Using Decision Tree Aggregation with Random Forest Model to Identify Gut Microbes Associated with Colorectal Cancer


Abstract
:1. Introduction
2. Materials and Methods
2.1. Mutual Information-Based Feature Selection
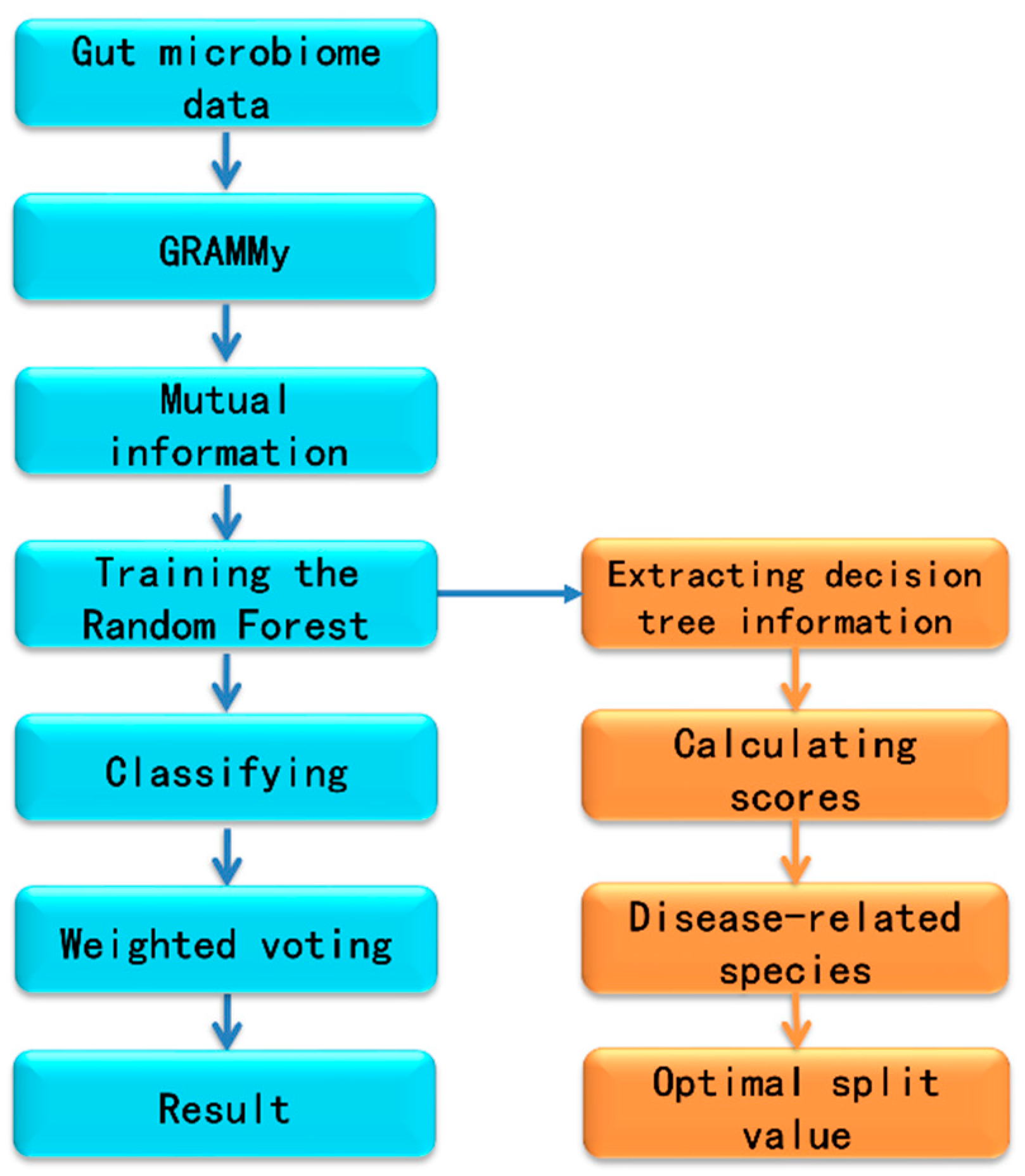
2.2. Workflow for Metagenomics Analysis
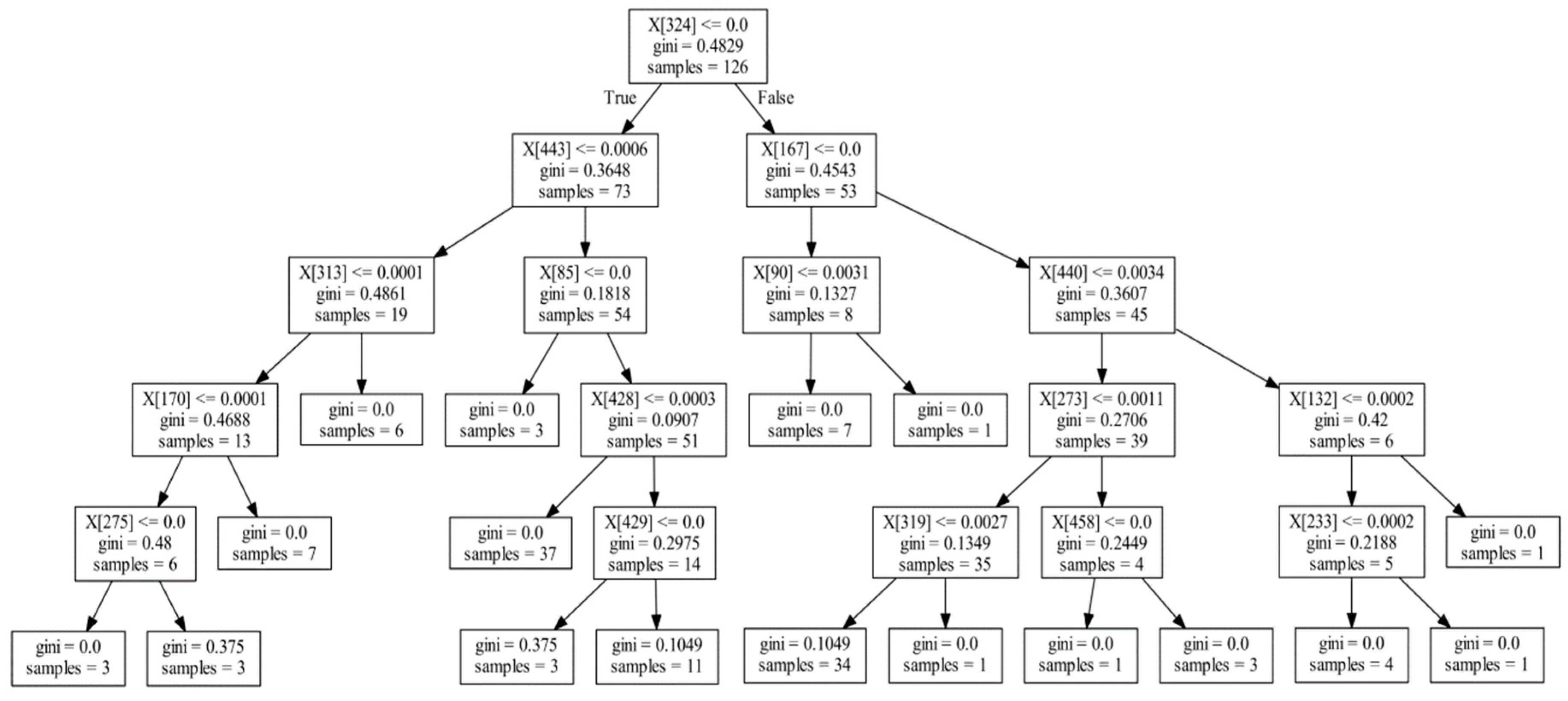
2.3. Extracting the Information of Decision Trees from Random Forest
2.4. Training and Testing Datasets
3. Results and Discussion
3.1. Informative Rank Extraction from Decision Tree
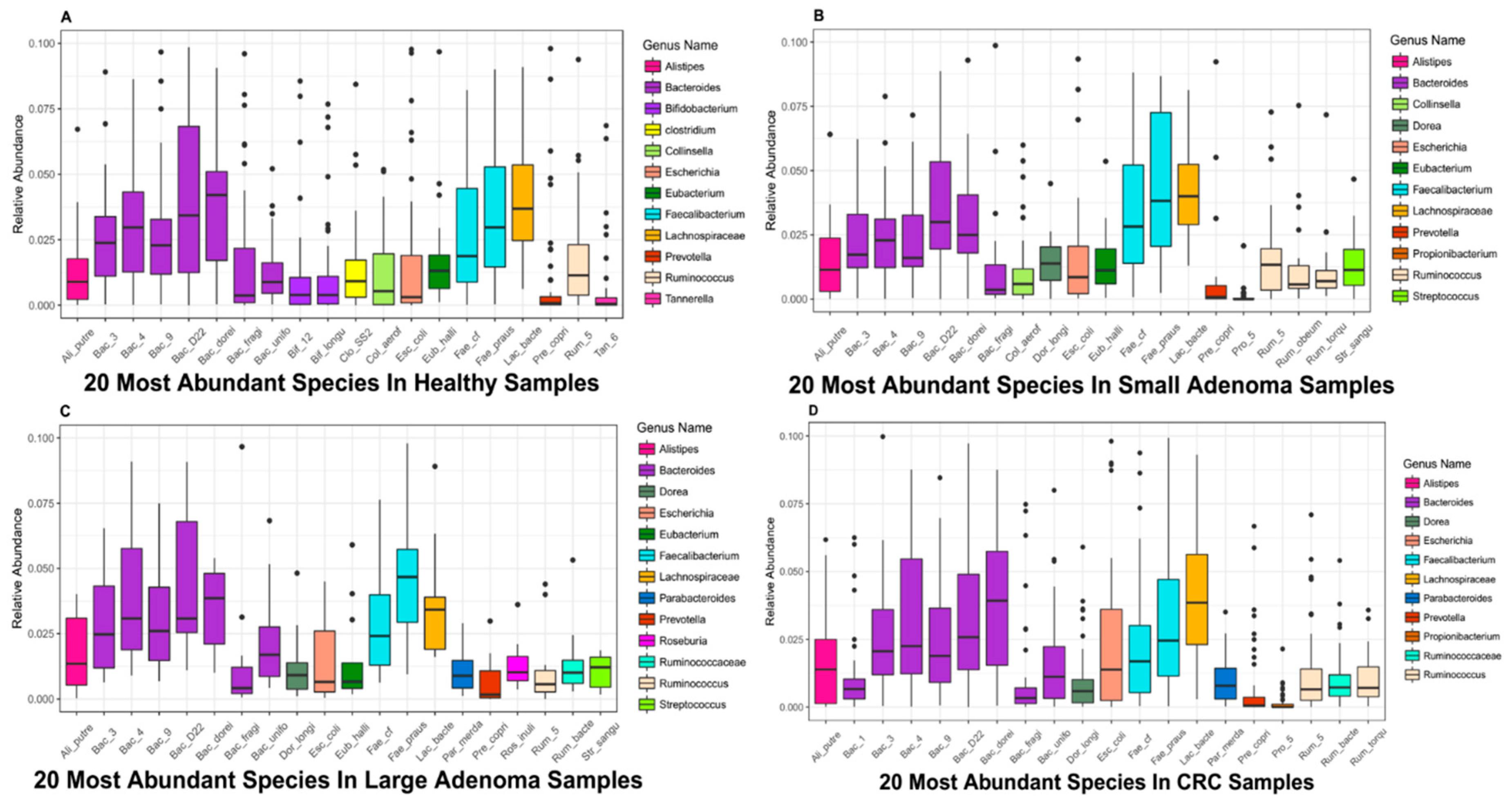
3.2. Top 20 Microbial Species with High Relative Abundance
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sommer, F.; Bäckhed, F. The gut microbiota-Masters of host development and physiology. Nat. Rev. Microbiol. 2013, 11, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yan, Q.; Xie, S.; Hu, W.; Yu, Y.; Hu, Z. Gut microbiota contributes to the growth of fast-growing transgenic common carp (Cyprinus carpio L.). PLoS ONE 2013, 8, e64577. [Google Scholar] [CrossRef] [PubMed]
- Vaarala, O. Human intestinal microbiota and type 1 diabetes. Curr. Diabetes Rep. 2013, 13, 601–607. [Google Scholar]
- Ghoshal, U.C.; Shukla, R.; Ghoshal, U.; Gwee, K.-A.; Ng, S.C.; Quigley, E.M. The gut microbiota and irritable bowel syndrome: Friend or foe? Int. J. Inflamm. 2012, 2012, 151085. [Google Scholar] [CrossRef] [PubMed]
- Manichanh, C.; Borruel, N.; Casellas, F.; Guarner, F. The gut microbiota in IBD. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, C.; Tang, C.; Li, N.; Li, J. Molecular-phylogenetic characterization of the microbiota in ulcerated and non-ulcerated regions in the patients with Crohn’s disease. PLoS ONE 2012, 7, e34939. [Google Scholar] [CrossRef] [PubMed]
- Kostic, A.D.; Gevers, D.; Pedamallu, C.S.; Michaud, M.; Duke, F.; Earl, A.M.; Ojesina, A.I.; Jung, J.; Bass, A.J.; Tabernero, J. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012, 22, 292–298. [Google Scholar] [CrossRef]
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef]
- Amieva, M.; Peek, R.M. Pathobiology of Helicobacter pylori–induced gastric cancer. Gastroenterology 2016, 150, 64–78. [Google Scholar] [CrossRef]
- Wiseman, M. The Second World Cancer Research Fund/American Institute for Cancer Research Expert Report. Food, Nutrition, Physical Activity, and the Prevention of Cancer: A Global Perspective: Nutrition Society and BAPEN Medical Symposium on ‘Nutrition support in cancer therapy’. Proc. Nutr. Soc. 2008, 67, 253–256. [Google Scholar]
- Parente, F.; Marino, B.; Ilardo, A.; Fracasso, P.; Zullo, A.; Hassan, C.; Moretti, R.; Cremaschini, M.; Ardizzoia, A.; Saracino, I. A combination of faecal tests for the detection of colon cancer: A new strategy for an appropriate selection of referrals to colonoscopy? A prospective multicentre Italian study. Eur. J. Gastroenterol. Hepatol. 2012, 24, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Becker, D.; Grapendorf, J.; Greving, H.; Sassenberg, K. Perceived Threat and Internet Use Predict Intentions to Get Bowel Cancer Screening (Colonoscopy): Longitudinal Questionnaire Study. J. Med. Internet Res. 2018, 20, e46. [Google Scholar] [CrossRef] [PubMed]
- Cam, N.B.; Cho, H.; Lee, Y.Y.; Suh, M.; Park, B.; Jun, J.K.; Kim, Y.; Choi, K.S. Stages of Adoption for FOBT and Colonoscopy Tests for Colorectal Cancer Screening in Korea. Cancer Res. Treat. 2018, 50, 416–427. [Google Scholar]
- Shah, M.S.; DeSantis, T.Z.; Weinmaier, T.; McMurdie, P.J.; Cope, J.L.; Altrichter, A.; Yamal, J.-M.; Hollister, E.B. Leveraging sequence-based faecal microbial community survey data to identify a composite biomarker for colorectal cancer. Gut 2018, 67, 882–891. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Matsumoto, S. Gut microbiota and colorectal cancer. Genes Environ. 2016, 38, 11. [Google Scholar] [CrossRef]
- Sobhani, I.; Tap, J.; Roudot-Thoraval, F.; Roperch, J.P.; Letulle, S.; Langella, P.; Corthier, G.; Van Nhieu, J.T.; Furet, J.P. Microbial dysbiosis in colorectal cancer (CRC) patients. PLoS ONE 2011, 6, e16393. [Google Scholar] [CrossRef]
- Zackular, J.P.; Rogers, M.A.; Ruffin, M.T.; Schloss, P.D. The human gut microbiome as a screening tool for colorectal cancer. Cancer Prev. Res. 2014. [Google Scholar] [CrossRef]
- Zeller, G.; Tap, J.; Voigt, A.Y.; Sunagawa, S.; Kultima, J.R.; Costea, P.I.; Amiot, A.; Böhm, J.; Brunetti, F.; Habermann, N. Potential of fecal microbiota for early-stage detection of colorectal cancer. Mol. Syst. Biol. 2014, 10, 766. [Google Scholar] [CrossRef]
- Hastie, T.; Tibshirani, R.; Wainwright, M. Statistical Learning with Sparsity: The Lasso and Generalizations; CRC Press: Boca Raton, FL, USA, 2015. [Google Scholar]
- Feng, Q.; Liang, S.; Jia, H.; Stadlmayr, A.; Tang, L.; Lan, Z.; Zhang, D.; Xia, H.; Xu, X.; Jie, Z.; et al. Gut microbiome development along the colorectal adenoma–carcinoma sequence. Nat. Commun. 2015, 6, 6528. [Google Scholar] [CrossRef]
- Ai, L.; Tian, H.; Chen, Z.; Chen, H.; Xu, J.; Fang, J.-Y. Systematic evaluation of supervised classifiers for fecal microbiota-based prediction of colorectal cancer. Oncotarget 2017, 8, 9546–9556. [Google Scholar] [CrossRef]
- Nassif, H.; Wu, Y.; Page, D.; Burnside, E. Logical Differential Prediction Bayes Net, improving breast cancer diagnosis for older women. In Proceedings of the AMIA Annual Symposium, Chicago, IL, USA, 3–7 November 2012; p. 1330. [Google Scholar]
- Díaz-Uriarte, R.; De Andres, S.A. Gene selection and classification of microarray data using random forest. BMC Bioinform. 2006, 7, 3. [Google Scholar] [CrossRef]
- Kong, Q.; Gong, H.; Ding, X.; Hou, R. Classification Application Based on Mutual Information and Random Forest Method for High Dimensional Data. In Proceedings of the 2017 9th International Conference on Intelligent Human-Machine Systems and Cybernetics (IHMSC), Hangzhou, China, 26–27 August 2017; pp. 171–174. [Google Scholar]
- Kraskov, A.; Stögbauer, H.; Andrzejak, R.G.; Grassberger, P. Hierarchical Clustering Based on Mutual Information. Physics 2012, arXiv:q-bio/0311039. [Google Scholar]
- Xia, L.C.; Cram, J.A.; Chen, T.; Fuhrman, J.A.; Sun, F. Accurate genome relative abundance estimation based on shotgun metagenomic reads. PLoS ONE 2011, 6, e27992. [Google Scholar] [CrossRef] [PubMed]
- Biers, E.J.; Sun, S.; Howard, E.C. Prokaryotic genomes and diversity in surface ocean waters: Interrogating the global ocean sampling metagenome. Appl. Environ. Microbiol. 2009, 75, 2221–2229. [Google Scholar] [CrossRef] [PubMed]
- Sender, R.; Fuchs, S.; Milo, R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.Y. Feature selection based on feature similarity measure. Comput. Eng. Appl. 2010, 46, 153–156. [Google Scholar]
- Cortez, P.; Embrechts, M.J. Using sensitivity analysis and visualization techniques to open black box data mining models. Inf. Sci. 2013, 225, 1–17. [Google Scholar] [CrossRef]
- Nguyen, T.-T.; Huang, J.Z.; Nguyen, T.T. Unbiased feature selection in learning random forests for high-dimensional data. Sci. World J. 2015, 2015, 471371. [Google Scholar] [CrossRef]
- Biau, G.; Scornet, E. A random forest guided tour. Test 2016, 25, 197–227. [Google Scholar] [CrossRef]
- O’Connell, J.B.; Maggard, M.A.; Ko, C.Y. Colon cancer survival rates with the new American Joint Committee on Cancer sixth edition staging. J. Natl. Cancer Inst. 2004, 96, 1420–1425. [Google Scholar] [CrossRef]
- Welling, S.H.; Refsgaard, H.H.; Brockhoff, P.B.; Clemmensen, L.H. Forest floor visualizations of random forests. arXiv, 2016; arXiv:1605.09196. [Google Scholar]
- Shang, F.-M.; Liu, H.-L. Fusobacterium nucleatum and colorectal cancer: A review. World J. Gastrointest. Oncol. 2018, 10, 71. [Google Scholar] [CrossRef] [PubMed]
- Mancabelli, L.; Milani, C.; Lugli, G.A.; Turroni, F.; Cocconi, D.; van Sinderen, D.; Ventura, M. Identification of universal gut microbial biomarkers of common human intestinal diseases by meta-analysis. FEMS Microbiol. Ecol. 2017, 93, fix153. [Google Scholar] [CrossRef] [PubMed]
- Wexler, H.M. Bacteroides: The good, the bad, and the nitty-gritty. Clin. Microbiol. Rev. 2007, 20, 593–621. [Google Scholar] [CrossRef] [PubMed]
- Pinzone, M.R.; Celesia, B.M.; Di Rosa, M.; Cacopardo, B.; Nunnari, G. Microbial translocation in chronic liver diseases. Int. J. Microbiol. 2012, 2012, 694629. [Google Scholar] [CrossRef] [PubMed]
- Ghouri, Y.A.; Richards, D.M.; Rahimi, E.F.; Krill, J.T.; Jelinek, K.A.; DuPont, A.W. Systematic review of randomized controlled trials of probiotics, prebiotics, and synbiotics in inflammatory bowel disease. Clin. Exp. Gastroenterol. 2014, 7, 473–487. [Google Scholar] [PubMed]
- Brüggemann, H.; Gottschalk, G. Clostridia: Molecular Biology in the Post-Genomic Era; Horizon Scientific Press: Norfolk, UK, 2009. [Google Scholar]
- Tyler, A.D.; Knox, N.; Kabakchiev, B.; Milgrom, R.; Kirsch, R.; Cohen, Z.; McLeod, R.S.; Guttman, D.S.; Krause, D.O.; Silverberg, M.S. Characterization of the gut-associated microbiome in inflammatory pouch complications following ileal pouch-anal anastomosis. PLoS ONE 2013, 8, e66934. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Tree ID | Node Index | Father | Layer | Microbe ID | Split Value | Gini Index | Sample Number |
|---|---|---|---|---|---|---|---|
| 0 | 0 | 0 | 0 | 324 | 0 | 0.4829 | 126 |
| 0 | 1 | 0 | 1 | 443 | 0.0006 | 0.3648 | 76 |
| 0 | 2 | 1 | 2 | 313 | 0.0001 | 0.4861 | 19 |
| 0 | 3 | 2 | 3 | 170 | 0.0001 | 0.4688 | 13 |
| 0 | 4 | 3 | 4 | 275 | 0 | 0.48 | 6 |
| 0 | 9 | 1 | 2 | 85 | 0 | 0.1818 | 54 |
| 0 | 11 | 9 | 3 | 428 | 0.0003 | 0.0907 | 51 |
| 0 | 13 | 11 | 4 | 429 | 0 | 0.2975 | 14 |
| 0 | 16 | 0 | 1 | 167 | 0 | 0.4543 | 53 |
| 0 | 17 | 16 | 2 | 90 | 0.0031 | 0.1327 | 8 |
| 0 | 20 | 16 | 2 | 440 | 0.0034 | 0.3607 | 45 |
| 0 | 21 | 20 | 3 | 273 | 0.0011 | 0.2706 | 39 |
| 0 | 22 | 21 | 4 | 319 | 0.0027 | 01349 | 35 |
| 0 | 25 | 21 | 4 | 458 | 0 | 0.2449 | 4 |
| 0 | 28 | 20 | 3 | 132 | 0.002 | 0.42 | 6 |
| 0 | 29 | 28 | 4 | 233 | 0.0002 | 0.2188 | 5 |
| Layer | 0 | 1 | 2 | 3 | 4 | Score | Microbial Species |
|---|---|---|---|---|---|---|---|
| Microbe ID | |||||||
| 334 | 213 | 262 | 270 | 239 | 181 | 232.437 | Porphyromonas asaccharolytica |
| 200 | 168 | 160 | 146 | 122 | 111 | 154.21 | Eubacterium hallii |
| 324 | 151 | 156 | 155 | 134 | 94 | 146.661 | Parvimonas oral |
| 220 | 177 | 127 | 129 | 117 | 80 | 145.268 | Fusobacterium 7 |
| 350 | 144 | 147 | 157 | 131 | 132 | 144.319 | Prevotella melaninogenica |
| 443 | 117 | 149 | 170 | 151 | 136 | 136.618 | Streptococcus vestibularis |
| 343 | 119 | 129 | 161 | 138 | 122 | 129.228 | Prevotella copri |
| 332 | 130 | 138 | 118 | 118 | 89 | 125.879 | Peptostreptococcus stomatis |
| 226 | 131 | 128 | 125 | 115 | 68 | 123.025 | Fusobacterium nucleatum |
| 323 | 135 | 122 | 81 | 133 | 107 | 122.101 | Parvimonas micra |
| 233 | 117 | 104 | 123 | 103 | 111 | 112.955 | Gemella morbillorum |
| 213 | 82 | 130 | 131 | 142 | 125 | 109.201 | Flavonifractor plautii |
| 217 | 135 | 103 | 82 | 78 | 47 | 107.802 | Fusobacterium 21 |
| 139 | 103 | 113 | 111 | 96 | 82 | 104.154 | Clostridium SS2 |
| Study Population | Healthy | Adenoma | Colorectal Cancer | Country of Residence | |||||
|---|---|---|---|---|---|---|---|---|---|
| Small (<1 cm) | Large (≥1 cm) | Early Stage | Late Stage | ||||||
| 0 | I | II | III | IV | |||||
| F (N = 156) | 61 | 27 | 15 | 0 | 15 | 7 | 10 | 21 | France |
| A (N = 156) | 63 | 47 | 46 | Austria | |||||
| Microbe ID | Microbial Species | Score | Abundance Thresholds |
|---|---|---|---|
| 334 | Porphyromonas asaccharolytica | 232.437 | 3.052 × 10−5 |
| 200 | Eubacterium hallii | 154.21 | 0.006662 |
| 324 | Parvimonas sp. | 146.661 | 1.391 × 10−5 |
| 220 | Fusobacterium 7 | 145.268 | 0 |
| 350 | Prevotella melaninogenica | 144.319 | 0 |
| 443 | Streptococcus vestibularis | 136.618 | 0.0006701 |
| 343 | Prevotellacopri | 129.228 | 0.000179 |
| 332 | Peptostreptococcus stomatis | 125.879 | 9.154 × 10−5 |
| 226 | Fusobacterium nucleatum | 123.025 | 9.15 × 10−5 |
| 323 | Parvimonas micra | 122.101 | 7.63 × 10−5 |
| 233 | Gemella morbillorum | 112.955 | 5.19 × 10−5 |
| 213 | Flavonifractor plautii | 109.201 | 9.83 × 10−5 |
| 217 | Fusobacterium 21 | 107.802 | 0 |
| 139 | Clostridium SS2 | 104.154 | 0.000912 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ai, D.; Pan, H.; Han, R.; Li, X.; Liu, G.; Xia, L.C. Using Decision Tree Aggregation with Random Forest Model to Identify Gut Microbes Associated with Colorectal Cancer. Genes 2019, 10, 112. https://doi.org/10.3390/genes10020112
Ai D, Pan H, Han R, Li X, Liu G, Xia LC. Using Decision Tree Aggregation with Random Forest Model to Identify Gut Microbes Associated with Colorectal Cancer. Genes. 2019; 10(2):112. https://doi.org/10.3390/genes10020112
Chicago/Turabian StyleAi, Dongmei, Hongfei Pan, Rongbao Han, Xiaoxin Li, Gang Liu, and Li C. Xia. 2019. "Using Decision Tree Aggregation with Random Forest Model to Identify Gut Microbes Associated with Colorectal Cancer" Genes 10, no. 2: 112. https://doi.org/10.3390/genes10020112
APA StyleAi, D., Pan, H., Han, R., Li, X., Liu, G., & Xia, L. C. (2019). Using Decision Tree Aggregation with Random Forest Model to Identify Gut Microbes Associated with Colorectal Cancer. Genes, 10(2), 112. https://doi.org/10.3390/genes10020112