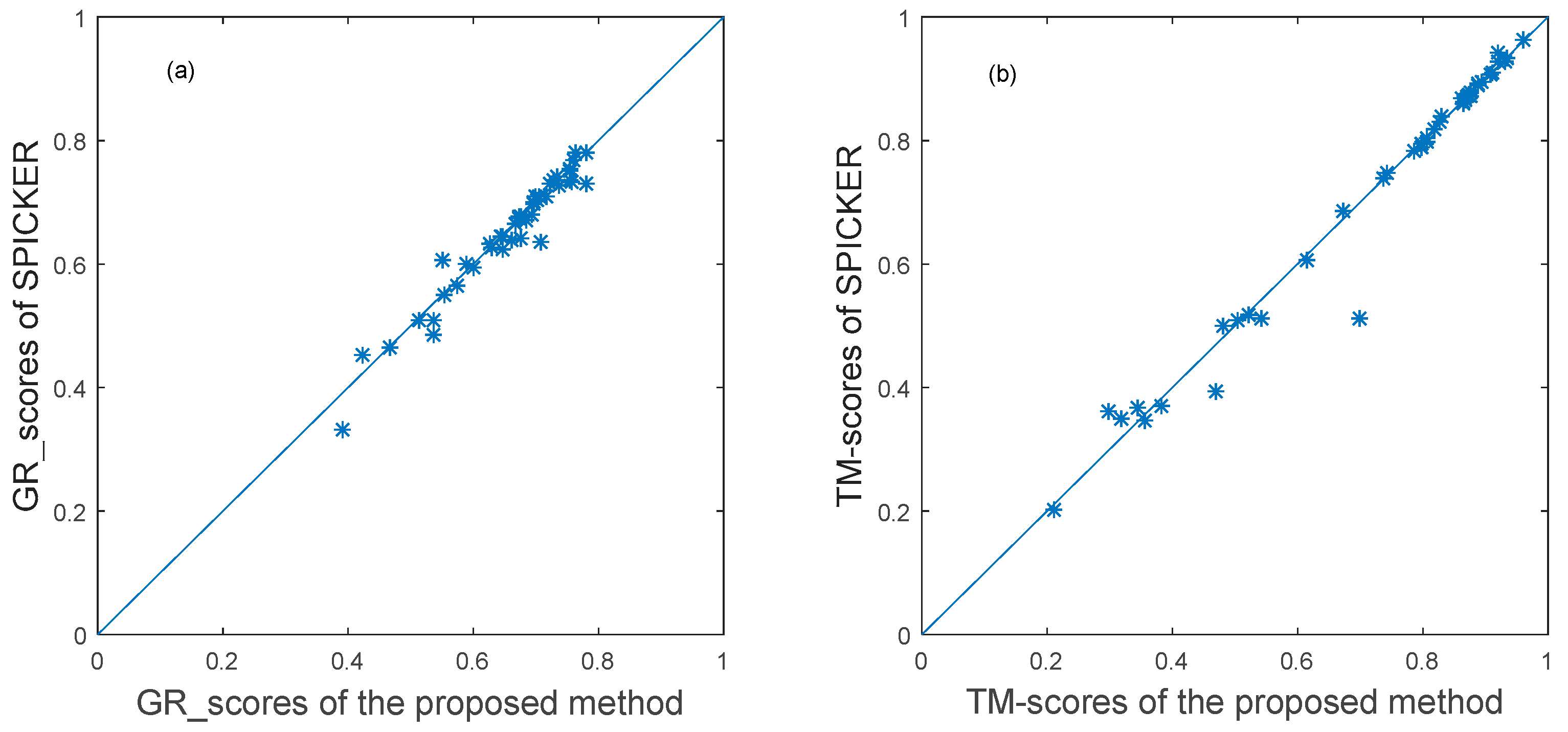
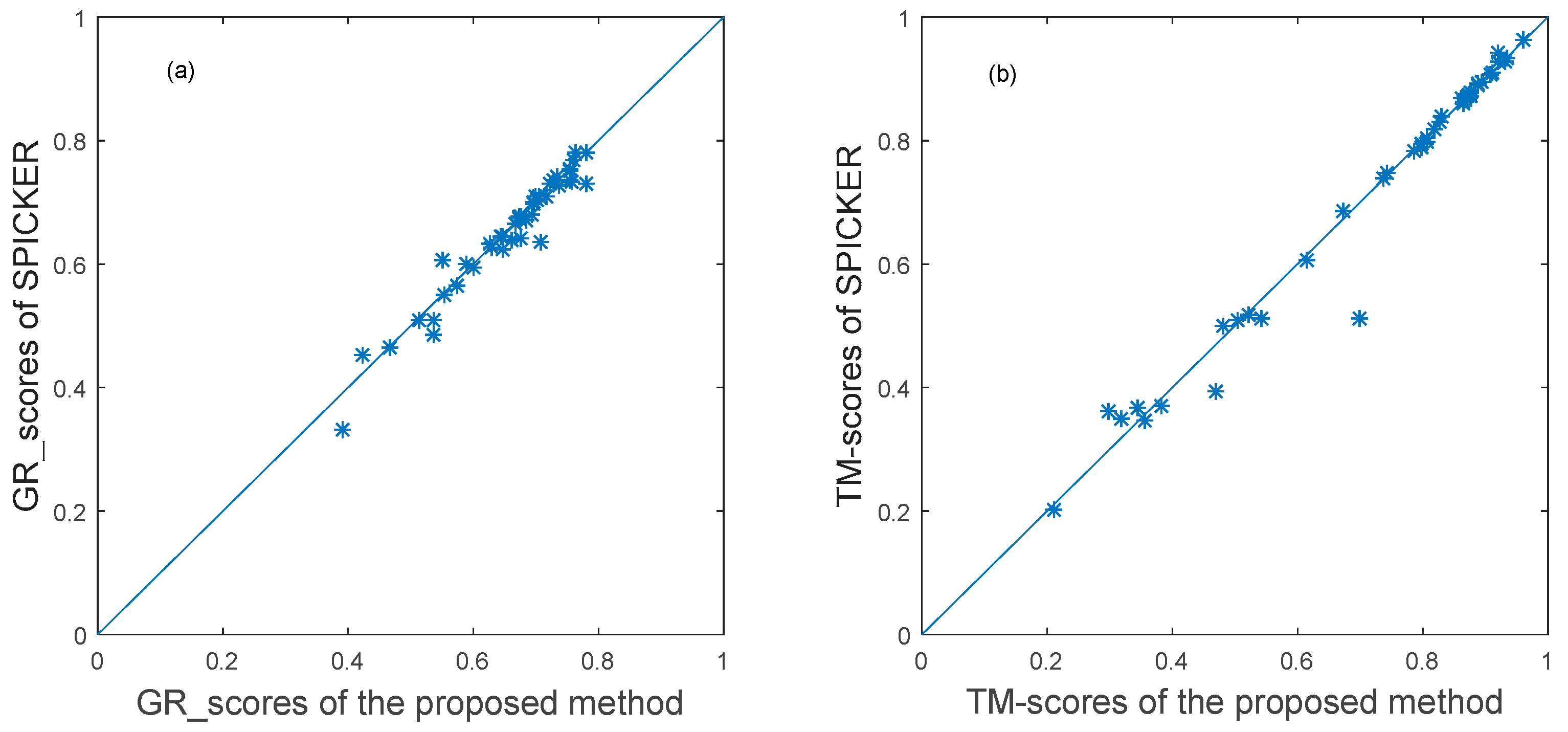
3.3. Experimental Results
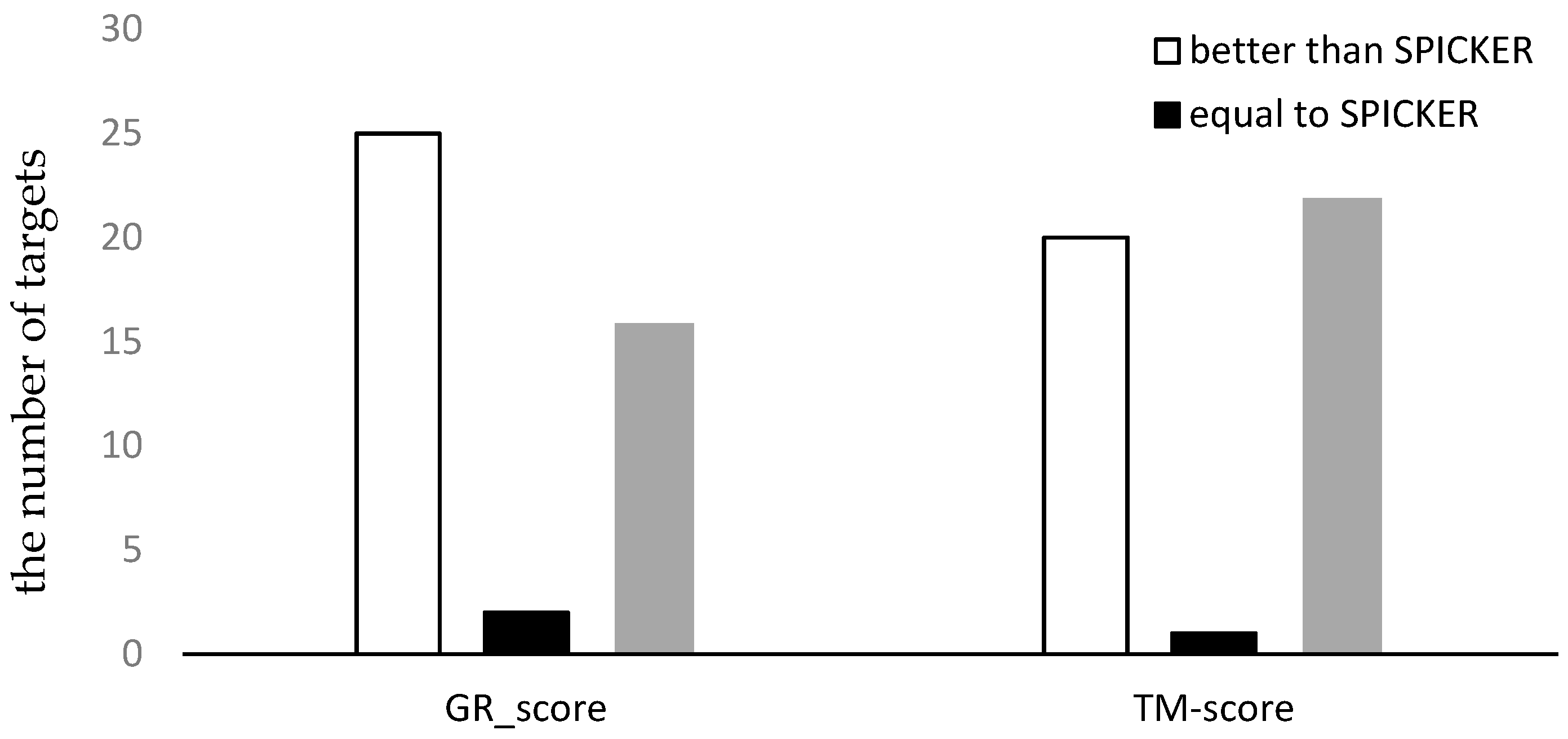
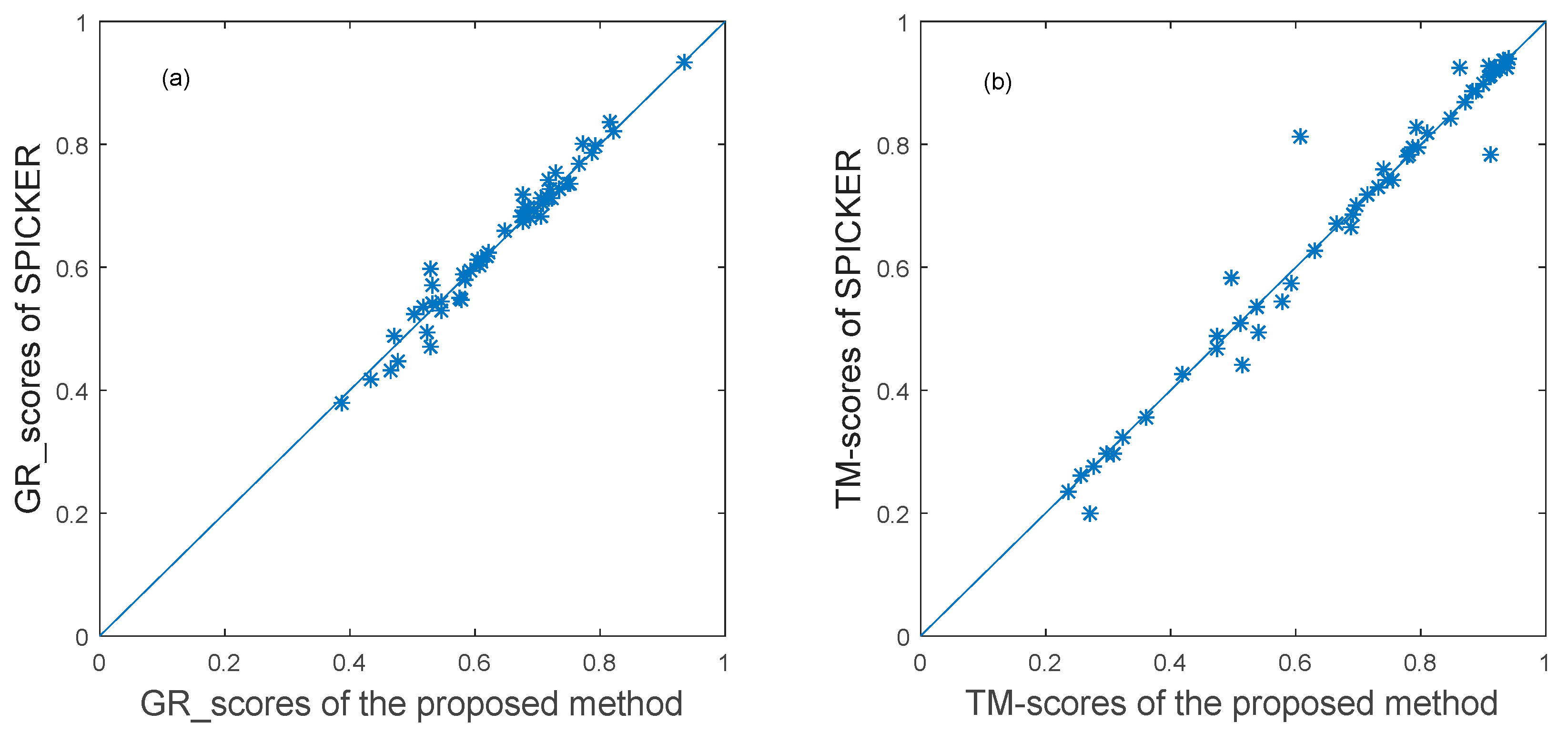
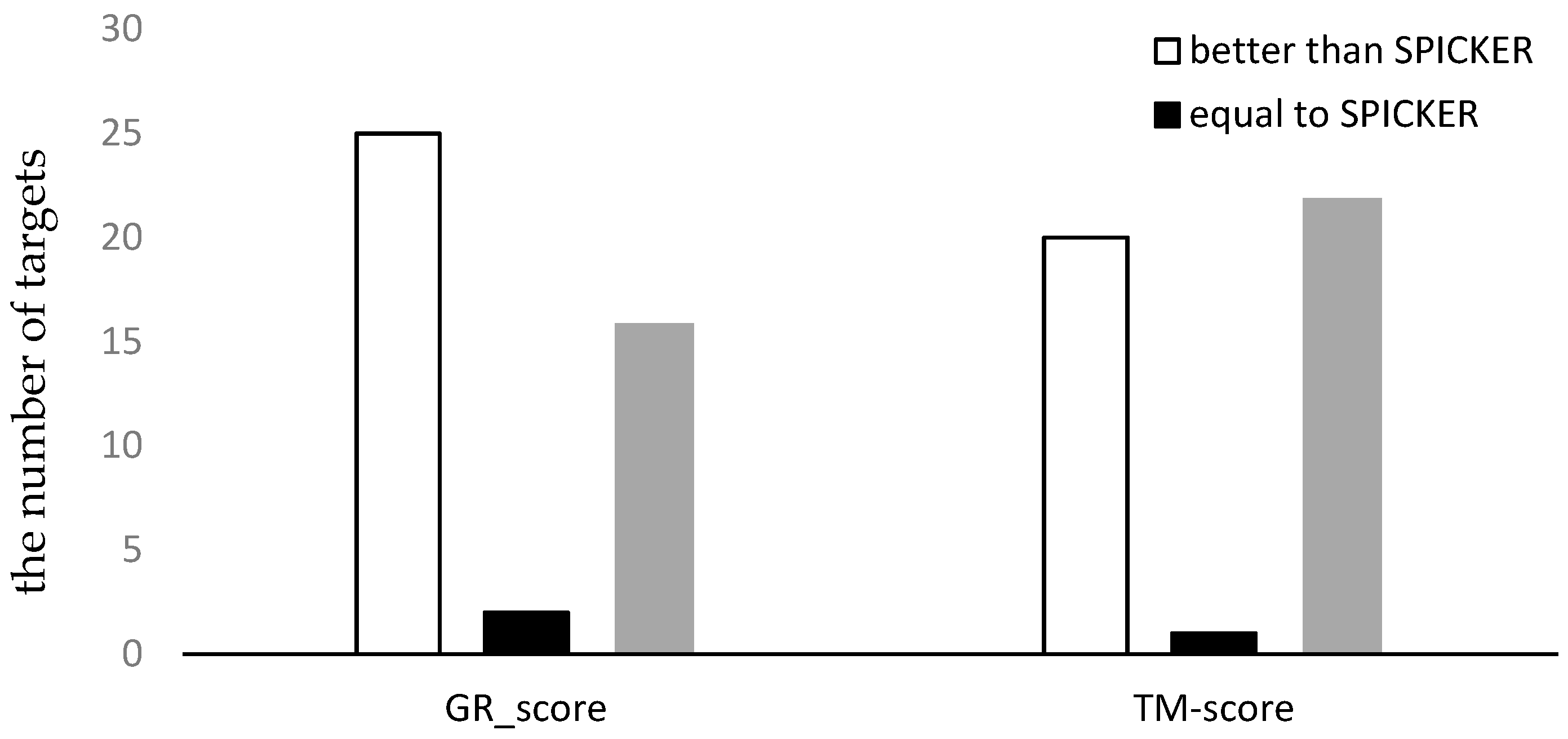
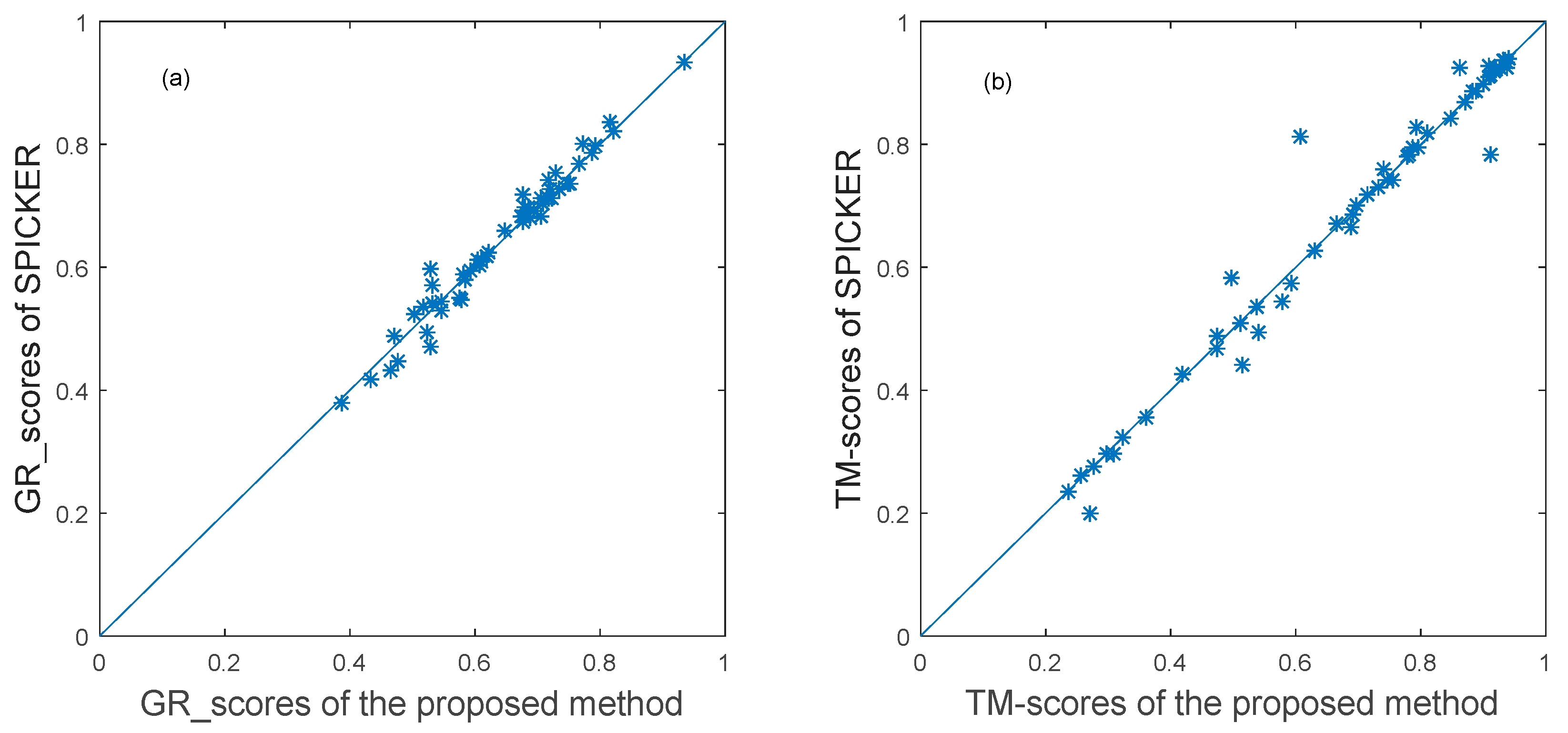
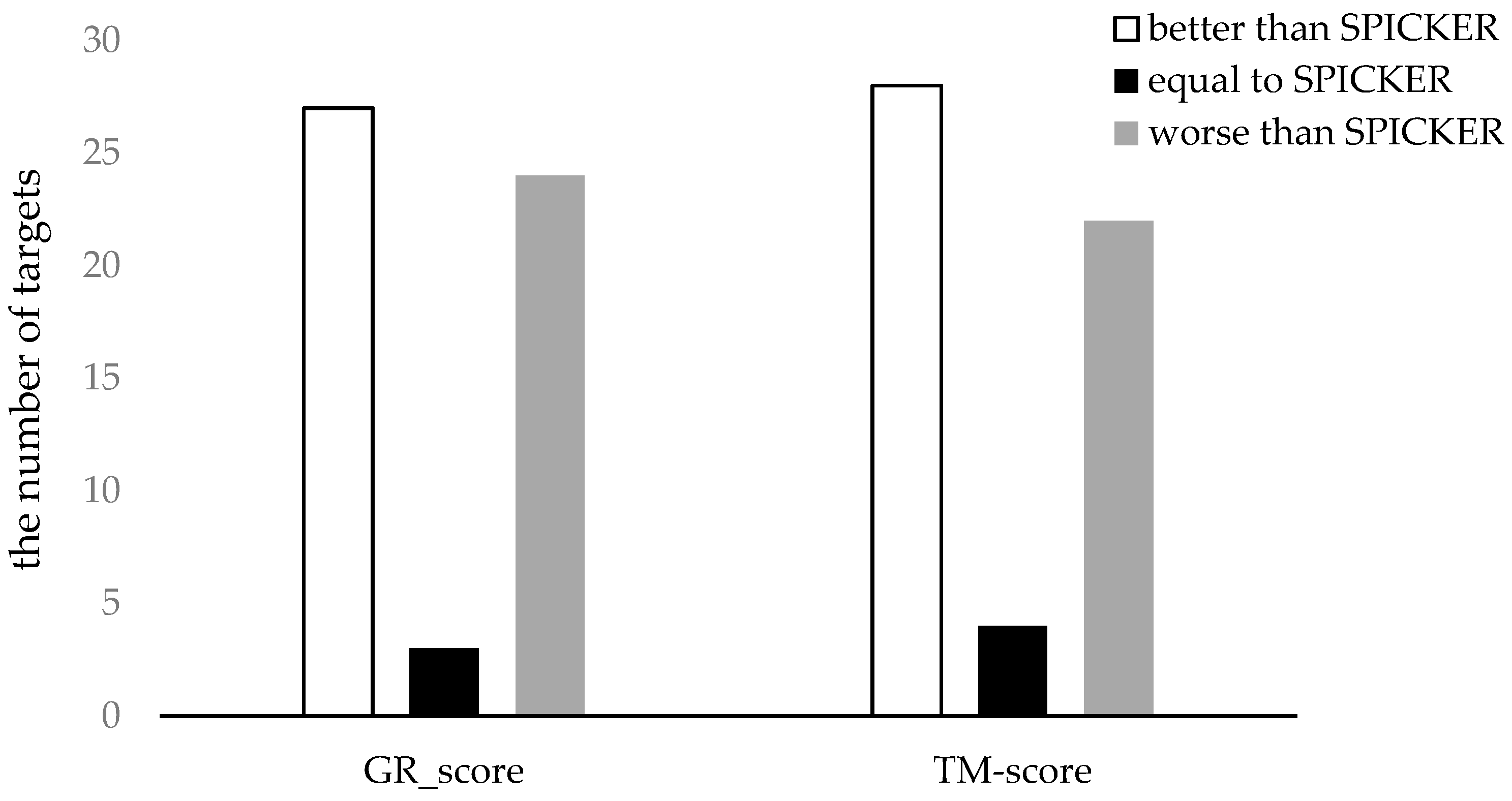
3.3.1. The Experimental Results for Datasets from CASP10.
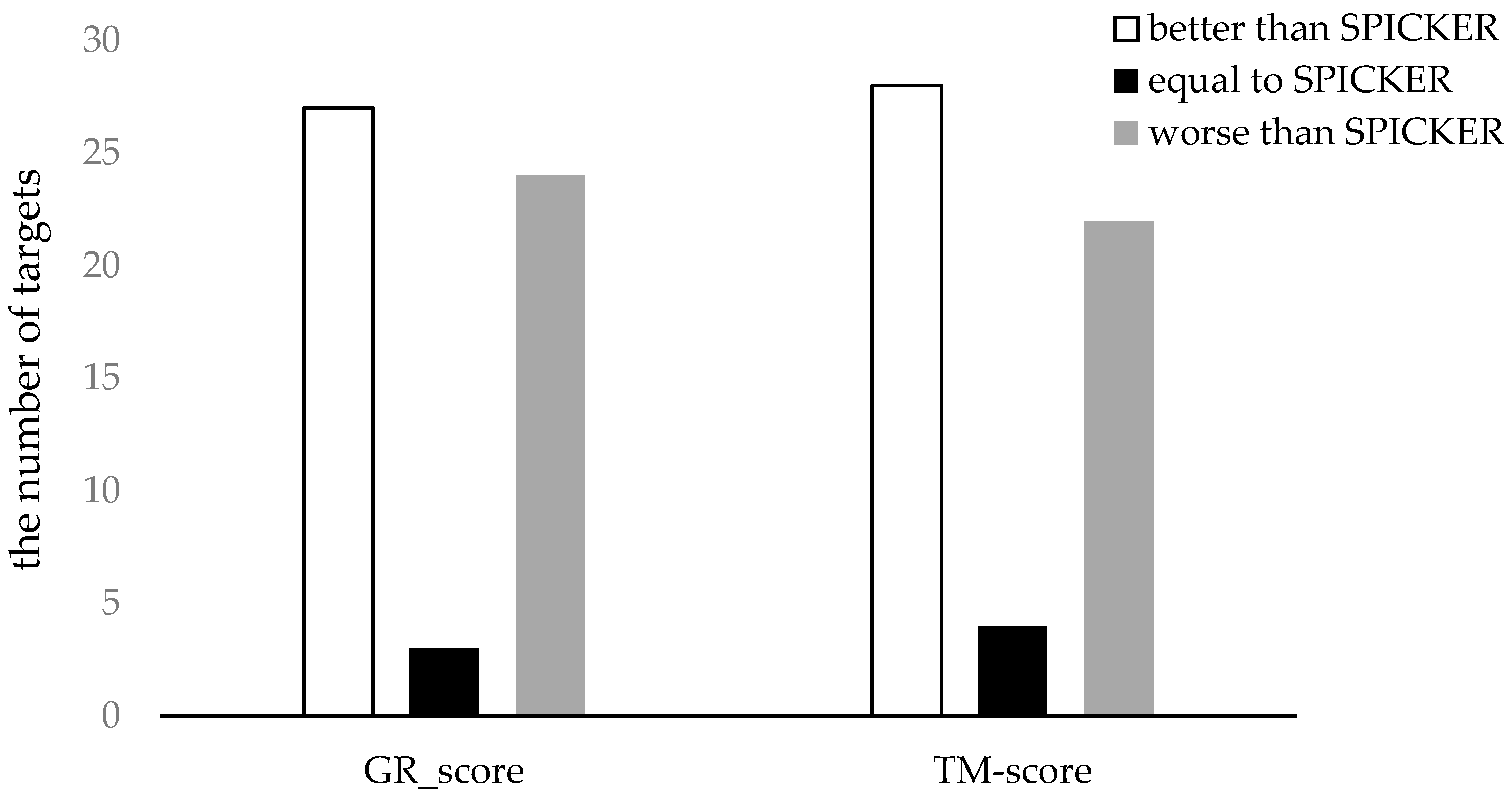
3.3.2. The Experimental Results for Datasets from CASP11.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Collins, F.S.; Michael, M.; Aristides, P. The human genome project: Lessons from large-scale biology. Science 2003, 300, 286. [Google Scholar] [CrossRef] [PubMed]
- Crick, F. Central dogma of molecular biology. Nature 1970, 227, 561–563. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, M.; Marcotte, E.M.; Thompson, M.J.; Eisenberg, D.; Yeates, T.O. Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. Proc. Natl. Acad. Sci. USA 1999, 96, 4285–4288. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucl. Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- UniProtKB/TrEMBL Protein database release statisics. Available online: http://www.ebi.ac.uk/uniprot/TrEMBLstats (accessed on 16 January 2019).
- Zhang, Z. An overview of protein structure prediction: From homology to ab initio. Bioc218 2002, 1–10. [Google Scholar]
- Hasegawa, H.; Holm, L. Advances and pitfalls of protein structural alignment. Curr. Opin. Struct. Biol. 2009, 19, 341–348. [Google Scholar] [CrossRef]
- Yang, Z.; Jeffrey, S. Automated structure prediction of weakly homologous proteins on a genomic scale. Proc. Natl. Acad. Sci. USA 2004, 101, 7594–7599. [Google Scholar]
- Wang, S.; Sun, S.; Li, Z.; Zhang, R.; Xu, J. Accurate de novo prediction of protein contact map by ultra-deep learning model. PLoS Comp. Biol. 2017, 13, e1005324. [Google Scholar] [CrossRef]
- Hamilton, N.; Burrage, K.; Ragan, M.A.; Huber, T. Protein contact prediction using patterns of correlation. Proteins 2004, 56, 679–684. [Google Scholar] [CrossRef]
- Moult, J.; Pedersen, J.T.; Judson, R.; Fidelis, K. A large-scale experiment to assess protein structure prediction methods. Proteins 1995, 23, ii–iv. [Google Scholar] [CrossRef]
- The 11th critical assessment of techniques for protein structure prediction. Available online: http://predictioncenter.org/casp11 (accessed on 7 December 2014).
- The 10th critical assessment of techniques for protein structure prediction. Available online: http://predictioncenter.org/casp10 (accessed on 7 December 2012).
- The Yang Zhang Lab. Available online: https://zhanglab.ccmb.med.umich.edu/decoys/ (accessed on 30 June 2018).
- Shortle, D.; Simons, K.T.; Baker, D. Clustering of low-energy conformations near the native structures of small proteins. Proc. Natl. Acad. Sci. USA 1998, 95, 11158–11162. [Google Scholar] [CrossRef] [PubMed]
- Godzik, A. The structural alignment between two proteins: Is there a unique answer? Protein Sci. 2010, 5, 1325–1338. [Google Scholar] [CrossRef] [PubMed]
- Shindyalov, I.N.; Bourne, P.E. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 1998, 11, 739–747. [Google Scholar] [CrossRef] [PubMed]
- Zemla, A.; Venclovas, C.; Moult, J.; Fidelis, K. Processing and analysis of CASP3 protein structure predictions. Proteins 2015, 37, 22–29. [Google Scholar]
- Zhang, Y.; Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins 2004, 57, 702–710. [Google Scholar] [PubMed]
- Ye, Y.; Godzik, A. Flexible structure alignment by chaining aligned fragment pairs allowing twists. Bioinformatics 2003, 19 (Suppl. 2), ii246. [Google Scholar] [CrossRef]
- Kliment, O.; Eleonora, K.; Ceslovas, V. CAD-score: A new contact area difference-based function for evaluation of protein structural models. Proteins 2013, 81, 149–162. [Google Scholar]
- Valerio, M.; Marco, B.; Alessandro, B.; Torsten, S. IDDT: A local superposition-free score for comparing protein structures and models using distance difference tests. Bioinformatics 2013, 29, 2722–2728. [Google Scholar]
- Manavalan, B.; Lee, J.; Lee, J. Random forest-based protein model quality assessment (RFMQA) using structural features and potential energy terms. PLoS ONE 2014, 9, e106542. [Google Scholar] [CrossRef]
- Manavalan, B.; Lee, J. SVMQA: Support-vector-machine-based protein single-model quality assessment. Bioinformatics 2017, 33, 2496. [Google Scholar] [CrossRef]
- Godzik, A.; Skolnick, J. Flexible algorithm for direct multiple alignment of protein structures and sequences. Bioinformatics 1994, 10, 587–596. [Google Scholar] [CrossRef]
- Przulj, N.; Corneil, D.G.; Jurisica, I. Modeling interactome: Scale-free or geometric? Bioinformatics 2004, 20, 3508–3515. [Google Scholar] [CrossRef] [PubMed]
- Malod-Dognin, N.; Przulj, N. GR-Align: Fast and flexible alignment of protein 3D structures using graphlet degree similarity. Bioinformatics 2014, 30, 1259–1265. [Google Scholar] [CrossRef] [PubMed]
- Murzin, A.G.; Brenner, S.E.; Hubbard, T.; Chothia, C. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995, 247, 536–540. [Google Scholar] [CrossRef]
- Zhang, Y.; Skolnick, J. SPICKER: A clustering approach to identify near-native protein folds. J. Comput. Chem. 2004, 25, 865–871. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Lu, Y.; Yan, H. Selecting near-native protein structures from ab initio models using ensemble clustering. Quant. Biol. 2018, 6, 307–312. [Google Scholar] [CrossRef]
- Needleman, S.B.; Wunsch, C.D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970, 48, 443–453. [Google Scholar] [CrossRef]
- Yang, J.Y.; Yan, R.X.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target ID | GR_Scores of the Proposed Method | GR_Scores of SPICKER | TM-Scores of the Proposed Method | TM-Scores of SPICKER |
|---|---|---|---|---|
| T0644 | 0.764 | 0.781 | 0.869 | 0.865 |
| T0645 | 0.666 | 0.664 | 0.932 | 0.929 |
| T0649 | 0.423 | 0.454 | 0.382 | 0.369 |
| T0650 | 0.702 | 0.703 | 0.876 | 0.877 |
| T0654 | 0.626 | 0.634 | 0.819 | 0.820 |
| T0655 | 0.672 | 0.677 | 0.743 | 0.749 |
| T0657 | 0.693 | 0.681 | 0.827 | 0.831 |
| T0659 | 0.753 | 0.754 | 0.909 | 0.906 |
| T0662 | 0.727 | 0.737 | 0.798 | 0.796 |
| T0664 | 0.684 | 0.671 | 0.936 | 0.934 |
| T0665 | 0.756 | 0.732 | 0.738 | 0.739 |
| T0667 | 0.643 | 0.646 | 0.807 | 0.803 |
| T0669 | 0.675 | 0.641 | 0.614 | 0.606 |
| T0672 | 0.590 | 0.601 | 0.785 | 0.784 |
| T0673 | 0.535 | 0.509 | 0.317 | 0.350 |
| T0675 | 0.552 | 0.606 | 0.356 | 0.346 |
| T0676 | 0.553 | 0.505 | 0.503 | 0.510 |
| T0678 | 0.599 | 0.594 | 0.297 | 0.362 |
| T0679 | 0.648 | 0.625 | 0.807 | 0.798 |
| T0680 | 0.709 | 0.637 | 0.699 | 0.513 |
| T0681 | 0.700 | 0.710 | 0.875 | 0.872 |
| T0683 | 0.660 | 0.639 | 0.888 | 0.889 |
| T0688 | 0.629 | 0.627 | 0.862 | 0.869 |
| T0689 | 0.734 | 0.742 | 0.919 | 0.927 |
| T0691 | 0.468 | 0.464 | 0.480 | 0.500 |
| T0692 | 0.704 | 0.710 | 0.921 | 0.942 |
| T0703 | 0.673 | 0.673 | 0.894 | 0.895 |
| T0704 | 0.675 | 0.677 | 0.831 | 0.838 |
| T0708 | 0.736 | 0.726 | 0.887 | 0.891 |
| T0714 | 0.781 | 0.781 | 0.911 | 0.911 |
| T0716 | 0.753 | 0.752 | 0.674 | 0.685 |
| T0721 | 0.716 | 0.710 | 0.870 | 0.872 |
| T0722 | 0.780 | 0.729 | 0.541 | 0.513 |
| T0723 | 0.697 | 0.702 | 0.866 | 0.859 |
| T0733 | 0.647 | 0.645 | 0.864 | 0.863 |
| T0749 | 0.755 | 0.737 | 0.961 | 0.963 |
| T0752 | 0.721 | 0.729 | 0.873 | 0.874 |
| T0753 | 0.696 | 0.698 | 0.797 | 0.790 |
| T0757 | 0.760 | 0.768 | 0.888 | 0.893 |
| R0001 | 0.390 | 0.333 | 0.212 | 0.202 |
| R0008 | 0.574 | 0.566 | 0.522 | 0.519 |
| R0014 | 0.536 | 0.486 | 0.469 | 0.393 |
| R0018 | 0.514 | 0.508 | 0.345 | 0.366 |
| Average | 0.657 | 0.651 | 0.729 | 0.726 |
| Target ID | GR_Scores of the Proposed Method | GR_Scores of SPICKER | TM-Scores of the Proposed Method | TM-Scores of SPICKER |
|---|---|---|---|---|
| T0759 | 0.547 | 0.530 | 0.362 | 0.356 |
| T0762 | 0.721 | 0.728 | 0.921 | 0.925 |
| T0763 | 0.432 | 0.416 | 0.272 | 0.198 |
| T0764 | 0.679 | 0.697 | 0.883 | 0.885 |
| T0765 | 0.530 | 0.597 | 0.740 | 0.761 |
| T0766 | 0.772 | 0.800 | 0.938 | 0.935 |
| T0768 | 0.547 | 0.544 | 0.629 | 0.626 |
| T0769 | 0.707 | 0.684 | 0.747 | 0.741 |
| T0773 | 0.729 | 0.754 | 0.608 | 0.812 |
| T0778 | 0.817 | 0.836 | 0.910 | 0.929 |
| T0782 | 0.580 | 0.589 | 0.691 | 0.687 |
| T0784 | 0.717 | 0.742 | 0.932 | 0.937 |
| T0785 | 0.387 | 0.380 | 0.257 | 0.261 |
| T0786 | 0.618 | 0.618 | 0.782 | 0.782 |
| T0787 | 0.594 | 0.593 | 0.235 | 0.235 |
| T0788 | 0.688 | 0.681 | 0.901 | 0.897 |
| T0792 | 0.750 | 0.735 | 0.665 | 0.672 |
| T0796 | 0.585 | 0.579 | 0.687 | 0.666 |
| T0797 | 0.934 | 0.934 | 0.794 | 0.826 |
| T0798 | 0.823 | 0.822 | 0.936 | 0.937 |
| T0800 | 0.523 | 0.495 | 0.592 | 0.575 |
| T0801 | 0.710 | 0.703 | 0.937 | 0.926 |
| T0803 | 0.464 | 0.431 | 0.475 | 0.467 |
| T0805 | 0.706 | 0.713 | 0.848 | 0.843 |
| T0807 | 0.693 | 0.691 | 0.911 | 0.913 |
| T0811 | 0.736 | 0.727 | 0.942 | 0.941 |
| T0812 | 0.503 | 0.525 | 0.539 | 0.536 |
| T0813 | 0.724 | 0.712 | 0.921 | 0.922 |
| T0815 | 0.794 | 0.798 | 0.888 | 0.885 |
| T0816 | 0.647 | 0.658 | 0.298 | 0.296 |
| T0817 | 0.678 | 0.675 | 0.715 | 0.718 |
| T0819 | 0.685 | 0.699 | 0.916 | 0.920 |
| T0820 | 0.472 | 0.488 | 0.325 | 0.324 |
| T0821 | 0.768 | 0.769 | 0.810 | 0.818 |
| T0822 | 0.528 | 0.470 | 0.514 | 0.442 |
| T0823 | 0.621 | 0.623 | 0.778 | 0.779 |
| T0824 | 0.477 | 0.446 | 0.308 | 0.296 |
| T0825 | 0.786 | 0.785 | 0.511 | 0.509 |
| T0829 | 0.603 | 0.611 | 0.496 | 0.584 |
| T0833 | 0.753 | 0.736 | 0.754 | 0.743 |
| T0835 | 0.531 | 0.541 | 0.697 | 0.700 |
| T0836 | 0.532 | 0.570 | 0.276 | 0.276 |
| T0837 | 0.608 | 0.604 | 0.418 | 0.427 |
| T0838 | 0.579 | 0.548 | 0.577 | 0.543 |
| T0841 | 0.715 | 0.715 | 0.861 | 0.926 |
| T0843 | 0.718 | 0.713 | 0.926 | 0.924 |
| T0847 | 0.673 | 0.683 | 0.788 | 0.788 |
| T0849 | 0.610 | 0.608 | 0.731 | 0.730 |
| T0851 | 0.678 | 0.717 | 0.913 | 0.782 |
| T0854 | 0.679 | 0.684 | 0.795 | 0.794 |
| T0855 | 0.576 | 0.551 | 0.541 | 0.494 |
| T0856 | 0.677 | 0.683 | 0.870 | 0.869 |
| T0857 | 0.516 | 0.534 | 0.475 | 0.487 |
| T0858 | 0.673 | 0.683 | 0.908 | 0.910 |
| Average | 0.644 | 0.645 | 0.688 | 0.688 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, X.; Li, L.; Lu, Y. Selecting Near-Native Protein Structures from Predicted Decoy Sets Using Ordered Graphlet Degree Similarity. Genes 2019, 10, 132. https://doi.org/10.3390/genes10020132
Han X, Li L, Lu Y. Selecting Near-Native Protein Structures from Predicted Decoy Sets Using Ordered Graphlet Degree Similarity. Genes. 2019; 10(2):132. https://doi.org/10.3390/genes10020132
Chicago/Turabian StyleHan, Xu, Li Li, and Yonggang Lu. 2019. "Selecting Near-Native Protein Structures from Predicted Decoy Sets Using Ordered Graphlet Degree Similarity" Genes 10, no. 2: 132. https://doi.org/10.3390/genes10020132
APA StyleHan, X., Li, L., & Lu, Y. (2019). Selecting Near-Native Protein Structures from Predicted Decoy Sets Using Ordered Graphlet Degree Similarity. Genes, 10(2), 132. https://doi.org/10.3390/genes10020132