Human Endometrial Microbiota at Term of Normal Pregnancies

 ,
,  ,
, 
 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Cohort and Sample Collection
2.2. DNA Extraction
2.3. Amplicon Library Preparation and Illumina-Based Sequencing
2.4. Bioinformatic Analysis
3. Results
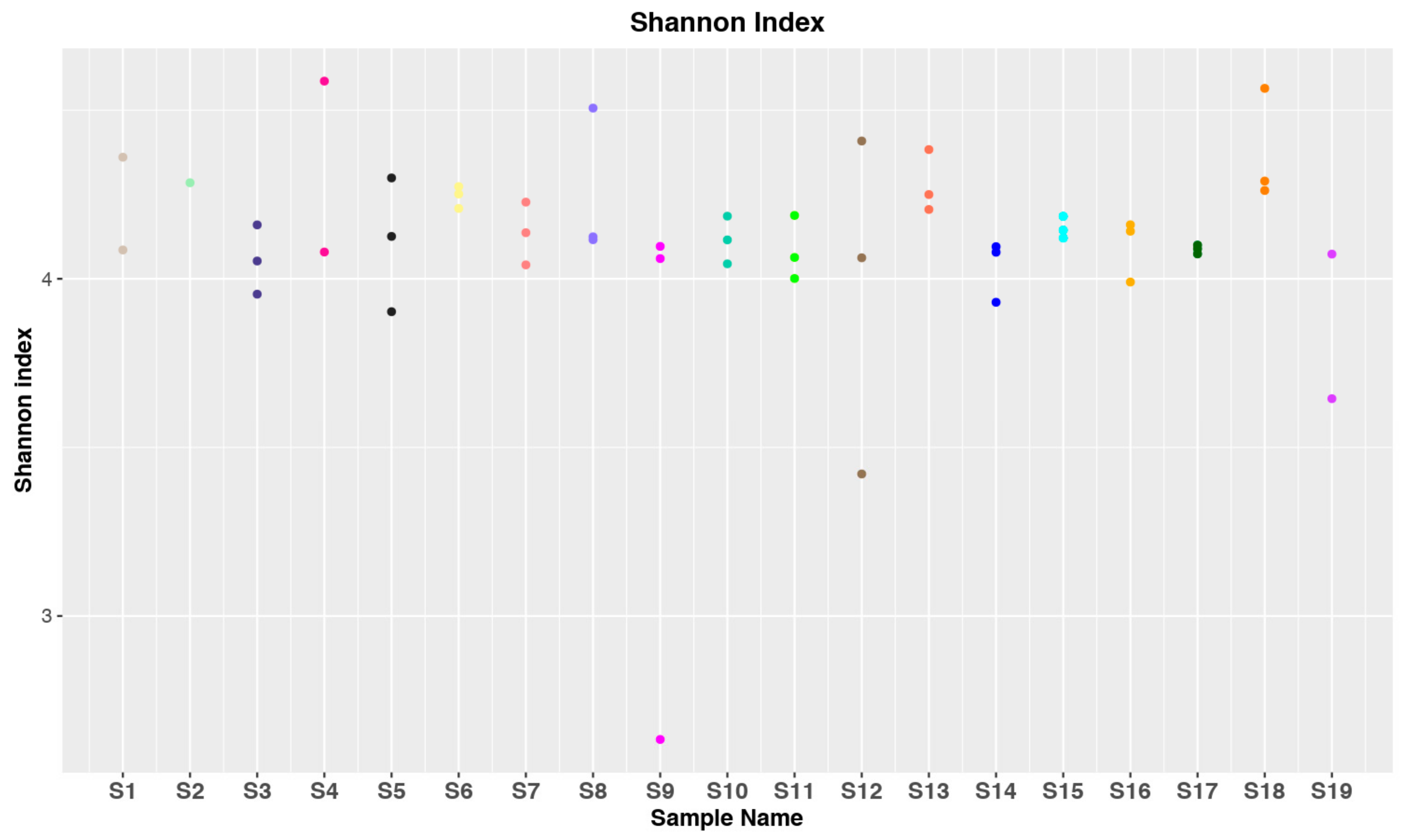
3.1. Data Collection and Statistical Analysis
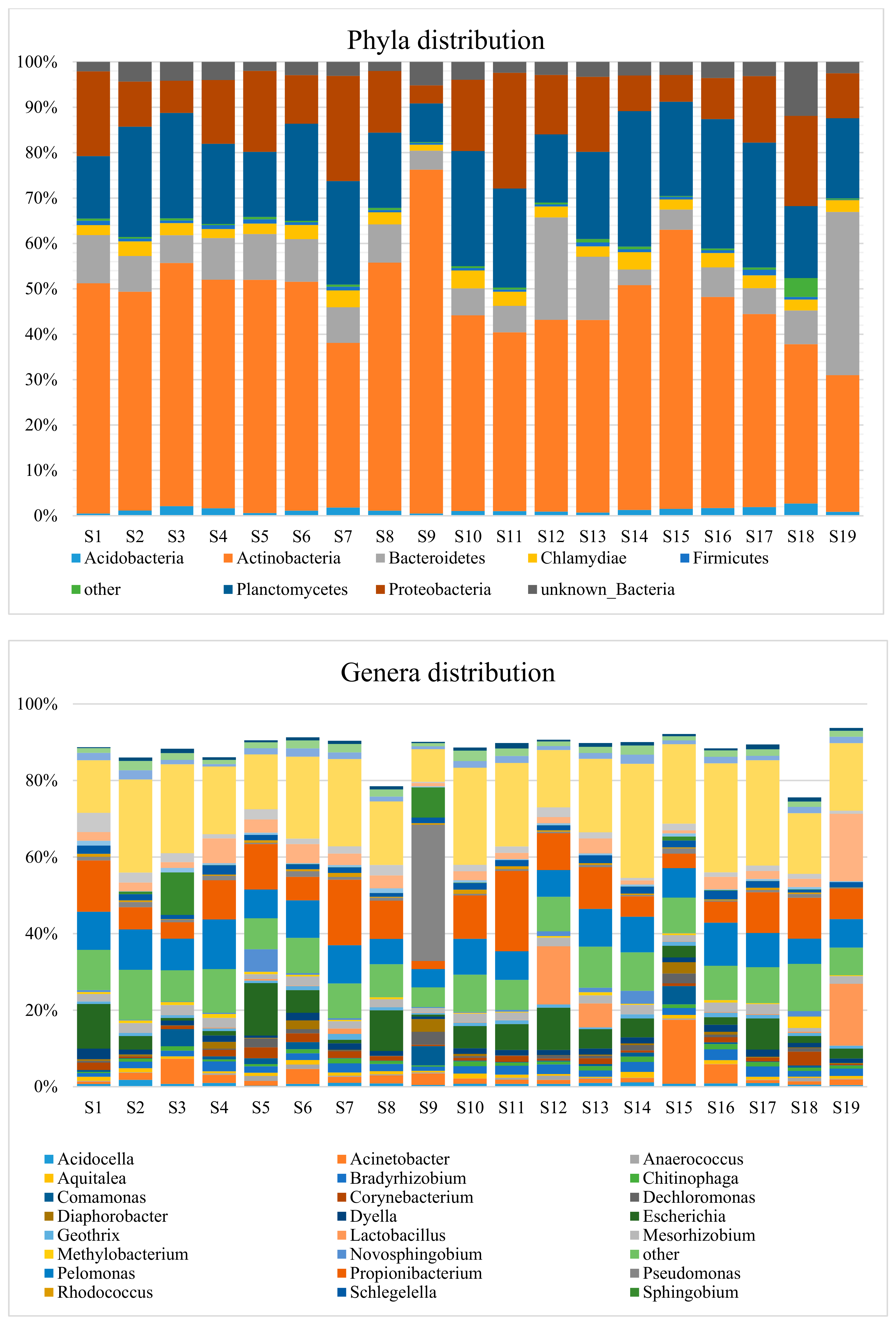
3.2. Taxonomic Distribution
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kim, Y.; Koh, I.; Rho, M. Deciphering the human microbiome using next-generation sequencing data and bioinformatics approaches. Methods 2015, 79–80, 52–59. [Google Scholar] [CrossRef]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.A.; Blaser, M.J.; Caporaso, J.G.; Jansson, J.K.; Lynch, S.V.; Knight, R. Current understanding of the human microbiome. Nat. Med. 2018, 24, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Hadrich, D. Microbiome Research Is Becoming the Key to Better Understanding Health and Nutrition. Front. Genet. 2018, 9, 212. [Google Scholar] [CrossRef] [PubMed]
- Franasiak, J.M.; Scott, R.T. Reproductive tract microbiome in assisted reproductive technologies. Fertil. Steril. 2015, 104, 1364–1371. [Google Scholar] [CrossRef]
- Moreno, I.; Franasiak, J.M. Endometrial microbiota—New player in town. Fertil. Steril. 2017, 108, 32–39. [Google Scholar] [CrossRef]
- Perez-Muñoz, M.E.; Arrieta, M.-C.; Ramer-Tait, A.E.; Walter, J. A critical assessment of the “sterile womb” and “in utero colonization” hypotheses: Implications for research on the pioneer infant microbiome. Microbiome 2017, 5, 48. [Google Scholar] [CrossRef]
- Altmäe, S. Commentary: Uterine Microbiota: Residents, Tourists, or Invaders? Front. Immunol. 2018, 9, 1874. [Google Scholar] [CrossRef]
- Baker, J.M.; Chase, D.M.; Herbst-Kralovetz, M.M. Uterine Microbiota: Residents, Tourists, or Invaders? Front. Immunol. 2018, 9, 208. [Google Scholar] [CrossRef]
- Benner, M.; Ferwerda, G.; Joosten, I.; van der Molen, R.G. How uterine microbiota might be responsible for a receptive, fertile endometrium. Hum. Reprod. Update 2018, 24, 393–415. [Google Scholar] [CrossRef]
- Koedooder, R.; Mackens, S.; Budding, A.; Fares, D.; Blockeel, C.; Laven, J.; Schoenmakers, S. Identification and evaluation of the microbiome in the female and male reproductive tracts. Hum. Reprod. Update 2019, 25, 298–325. [Google Scholar] [CrossRef] [PubMed]
- Peric, A.; Weiss, J.; Vulliemoz, N.; Baud, D.; Stojanov, M. Bacterial Colonization of the Female Upper Genital Tract. IJMS 2019, 20, 3405. [Google Scholar] [CrossRef] [PubMed]
- Franasiak, J.M.; Werner, M.D.; Juneau, C.R.; Tao, X.; Landis, J.; Zhan, Y.; Treff, N.R.; Scott, R.T. Endometrial microbiome at the time of embryo transfer: Next-generation sequencing of the 16S ribosomal subunit. J. Assist. Reprod. Genet. 2016, 33, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Franasiak, J.M.; Zhan, Y.; Scott, R.T.; Rajchel, J.; Bedard, J.; Newby, R.; Scott, R.T.; Treff, N.R.; Chu, T. Characterizing the endometrial microbiome by analyzing the ultra-low bacteria from embryo transfer catheter tips in IVF cycles: Next generation sequencing (NGS) analysis of the 16S ribosomal gene. Hum. Microbiome J. 2017, 3, 15–21. [Google Scholar] [CrossRef]
- Moreno, I.; Codoñer, F.M.; Vilella, F.; Valbuena, D.; Martinez-Blanch, J.F.; Jimenez-Almazán, J.; Alonso, R.; Alamá, P.; Remohí, J.; Pellicer, A.; et al. Evidence that the endometrial microbiota has an effect on implantation success or failure. Am. J. Obstet. Gynecol. 2016, 215, 684–703. [Google Scholar] [CrossRef]
- Verstraelen, H.; Vilchez-Vargas, R.; Desimpel, F.; Jauregui, R.; Vankeirsbilck, N.; Weyers, S.; Verhelst, R.; De Sutter, P.; Pieper, D.H.; Van De Wiele, T. Characterisation of the human uterine microbiome in non-pregnant women through deep sequencing of the V1-2 region of the 16S rRNA gene. PeerJ 2016, 4, e1602. [Google Scholar] [CrossRef]
- Kitaya, K.; Nagai, Y.; Arai, W.; Sakuraba, Y.; Ishikawa, T. Characterization of Microbiota in Endometrial Fluid and Vaginal Secretions in Infertile Women with Repeated Implantation Failure. Mediat. Inflamm. 2019, 2019, 4893437. [Google Scholar] [CrossRef]
- Khan, K.N.; Fujishita, A.; Masumoto, H.; Muto, H.; Kitajima, M.; Masuzaki, H.; Kitawaki, J. Molecular detection of intrauterine microbial colonization in women with endometriosis. Eur. J. Obstet. Gynecol. Reprod. Biol. 2016, 199, 69–75. [Google Scholar] [CrossRef]
- Fang, R.-L.; Chen, L.-X.; Shu, W.-S.; Yao, S.-Z.; Wang, S.-W.; Chen, Y.-Q. Barcoded sequencing reveals diverse intrauterine microbiomes in patients suffering with endometrial polyps. Am. J. Transl. Res. 2016, 8, 1581–1592. [Google Scholar]
- Walther-António, M.R.S.; Chen, J.; Multinu, F.; Hokenstad, A.; Distad, T.J.; Cheek, E.H.; Keeney, G.L.; Creedon, D.J.; Nelson, H.; Mariani, A.; et al. Potential contribution of the uterine microbiome in the development of endometrial cancer. Genome Med. 2016, 8, 122. [Google Scholar] [CrossRef]
- Chen, C.; Song, X.; Wei, W.; Zhong, H.; Dai, J.; Lan, Z.; Li, F.; Yu, X.; Feng, Q.; Wang, Z.; et al. The microbiota continuum along the female reproductive tract and its relation to uterine-related diseases. Nat. Commun. 2017, 8, 875. [Google Scholar] [CrossRef] [PubMed]
- Miles, S.M.; Hardy, B.L.; Merrell, D.S. Investigation of the microbiota of the reproductive tract in women undergoing a total hysterectomy and bilateral salpingo-oopherectomy. Fertil. Steril. 2017, 107, 813–820.e1. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Chen, C.; Wei, W.; Wang, Z.; Dai, J.; Hao, L.; Song, L.; Zhang, X.; Zeng, L.; Du, H.; et al. The metagenome of the female upper reproductive tract. GigaScience 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Winters, A.D.; Romero, R.; Gervasi, M.T.; Gomez-Lopez, N.; Tran, M.R.; Garcia-Flores, V.; Pacora, P.; Jung, E.; Hassan, S.S.; Hsu, C.-D.; et al. Does the endometrial cavity have a molecular microbial signature? Sci. Rep. 2019, 9, 9905. [Google Scholar] [CrossRef]
- Chakravorty, S.; Helb, D.; Burday, M.; Connell, N.; Alland, D. A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria. J. Microbiol. Methods 2007, 69, 330–339. [Google Scholar] [CrossRef]
- Stecher, B.; Chaffron, S.; Käppeli, R.; Hapfelmeier, S.; Freedrich, S.; Weber, T.C.; Kirundi, J.; Suar, M.; McCoy, K.D.; von Mering, C.; et al. Like will to like: Abundances of closely related species can predict susceptibility to intestinal colonization by pathogenic and commensal bacteria. PLoS Pathog. 2010, 6, e1000711. [Google Scholar] [CrossRef]
- Manzari, C.; Fosso, B.; Marzano, M.; Annese, A.; Caprioli, R.; D’Erchia, A.M.; Gissi, C.; Intranuovo, M.; Picardi, E.; Santamaria, M.; et al. The influence of invasive jellyfish blooms on the aquatic microbiome in a coastal lagoon (Varano, SE Italy) detected by an Illumina-based deep sequencing strategy. Biol. Invasions 2015, 17, 923–940. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Fosso, B.; Santamaria, M.; Marzano, M.; Alonso-Alemany, D.; Valiente, G.; Donvito, G.; Monaco, A.; Notarangelo, P.; Pesole, G. BioMaS: A modular pipeline for Bioinformatic analysis of Metagenomic AmpliconS. BMC Bioinform. 2015, 16, 203. [Google Scholar] [CrossRef]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal Database Project: Data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014, 42, D633–D642. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Alemany, D.; Barre, A.; Beretta, S.; Bonizzoni, P.; Nikolski, M.; Valiente, G. Further Steps in TANGO: Improved taxonomic assignment in metagenomics. Bioinformatics 2014, 30, 17–23. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mende, D.R.; Sunagawa, S.; Zeller, G.; Bork, P. Accurate and universal delineation of prokaryotic species. Nat. Methods 2013, 10, 881–884. [Google Scholar] [CrossRef] [PubMed]
- Davis, N.M.; Proctor, D.M.; Holmes, S.P.; Relman, D.A.; Callahan, B.J. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 2018, 6, 226. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.; Kindt, R.; Legendre, P.; Minchin, P.; O’Hara, R.; Simpson, G.; Stevens, M.; Wagner, H. Vegan: Community Ecology Package. R Package Version 2.2-1. Available online: http://outputs.worldagroforestry.org/cgi-bin/koha/opac-detail.pl?biblionumber=39154 (accessed on 29 July 2019).
- McMurdie, P.J.; Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Grennan, K.; Badner, J.; Zhang, D.; Gershon, E.; Jin, L.; Liu, C. Removing batch effects in analysis of expression microarray data: An evaluation of six batch adjustment methods. PLoS ONE 2011, 6, e17238. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, S.M.; Duvallet, C.; Alm, E.J. Correcting for batch effects in case-control microbiome studies. PLoS Comput. Biol. 2018, 14, e1006102. [Google Scholar] [CrossRef]
- Faith, D.P. Conservation evaluation and phylogenetic diversity. Biol. Conserv. 1992, 61, 1–10. [Google Scholar] [CrossRef]
- Marzano, M.; Fosso, B.; Manzari, C.; Grieco, F.; Intranuovo, M.; Cozzi, G.; Mulè, G.; Scioscia, G.; Valiente, G.; Tullo, A.; et al. Complexity and Dynamics of the Winemaking Bacterial Communities in Berries, Musts, and Wines from Apulian Grape Cultivars through Time and Space. PLoS ONE 2016, 11, e0157383. [Google Scholar] [CrossRef]
- Shannon, C.E. A Mathematical Theory of Communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef]
- Laurence, M.; Hatzis, C.; Brash, D.E. Common Contaminants in Next-Generation Sequencing that Hinder Discovery of Low-Abundance Microbes. PLoS ONE 2014, 9, e97876. [Google Scholar] [CrossRef] [PubMed]
- Aagaard, K.; Ma, J.; Antony, K.M.; Ganu, R.; Petrosino, J.; Versalovic, J. The Placenta Harbors a Unique Microbiome. Sci. Transl. Med. 2014, 6, ra65–ra237. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.C.; Rautava, S.; Aakko, J.; Isolauri, E.; Salminen, S. Human gut colonisation may be initiated in utero by distinct microbial communities in the placenta and amniotic fluid. Sci. Rep. 2016, 6, 23129. [Google Scholar] [CrossRef] [PubMed]
- Pelzer, E.S.; Willner, D.; Buttini, M.; Huygens, F. A role for the endometrial microbiome in dysfunctional menstrual bleeding. Antonie van Leeuwenhoek 2018, 111, 933–943. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| ID | Age | BMI | Weeks at Birth | Previous Births/Abortions | Indication for Cesarean Section |
|---|---|---|---|---|---|
| S1 | 35 | 27.34 | 39 | 1/0 | Previous cesarean section |
| S2 | 49 | 20.76 | 40 | 0/2 | Voluminous Cervical Leiomyoma |
| S3 | 33 | 23.88 | 40 | 0/0 | Previous laparoscopic myomectomy |
| S4 | 22 | 21.48 | 39 | 1/0 | Previous cesarean section |
| S5 | 39 | 24.97 | 39 | 1/1 | Previous cesarean section |
| S6 | 25 | 31.49 | 41 | 0/0 | Hereditary angioedema |
| S7 | 27 | 19.47 | 39 | 0/0 | Previous cerebral hemorrhage |
| S8 | 32 | 17.93 | 39 | 1/0 | Severe myopia |
| S9 | 25 | 28.16 | 40 | 2/1 | Previous cesarean sections |
| S10 | 44 | 17.72 | 40 | 0/2 | Tocophobia |
| S11 | 39 | 24.02 | 39 | 3/2 | Hip dysplasia |
| S12 | 34 | 21.19 | 39 | 0/0 | Previous laparoscopic myomectomy |
| S13 | 35 | 24.28 | 39 | 1/1 | Placenta previa |
| S14 | 26 | 24.24 | 39 | 1/0 | Previous cesarean sections |
| S15 | 20 | 17.99 | 40 | 0/0 | Breech presentation |
| S16 | 37 | 24.61 | 39 | 1/1 | Previous cesarean section |
| S17 | 34 | 19.49 | 40 | 0/2 | Fetal macrosomia |
| S18 | 32 | 29.30 | 42 | 0/0 | Fetal macrosomia |
| S19 | 25 | 17.78 | 39 | 0/0 | Fetal malformation (neck hemangioma) |
| Genus | Abundance % (*) | Representation % (**) |
|---|---|---|
| Cutibacterium | 9.35 | 100 |
| Pelomonas | 8.70 | 100 |
| Escherichia | 5.27 | 84 |
| Staphylococcus | 3.41 | 89 |
| Acinetobacter | 2.82 | 84 |
| Mesorhizobium | 2.07 | 95 |
| Bradyrhizobium | 1.96 | 95 |
| Streptococcus | 1.82 | 89 |
| Schlegelella | 1.60 | 89 |
| Dyella | 1.46 | 95 |
| Corynebacterium | 1.34 | 53 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leoni, C.; Ceci, O.; Manzari, C.; Fosso, B.; Volpicella, M.; Ferrari, A.; Fiorella, P.; Pesole, G.; Cicinelli, E.; Ceci, L.R. Human Endometrial Microbiota at Term of Normal Pregnancies. Genes 2019, 10, 971. https://doi.org/10.3390/genes10120971
Leoni C, Ceci O, Manzari C, Fosso B, Volpicella M, Ferrari A, Fiorella P, Pesole G, Cicinelli E, Ceci LR. Human Endometrial Microbiota at Term of Normal Pregnancies. Genes. 2019; 10(12):971. https://doi.org/10.3390/genes10120971
Chicago/Turabian StyleLeoni, Claudia, Oronzo Ceci, Caterina Manzari, Bruno Fosso, Mariateresa Volpicella, Alessandra Ferrari, Paola Fiorella, Graziano Pesole, Ettore Cicinelli, and Luigi Ruggiero Ceci. 2019. "Human Endometrial Microbiota at Term of Normal Pregnancies" Genes 10, no. 12: 971. https://doi.org/10.3390/genes10120971
APA StyleLeoni, C., Ceci, O., Manzari, C., Fosso, B., Volpicella, M., Ferrari, A., Fiorella, P., Pesole, G., Cicinelli, E., & Ceci, L. R. (2019). Human Endometrial Microbiota at Term of Normal Pregnancies. Genes, 10(12), 971. https://doi.org/10.3390/genes10120971