Transcriptome and DNA Methylation Analyses of the Molecular Mechanisms Underlying with Longissimus dorsi Muscles at Different Stages of Development in the Polled Yak

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Tissue Collection and Library Preparation
2.2. cDNA Library Construction and Sequencing
2.3. RNA-Seq Data Analysis
2.4. Time-series Expression Profile Clustering
2.5. Quantitative PCR Assay
2.6. MethylRAD Library Sequencing
2.7. MethylRAD Data Analysis
3. Results
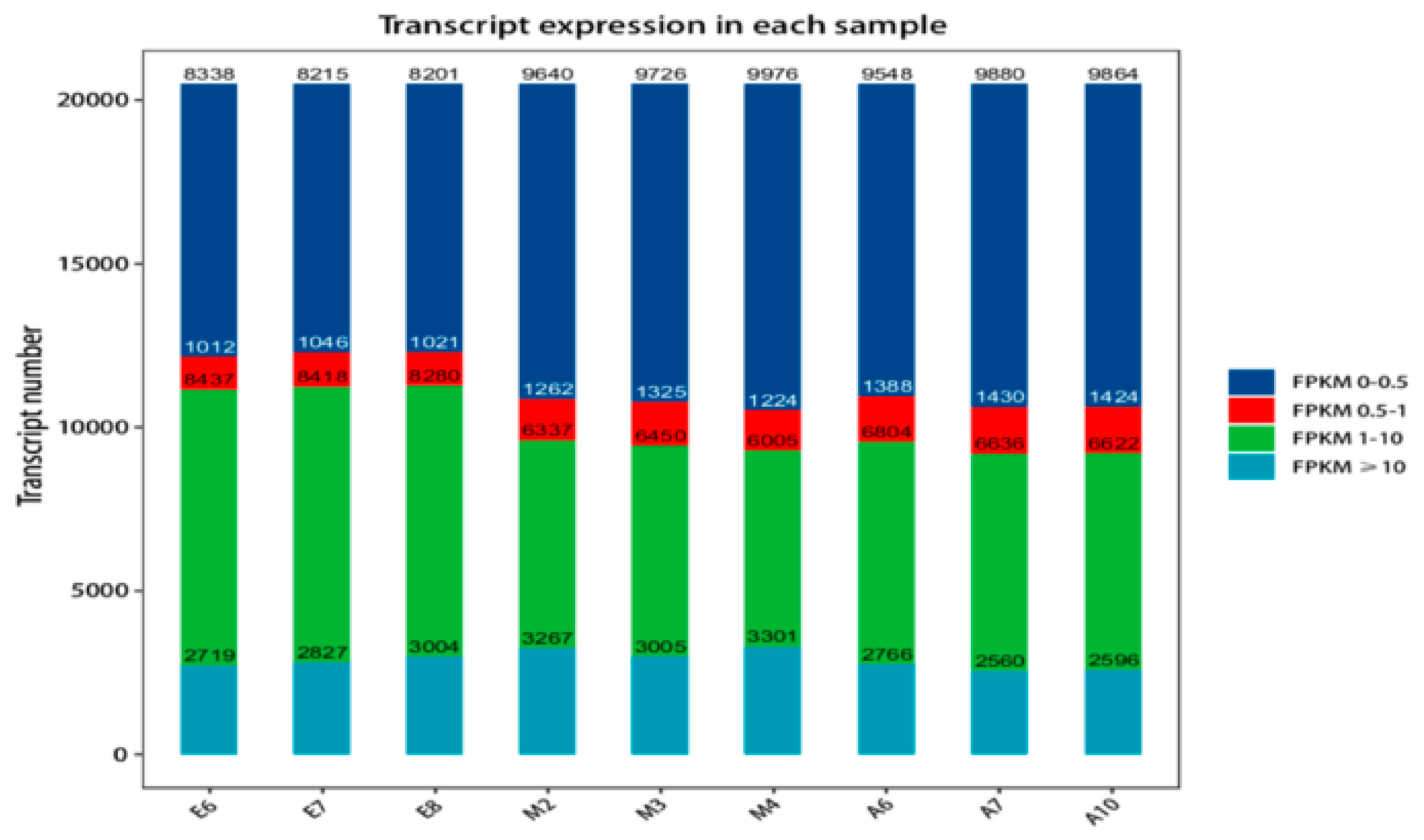
3.1. Alignment to the Reference Genome
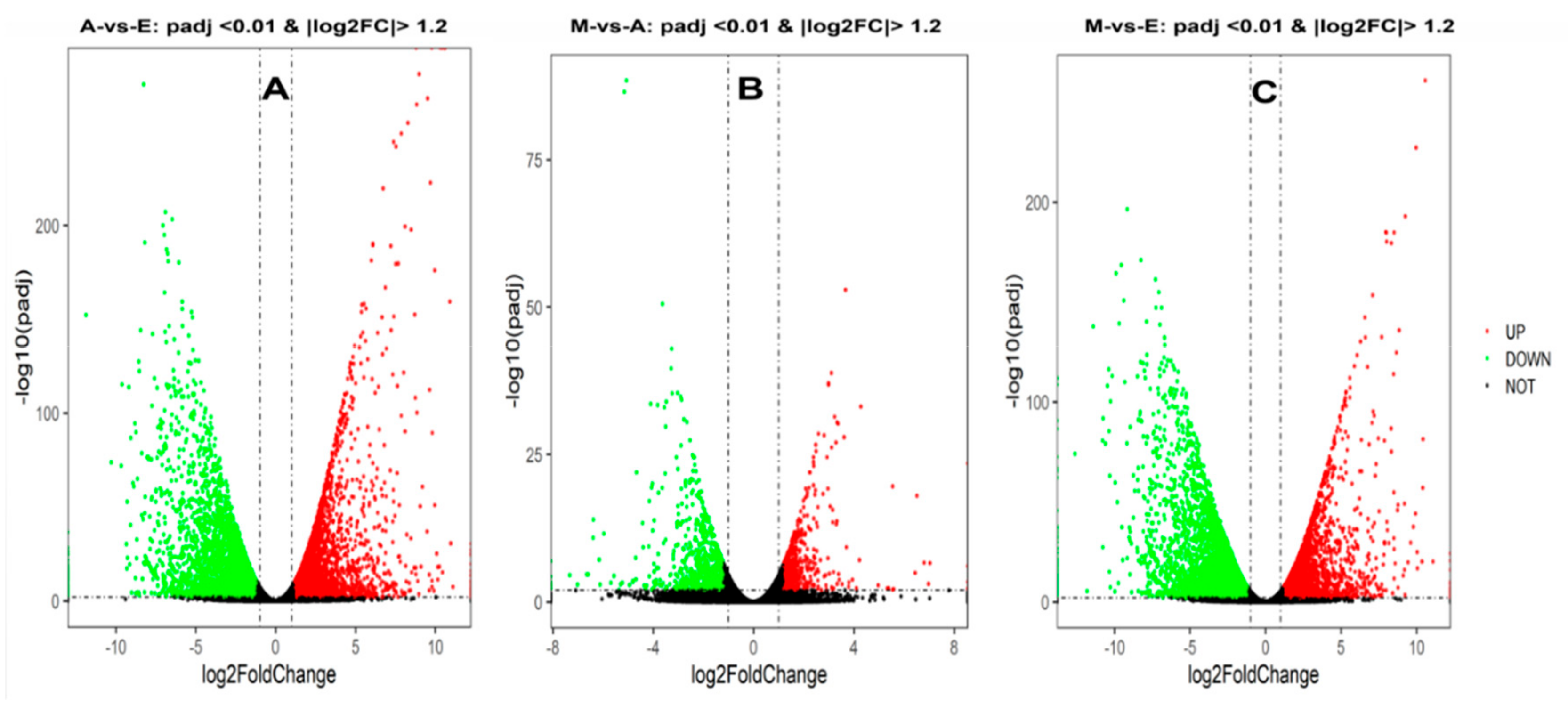
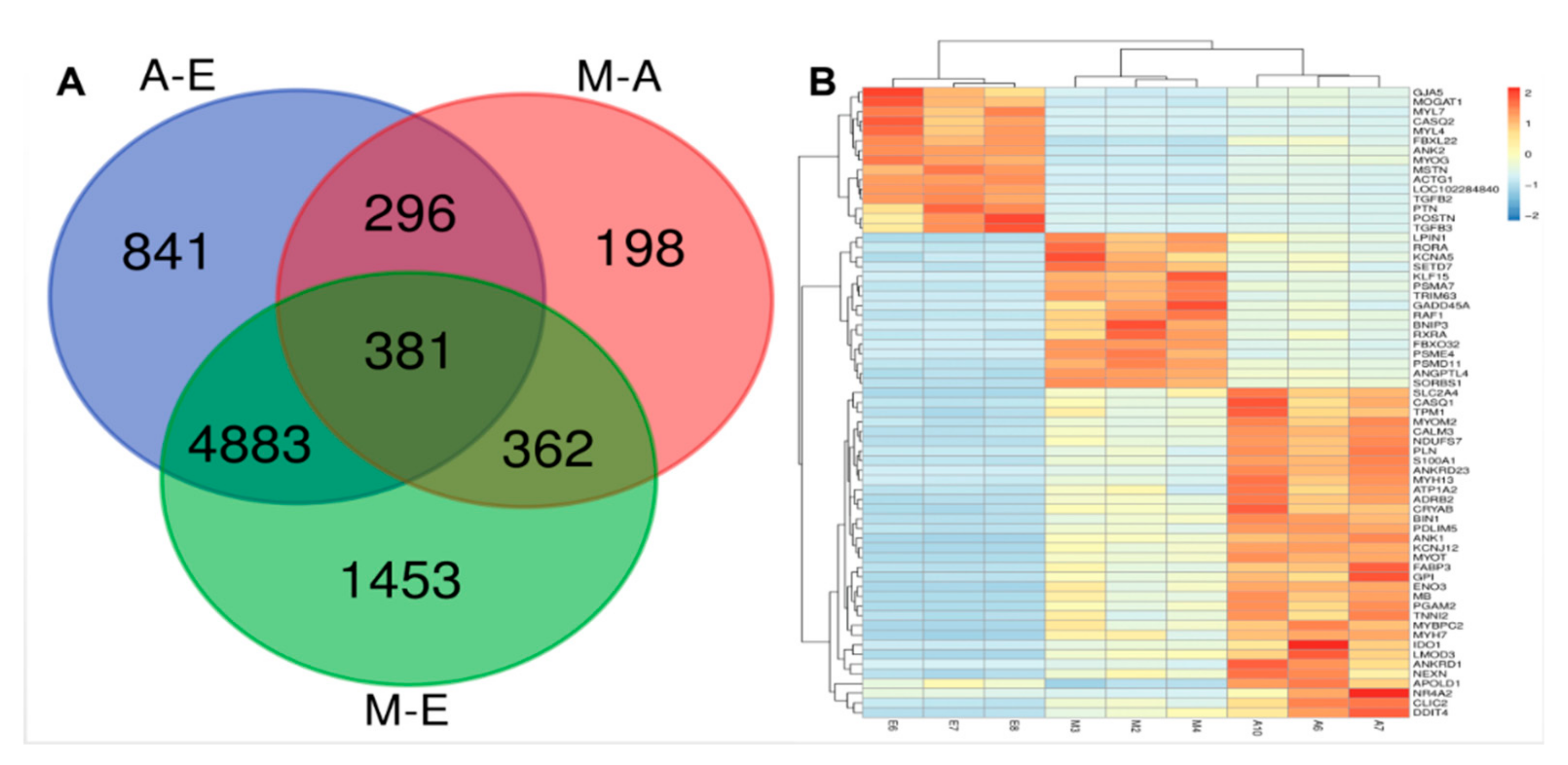
3.2. Differentially Expressed Genes Among the Three Combinations of Groups
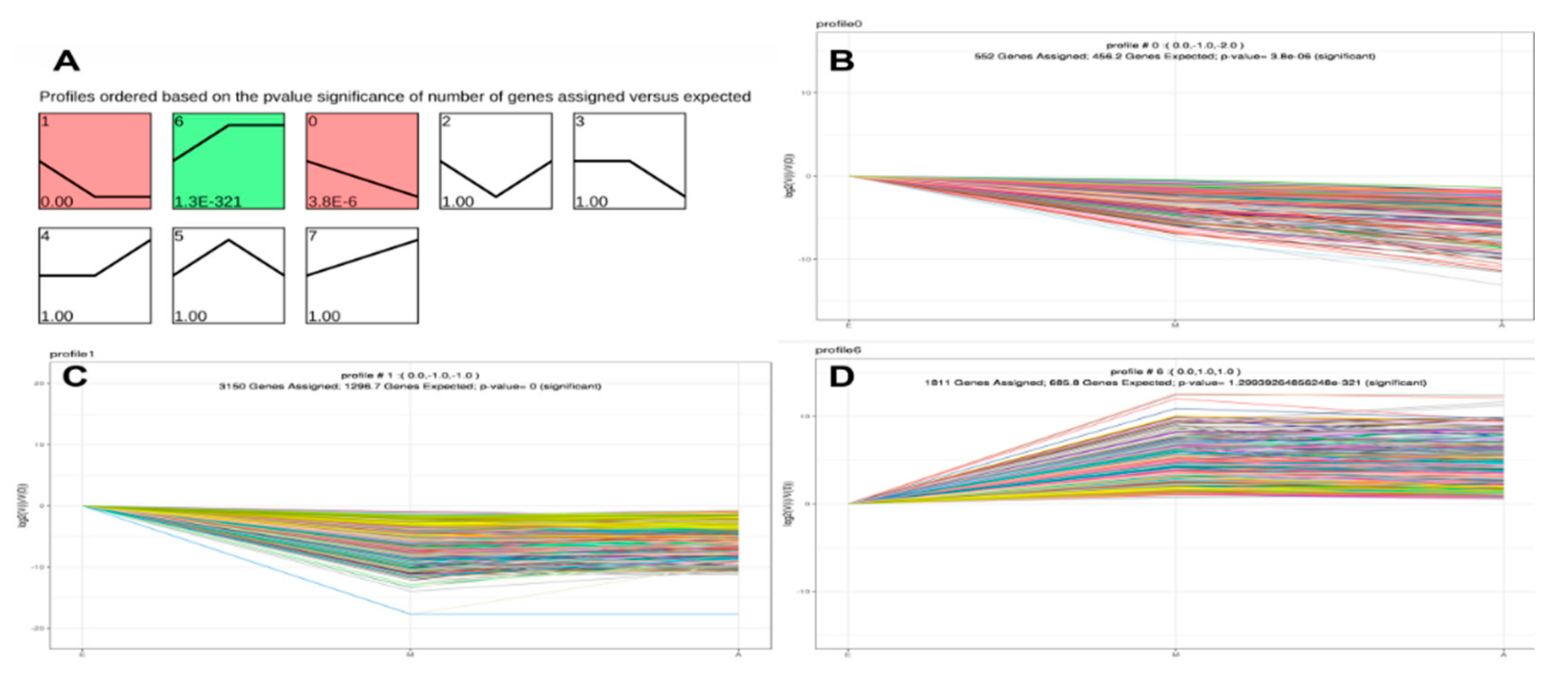
3.3. STEM Analysis of DEG Expression Profiles
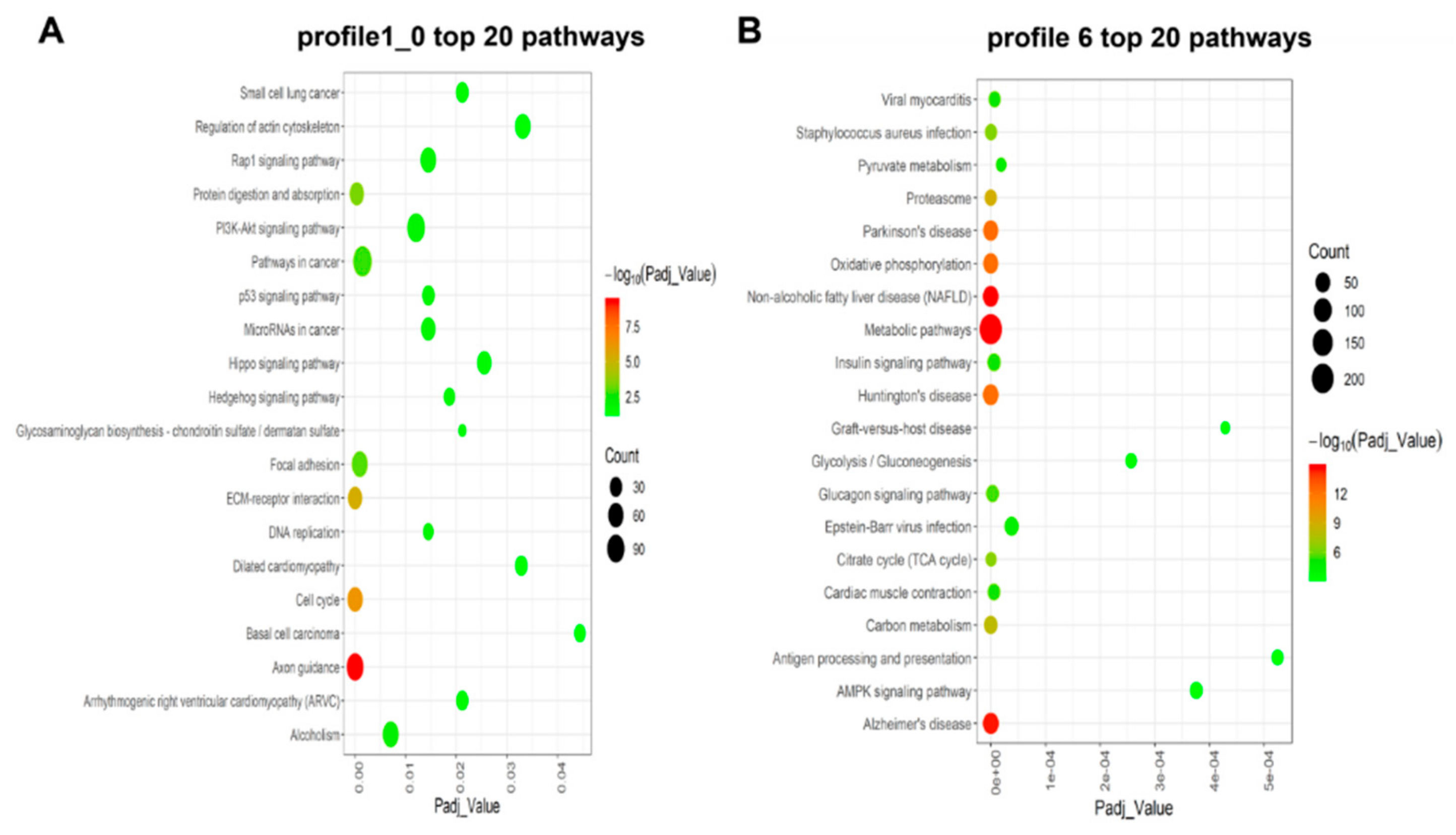
3.4. KEGG Enrichment Analysis for Time-series Expression Profile Clustering
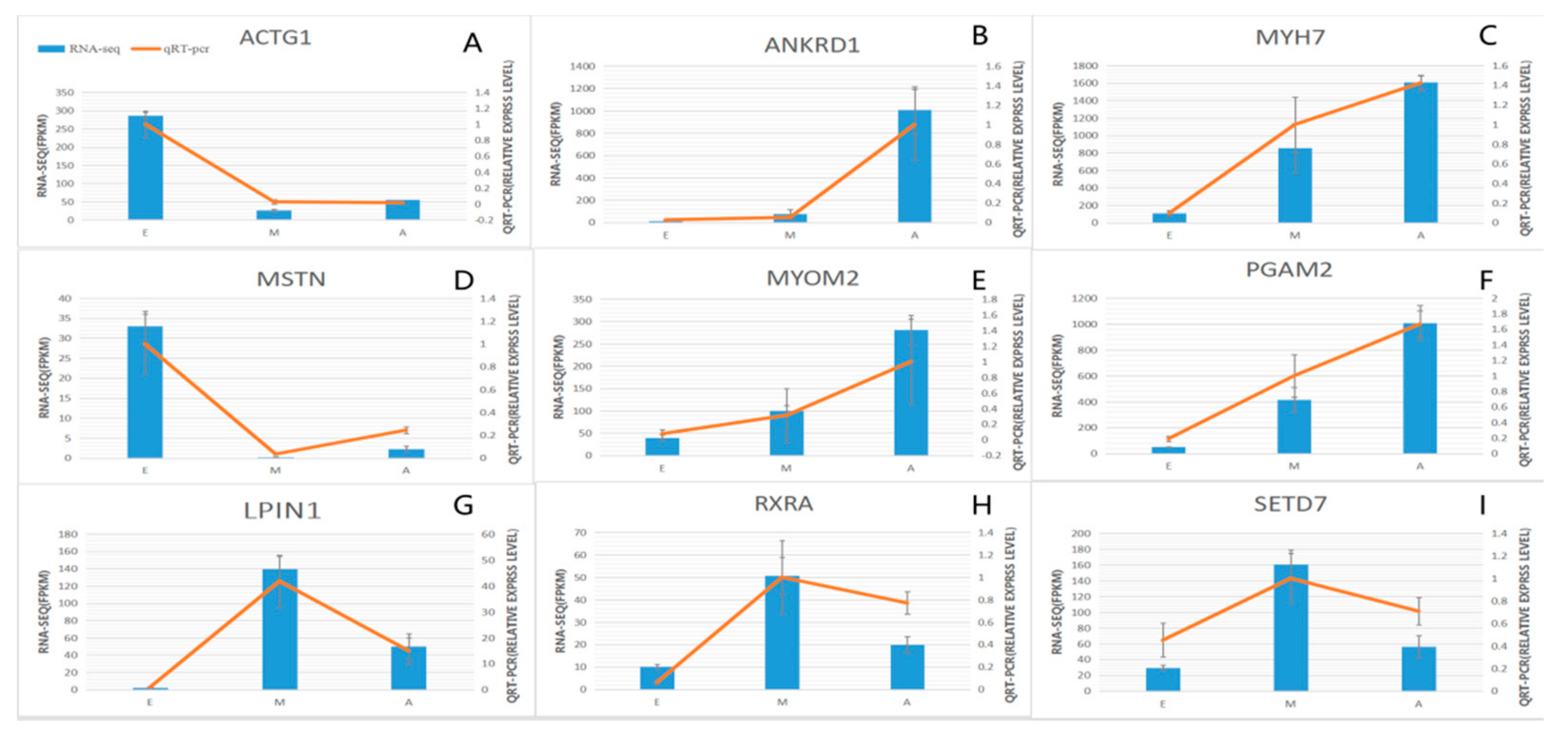
3.5. qPCR Validation of the Three Developmental Stages of Common DEGs
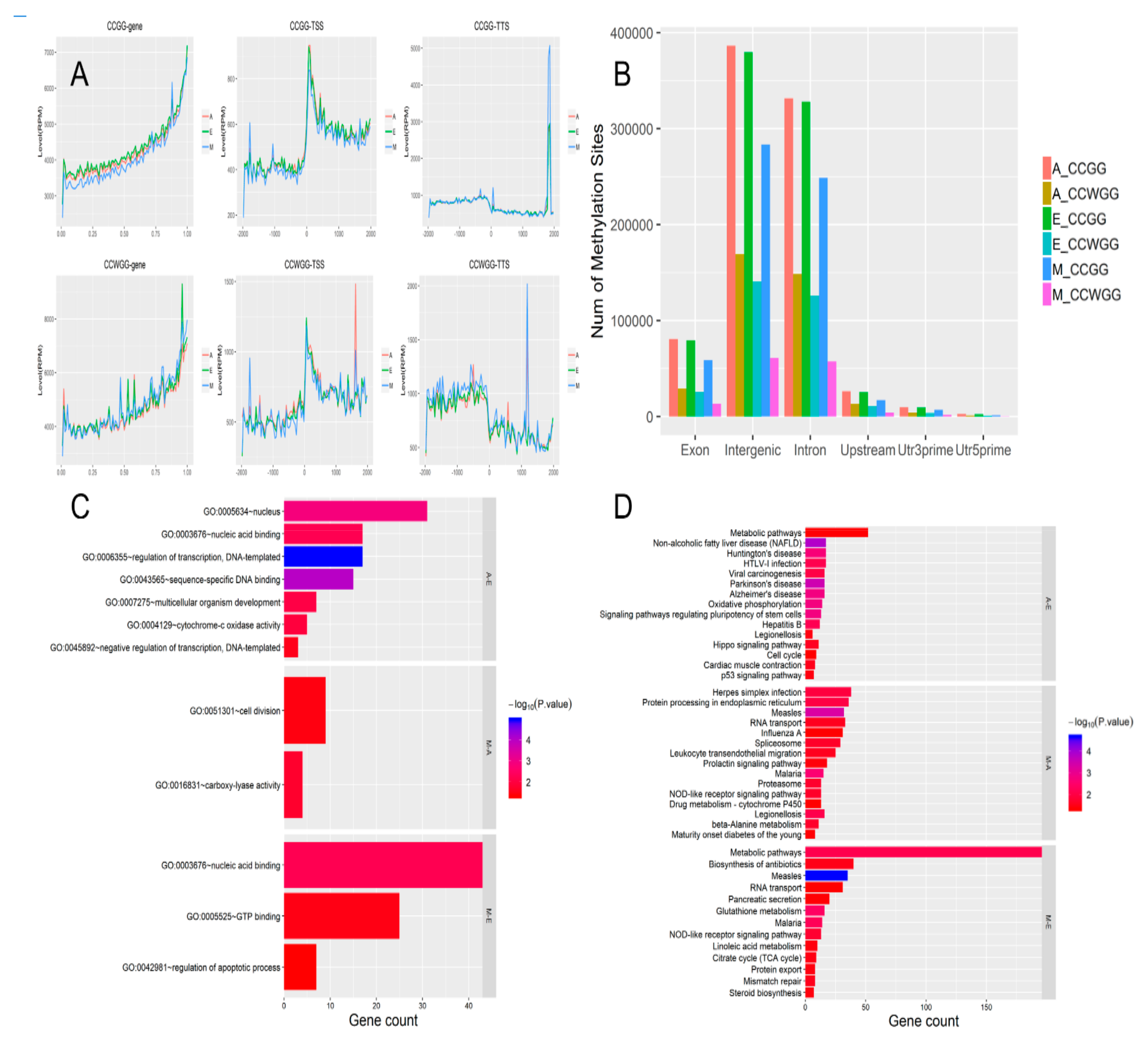
3.6. Analysis of DNA Methylation and Its Distribution
3.7. Differential Methylation Regions Among the Three Groups
3.8. Enrichment Analysis of DMRs
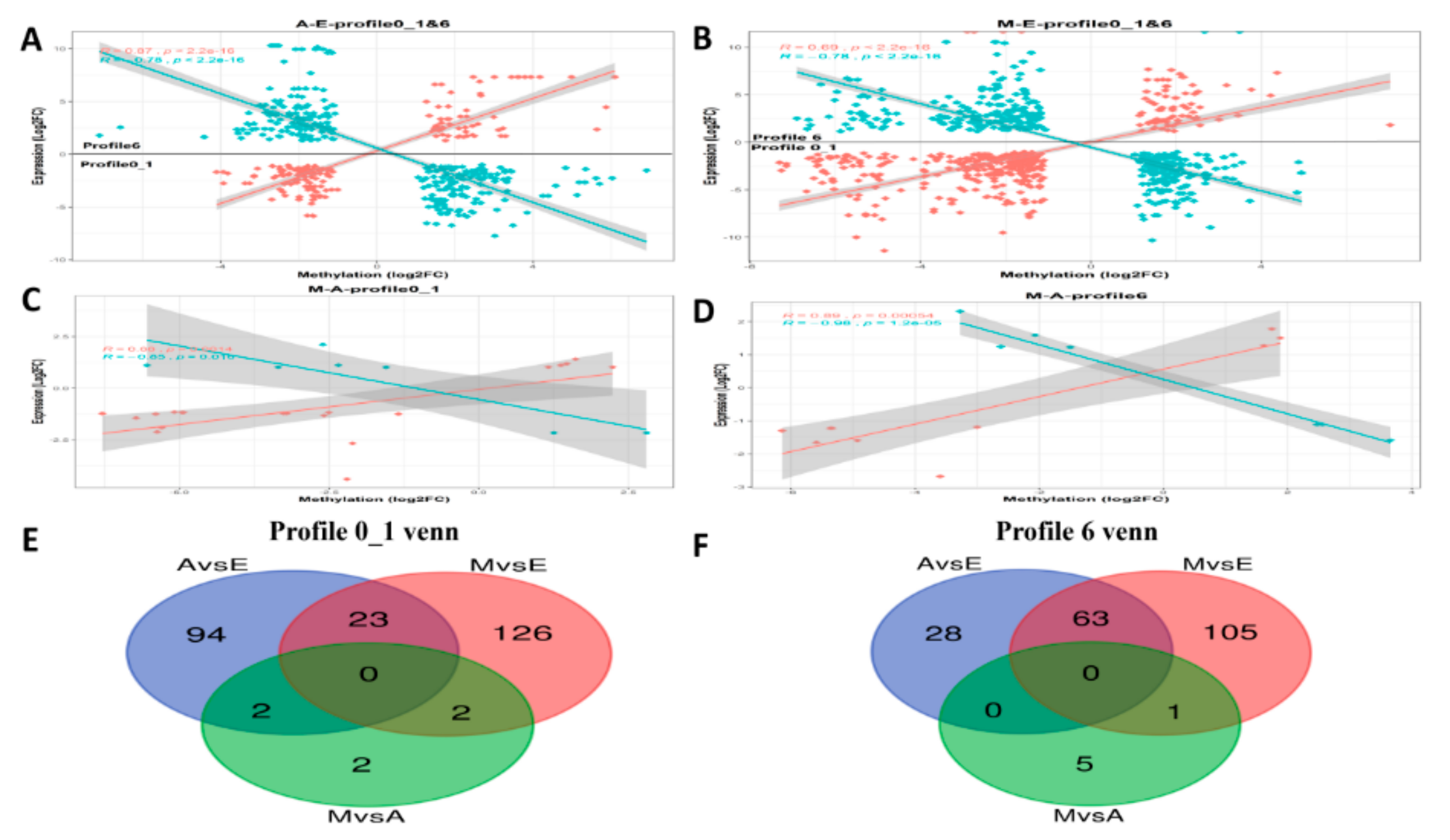
3.9. Integrated Analysis of DEG and DMP Results
4. Discussion
4.1. KEGG Analysis Based on Time-series Expression Profile Clustering
4.2. Longissimus Dorsi Muscle Methylation Profile and Correlation Analysis of DMR Signaling Pathway
4.3. Negative Correlation Between Methylation Levels of DMPs and Differential Expression of DEGs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Qi, X.; Zhang, Q.; He, Y.; Yang, L.; Zhang, X.; Shi, P.; Yang, L.; Liu, Z.; Zhang, F.; Liu, F.; et al. The Transcriptomic Landscape of Yaks Reveals Molecular Pathways for High Altitude Adaptation. Genome Biol. Evol. 2019, 11, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Cui, C.; Peng, Y.; Zhang, X.; Yang, Z.; Zhong, H.; Zhang, H.; Xiang, K.; Cao, X.; Wang, Y.; et al. Genetic Evidence of Paleolithic Colonization and Neolithic Expansion of Modern Humans on the Tibetan Plateau. Mol. Biol. Evol. 2013, 30, 1761–1778. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.J.; Chen, S.Y.; Liu, Y.P.; Yao, Y.G. Mitochondrial DNA sequence diversity and origin of Chinese domestic yak. Anim. Genet. 2007, 38, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Long, R.; Kreuzer, M.; Ding, L.; Shang, Z.; Zhang, Y.; Yang, Y.; Cui, G. Importance of Functional Ingredients in Yak Milk-Derived Food on Health of Tibetan Nomads Living Under High-Altitude Stress: A Review. Crit. Rev. Food Sci. Nutr. 2014, 54, 292–302. [Google Scholar] [CrossRef]
- Zhu, C.; Song, W.; Tao, Z.; Liu, H.; Xu, W.; Zhang, S.; Li, H. Deep RNA sequencing of pectoralis muscle transcriptomes during late-term embryonic to neonatal development in indigenous Chinese duck breeds. PLoS ONE 2017, 12, e0180403. [Google Scholar] [CrossRef]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33 (Suppl. 3), 245–254. [Google Scholar] [CrossRef]
- Lockhart, D.J.; Winzeler, E.A. Genomics, gene expression and DNA arrays. Nature 2000, 405, 827–836. [Google Scholar] [CrossRef]
- Lindberg, J.; Lundeberg, J. The plasticity of the mammalian transcriptome. Genomics 2010, 95, 1–6. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57. [Google Scholar] [CrossRef]
- Bryson-Richardson, R.J.; Currie, P.D. The genetics of vertebrate myogenesis. Nat. Rev. Genet. 2008, 9, 632. [Google Scholar] [CrossRef]
- Gent, J.; van den Eijnden, M.; van Kerkhof, P.; Strous, G.J. Dimerization and signal transduction of the growth hormone receptor. Mol. Endocrinol. 2003, 17, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Duan, C.; Ren, H.; Gao, S. Insulin-like growth factors (IGFs), IGF receptors, and IGF-binding proteins: Roles in skeletal muscle growth and differentiation. Gen. Comp. Endocrinol. 2010, 167, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Holliday, R.; Pugh, J.E. DNA modification mechanisms and gene activity during development. Science 1975, 187, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Singal, R.; Ginder, G.D. DNA methylation. Blood 1999, 93, 4059–4070. [Google Scholar] [CrossRef]
- Mohandas, T.; Sparkes, R.; Shapiro, L. Reactivation of an inactive human X chromosome: Evidence for X inactivation by DNA methylation. Science 1981, 211, 393–396. [Google Scholar] [CrossRef]
- Reik, W.; Collick, A.; Norris, M.L.; Barton, S.C.; Surani, M.A. Genomic imprinting determines methylation of parental alleles in transgenic mice. Nature 1987, 328, 248. [Google Scholar] [CrossRef]
- Gaudet, F.; Hodgson, J.G.; Eden, A.; Jackson-Grusby, L.; Dausman, J.; Gray, J.W.; Leonhardt, H.; Jaenisch, R. Induction of tumors in mice by genomic hypomethylation. Science 2003, 300, 489–492. [Google Scholar] [CrossRef]
- Hsieh, C.-L. Dependence of transcriptional repression on CpG methylation density. Mol. Cell. Biol. 1994, 14, 5487–5494. [Google Scholar] [CrossRef]
- Gelfman, S.; Cohen, N.; Yearim, A.; Ast, G. DNA-methylation effect on cotranscriptional splicing is dependent on GC architecture of the exon–intron structure. Genome Res. 2013, 23, 789–799. [Google Scholar] [CrossRef]
- Stadler, M.B.; Murr, R.; Burger, L.; Ivanek, R.; Lienert, F.; Schöler, A.; van Nimwegen, E.; Wirbelauer, C.; Oakeley, E.J.; Gaidatzis, D. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 2011, 480, 490. [Google Scholar] [CrossRef]
- Carrió Gaspar, E.; Suelves, M. DNA methylation dynamics in muscle development and disease. Front. Aging Neurosci. 2015, 7, 19. [Google Scholar]
- Brunk, B.P.; Goldhamer, D.J.; Emerson, C.P., Jr. Regulated Demethylation of themyoDDistal Enhancer during Skeletal Myogenesis. Dev. Biol. 1996, 177, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Salilewwondim, D.; Fournier, E.; Hoelker, M.; Saeedzidane, M.; Tholen, E.; Looft, C.; Neuhoff, C.; Besenfelder, U.; Havlicek, V.; Rings, F. Genome-Wide DNA Methylation Patterns of Bovine Blastocysts Developed In Vivo from Embryos Completed Different Stages of Development In Vitro. PLoS ONE 2015, 10, e0140467. [Google Scholar]
- Huang, Y.Z.; Sun, J.J.; Zhang, L.Z.; Li, C.J.; Womack, J.E.; Li, Z.J.; Lan, X.Y.; Lei, C.Z.; Zhang, C.L.; Zhao, X. Genome-wide DNA Methylation Profiles and Their Relationships with mRNA and the microRNA Transcriptome in Bovine Muscle Tissue (Bos taurine). Sci. Rep. 2014, 4, 6546. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wu, H.; Luo, Z.; Xia, Y.; Guan, J.; Wang, T.; Gu, Y.; Chen, L.; Zhang, K.; Ma, J. An atlas of DNA methylomes in porcine adipose and muscle tissues. Nat. Commun. 2012, 3, 850. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Jin, H.G.; Ma, H.H.; Zhao, Z.H. Comparative analysis on genome-wide DNA methylation in longissimus dorsi muscle between Small Tailed Han and Dorper×Small Tailed Han crossbred sheep. Asian Australas. J. Anim. Sci. 2017, 30, 1529–1539. [Google Scholar] [CrossRef] [PubMed]
- Richardson, C.; Jones, P.C.; Barnard, V.; Hebert, C.N.; Terlecki, S.; Wijeratne, W.V. Estimation of the developmental age of the bovine fetus and newborn calf. Vet. Rec. 1990, 126, 279–284. [Google Scholar]
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. In Babraham Bioinformatics; Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Daehwan, K.; Ben, L.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar]
- Simon, A.; Paul Theodor, P.; Wolfgang, H. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar]
- Trapnell, C. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.; Bar-Joseph, Z. STEM: A tool for the analysis of short time series gene expression data. BMC Bioinform. 2006, 7, 191. [Google Scholar] [CrossRef]
- Wang, S.; Lv, J.; Zhang, L.; Dou, J.; Sun, Y.; Li, X.; Fu, X.; Dou, H.; Mao, J.; Hu, X. MethylRAD: A simple and scalable method for genome-wide DNA methylation profiling using methylation-dependent restriction enzymes. Open Biol. 2015, 5, 150130. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- D Robinson, M.; McCarthy, D.; Smyth, G. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Ginestet, C. ggplot2: Elegant graphics for data analysis. J. R. Stat. Soc. Ser. A Stat. Soc. 2011, 174, 245–246. [Google Scholar] [CrossRef]
- Roberts, A. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 2011, 12, R22. [Google Scholar] [CrossRef]
- Goodrich, J.A.; Tjian, R. Unexpected roles for core promoter recognition factors in cell-type-specific transcription and gene regulation. Nat. Rev. Genet. 2010, 11, 549. [Google Scholar] [CrossRef] [PubMed]
- Schübeler, D. Function and information content of DNA methylation. Nature 2015, 517, 321. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Kim, H.; Zhong, S.; Chen, H.; Hu, Z.; Zhou, B. De novo transcriptome assembly for rudimentary leaves in Litchi chinesis Sonn. and identification of differentially expressed genes in response to reactive oxygen species. BMC Genom. 2014, 15, 805. [Google Scholar] [CrossRef] [PubMed]
- Mai, M.; Jin, L.; Tian, S.; Liu, R.; Huang, W.; Tang, Q.; Ma, J.; Wang, X.; Hu, Y.; Wang, D. Deciphering the microRNA transcriptome of skeletal muscle during porcine development. PeerJ 2016, 4, e1504. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, C.; Plath, M.; Fang, X.; Lan, X.; Zhou, Y.; Chen, H. Global transcriptional profiling of longissimus thoracis muscle tissue in fetal and juvenile domestic goat using RNA sequencing. Anim. Genet. 2015, 46, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Li, M.; Sun, Y.; Cai, H.; Li, R.; Wei, X.; Lan, X.; Huang, Y.; Lei, C.; Chen, H. The developmental transcriptome landscape of bovine skeletal muscle defined by Ribo-Zero ribonucleic acid sequencing. J. Anim. Sci. 2015, 93, 5648–5658. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, K.; Kautz, S.; Hajdu, G.; Winter, C.; Whalen, R.G.; Starzinski-Powitz, A. Heterogenic mRNAs with an identical protein-coding region of the human embryonic myosin alkali light chain in skeletal muscle cells. J. Mol. Biol. 1990, 211, 505–513. [Google Scholar] [CrossRef]
- Timson, D.J.; Trayer, H.R.; Trayer, I.P. The N-terminus of A1-type myosin essential light chains binds actin and modulates myosin motor function. Eur. J. Biochem. 1998, 255, 654–662. [Google Scholar] [CrossRef]
- Tureckova, J.; Wilson, E.M.; Cappalonga, J.L.; Rotwein, P. Insulin-like growth factor-mediated muscle differentiation collaboration between phosphatidylinositol 3-kinase-Akt-signaling pathways and myogenin. J. Biol. Chem. 2001, 276, 39264–39270. [Google Scholar] [CrossRef]
- Liu, S.; Gao, F.; Wen, L.; Ouyang, M.; Wang, Y.; Wang, Q.; Luo, L.; Jian, Z. Osteocalcin induces proliferation via positive activation of the PI3K/Akt, P38 MAPK pathways and promotes differentiation through activation of the GPRC6A-ERK1/2 pathway in C2C12 myoblast cells. Cell. Physiol. Biochem. 2017, 43, 1100–1112. [Google Scholar] [CrossRef]
- Jiang, B.-H.; Aoki, M.; Zheng, J.Z.; Li, J.; Vogt, P.K. Myogenic signaling of phosphatidylinositol 3-kinase requires the serine-threonine kinase Akt/protein kinase B. Proc. Natl. Acad. Sci. USA 1999, 96, 2077–2081. [Google Scholar] [CrossRef] [PubMed]
- Lynch, G.S.; Ryall, J.G. Role of β-adrenoceptor signaling in skeletal muscle: Implications for muscle wasting and disease. Physiol. Rev. 2008, 88, 729–767. [Google Scholar] [CrossRef] [PubMed]
- Pizon, V.; Cifuentes-Diaz, C.; Mège, R.M.; Baldacci, G.; Rieger, F. Expression and localization of RAP1 proteins during myogenic differentiation. Eur. J. Cell Biol. 1996, 69, 224–235. [Google Scholar]
- Louis, M.; Van Beneden, R.; Dehoux, M.; Thissen, J.P.; Francaux, M. Creatine increases IGF-I and myogenic regulatory factor mRNA in C2C12 cells. FEBS Lett. 2004, 557, 243–247. [Google Scholar] [CrossRef]
- Ploug, T.; Van Deurs, B.; Ai, H.; Cushman, S.W.; Ralston, E. Analysis of GLUT4 distribution in whole skeletal muscle fibers: Identification of distinct storage compartments that are recruited by insulin and muscle contractions. J. Cell Biol. 1998, 142, 1429–1446. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, H.M.; Maarbjerg, S.J.; Crane, J.D.; Jeppesen, J.; Jørgensen, S.B.; Schertzer, J.D.; Shyroka, O.; Kiens, B.; Van Denderen, B.J.; Tarnopolsky, M.A. AMP-activated protein kinase (AMPK) β1β2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc. Natl. Acad. Sci. USA 2011, 108, 16092–16097. [Google Scholar] [CrossRef] [PubMed]
- Egan, B.; Zierath, J.R. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab. 2013, 17, 162–184. [Google Scholar] [CrossRef]
- Hevener, A.L.; He, W.; Barak, Y.; Le, J.; Bandyopadhyay, G.; Olson, P.; Wilkes, J.; Evans, R.M.; Olefsky, J. Muscle-specific Pparg deletion causes insulin resistance. Nat. Med. 2003, 9, 1491. [Google Scholar] [CrossRef]
- Norris, A.W.; Chen, L.; Fisher, S.J.; Szanto, I.; Ristow, M.; Jozsi, A.C.; Hirshman, M.F.; Rosen, E.D.; Goodyear, L.J.; Gonzalez, F.J. Muscle-specific PPARγ-deficient mice develop increased adiposity and insulin resistance but respond to thiazolidinediones. J. Clin. Investig. 2003, 112, 608–618. [Google Scholar] [CrossRef]
- Yau, M.-H.; Wang, Y.; Lam, K.S.; Zhang, J.; Wu, D.; Xu, A. A highly conserved motif within the NH2-terminal coiled-coil domain of angiopoietin-like protein 4 confers its inhibitory effects on lipoprotein lipase by disrupting the enzyme dimerization. J. Biol. Chem. 2009, 284, 11942–11952. [Google Scholar] [CrossRef]
- Mangelsdorf, D.J.; Evans, R.M. The RXR heterodimers and orphan receptors. Cell 1995, 83, 841–850. [Google Scholar] [CrossRef]
- Eckhardt, F.; Lewin, J.; Cortese, R.; Rakyan, V.K.; Attwood, J.; Burger, M.; Burton, J.; Cox, T.V.; Davies, R.; Down, T.A. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat. Genet. 2006, 38, 1378. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yazaki, J.; Sundaresan, A.; Cokus, S.; Chan, S.W.-L.; Chen, H.; Henderson, I.R.; Shinn, P.; Pellegrini, M.; Jacobsen, S.E. Genome-wide high-resolution mapping and functional analysis of DNA methylation in Arabidopsis. Cell 2006, 126, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Wend, P.; Wend, K.; Krum, S.; Miranda-Carboni, G. The role of WNT10B in physiology and disease. Acta Physiol. 2012, 204, 34–51. [Google Scholar] [CrossRef]
- Brun, J.; Fromigué, O.; Dieudonné, F.-X.; Marty, C.; Chen, J.; Dahan, J.; Wei, Y.; Marie, P.J. The LIM-only protein FHL2 controls mesenchymal cell osteogenic differentiation and bone formation through Wnt5a and Wnt10b. Bone 2013, 53, 6–12. [Google Scholar] [CrossRef]
- Jeong, J.; Kim, J.; Nguyen, T.; Lee, H.J.; Baik, M. Wnt/β-catenin signaling and adipogenic genes are associated with intramuscular fat content in the longissimus dorsi muscle of Korean cattle. Anim. Genet. 2013, 44, 627–635. [Google Scholar] [CrossRef]
- Von Maltzahn, J.; Bentzinger, C.F.; Rudnicki, M.A. Wnt7a–Fzd7 signalling directly activates the Akt/mTOR anabolic growth pathway in skeletal muscle. Nat. Cell Biol. 2012, 14, 186. [Google Scholar] [CrossRef]
- Júnior, G.F.; Costa, R.; De Camargo, G.; Carvalheiro, R.; Rosa, G.; Baldi, F.; Garcia, D.; Gordo, D.; Espigolan, R.; Takada, L. Genome scan for postmortem carcass traits in Nellore cattle. J. Anim. Sci. 2016, 94, 4087–4095. [Google Scholar] [CrossRef]
- Sorbolini, S.; Bongiorni, S.; Cellesi, M.; Gaspa, G.; Dimauro, C.; Valentini, A.; Macciotta, N. Genome wide association study on beef production traits in Marchigiana cattle breed. J. Anim. Breed. Genet. 2017, 134, 43–48. [Google Scholar] [CrossRef]
- DeChiara, T.M.; Efstratiadis, A.; Robertsen, E.J. A growth-deficiency phenotype in heterozygous mice carrying an insulin-like growth factor II gene disrupted by targeting. Nature 1990, 345, 78. [Google Scholar] [CrossRef]
- Andersson, L.; Haley, C.S.; Ellegren, H.; Knott, S.A.; Johansson, M.; Andersson, K.; Andersson-Eklund, L.; Edfors-Lilja, I.; Fredholm, M.; Hansson, I. Genetic mapping of quantitative trait loci for growth and fatness in pigs. Science 1994, 263, 1771–1774. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Li, Y.; Wan, P.; Li, X.; Zhao, S.; Liu, B.; Fan, B.; Zhu, M.; Yu, M.; Li, K. LongSAGE analysis of skeletal muscle at three prenatal stages in Tongcheng and Landrace pigs. Genome Biol. 2007, 8, R115. [Google Scholar] [CrossRef]
- Vykoukalova, Z.; Knoll, A.; Dvořák, J.; Čepica, S. New SNPs in the IGF2 gene and association between this gene and backfat thickness and lean meat content in Large White pigs. J. Anim. Breed. Genet. 2006, 123, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-Z.; Zhang, L.-Z.; Lai, X.-S.; Li, M.-X.; Sun, Y.-J.; Li, C.-J.; Lan, X.-Y.; Lei, C.-Z.; Zhang, C.-L.; Zhao, X. Transcription factor ZBED6 mediates IGF2 gene expression by regulating promoter activity and DNA methylation in myoblasts. Sci. Rep. 2014, 4, 4570. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Li, N.; Deng, X.; Hu, X.; Liu, Z.; Zhao, X.; Lian, Z.; Wu, C. Single nucleotide polymorphism analysis in chicken insulin-like growth factor-II gene and its associations with growth and carcass traits. Yi Chuan Xue Bao Acta Genet. Sin. 2002, 29, 30–33. [Google Scholar]
- Millay, D.; O’Rourke, J.; Sutherland, L.; Bezprozvannaya, S.; Shelton, J.; Bassel-Duby, R.; Olson, E. Myomaker: A membrane activator of myoblast fusion and muscle formation. Nature 2013, 499, 301. [Google Scholar] [CrossRef]
- Miretti, S.; Volpe, M.; Martignani, E.; Accornero, P.; Baratta, M. Temporal correlation between differentiation factor expression and microRNAs in Holstein bovine skeletal muscle. Animal 2016, 11, 227–235. [Google Scholar] [CrossRef]
- Huang, Y.; Wu, S.; Zhang, J.; Wen, H.; Zhang, M.; He, F. Methylation status and expression patterns of myomaker gene play important roles in postnatal development in the Japanese flounder (Paralichthys olivaceus). Gen. Comp. Endocrinol. 2019, 280. [Google Scholar] [CrossRef]
- Renganathan, M.; Messi, M.; Delbono, O. Dihydropyridine receptor-ryanodine receptor uncoupling in aged skeletal muscle. J. Membr. Biol. 1997, 157, 247–253. [Google Scholar] [CrossRef]
- Renganathan, M.; Messi, M.A.L.; Delbono, O. Overexpression of IGF-1 exclusively in skeletal muscle prevents age-related decline in the number of dihydropyridine receptors. J. Biol. Chem. 1998, 273, 28845–28851. [Google Scholar] [CrossRef]
- Chaudhari, N.; Beam, K.G. mRNA for cardiac calcium channel is expressed during development of skeletal muscle. Dev. Biol. 1993, 155, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Péréon, Y.; Sorrentino, V.; Dettbarn, C.; Noireaud, J.; Palade, P. Dihydropyridine receptor and ryanodine receptor gene expression in long-term denervated rat muscles. Biochem. Biophys. Res. Commun. 1997, 240, 612–617. [Google Scholar] [CrossRef] [PubMed]
- Frönicke, L.; Wienberg, J. Comparative chromosome painting defines the high rate of karyotype changes between pigs and bovids. Mamm. Genome 2001, 12, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Zhao, X.; Guo, X. Mapping CACNA1S gene in porcine by radiation hybrid panel. Acta Vet. Zootech. Sin. 2006, 37, 309. [Google Scholar]
- Hadjiargyrou, M.; Lombardo, F.; Zhao, S.; Ahrens, W.; Joo, J.; Ahn, H.; Jurman, M.; W White, D.; Rubin, C. Transcriptional profiling of bone regeneration—Insight into the molecular complexity of wound repair. J. Biol. Chem. 2002, 277, 30177–30182. [Google Scholar] [CrossRef]
- Liu, C.; Hadjiargyrou, M. Identification and characterization of the Mustang promoter: Regulation by AP-1 during myogenic differentiation. Bone 2006, 39, 815–824. [Google Scholar] [CrossRef]
- Gao, J.; Zengkui, L.U.; Youji, M.A.; Taotao, L.I.; Zhao, X. Cloning, sequence analysis and tissue expression of MUSTN1 gene in sheep (Ovis aires). Acta Agric. Zhejiangensis 2017, 29, 1661–1668. [Google Scholar] [CrossRef]
- Du, M.; Zhu, M.; J Means, W.; Hess, B.; Ford, S. Nutrient restriction differentially modulates the mammalian target of rapamycin signaling and the ubiquitin–proteasome system in skeletal muscle of cows and their fetuses. J. Anim. Sci. 2005, 83, 117–123. [Google Scholar] [CrossRef]
- Abe, T.; Kearns, C.F.; Fukunaga, T. Sex differences in whole body skeletal muscle mass measured by magnetic resonance imaging and its distribution in young Japanese adults. Br. J. Sports Med. 2003, 37, 436–440. [Google Scholar] [CrossRef]
- Wu, G.Y.; Bazer, F.W.; Wallace, J.M.; Spencer, T. Board-Invited Review: Intrauterine growth retardation: Implications for the animal sciences. J. Anim. Sci. 2006, 84, 2316–2337. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw_Reads | Raw_Bases | Clean_Reads | Clean_Bases | Valid_Bases | Q30 1 | GC |
|---|---|---|---|---|---|---|---|
| E6 | 98.03M | 14.70G | 93.46M | 13.67G | 92.96% | 94.20% | 46.73% |
| E7 | 98.27M | 14.74G | 94.05M | 13.77G | 93.45% | 94.57% | 47.06% |
| E8 | 99.04M | 14.86G | 93.91M | 13.73G | 92.43% | 93.74% | 47.61% |
| M2 | 97.85M | 14.68G | 95.03M | 13.67G | 93.16% | 94.91% | 49.89% |
| M3 | 98.12M | 14.72G | 95.08M | 13.78G | 93.64% | 95.01% | 48.90% |
| M4 | 99.61M | 14.94G | 96.08M | 13.86G | 92.75% | 94.70% | 51.32% |
| A6 | 98.22M | 14.73G | 93.56M | 13.68G | 92.87% | 94.33% | 48.93% |
| A7 | 99.56M | 14.93G | 94.64M | 13.86G | 92.83% | 94.00% | 47.78% |
| A10 | 99.28M | 14.89G | 94.57M | 13.86G | 93.07% | 94.27% | 48.58% |
| Sample | Raw Reads | Enzyme Reads | Mapping Reads | Ratio |
|---|---|---|---|---|
| E6 | 139,680,224 | 64,643,466 | 30,210,658 | 46.73% |
| E7 | 142,659,082 | 65,566,251 | 38,487,733 | 58.70% |
| E8 | 142,659,082 | 75,344,977 | 35,403,055 | 46.99% |
| M2 | 36,447,959 | 21,083,527 | 9,529,418 | 45.20% |
| M3 | 36,447,959 | 22,576,370 | 10,604,071 | 46.97% |
| M4 | 36,447,959 | 23,206,057 | 11,126,778 | 47.95% |
| A6 | 139,680,224 | 57,187,481 | 30,914,358 | 54.06% |
| A7 | 139,680,224 | 65,584,779 | 30,544,305 | 46.57% |
| A10 | 139,680,224 | 65,816,098 | 32,854,859 | 49.92% |
| Average | 105,931,437 | 51,223,223 | 25,519,471 | 49.23% |
| Profile | Profile Number | DMP Group | Two Set Data Overlap Number of DMPs |
|---|---|---|---|
| profile1_0 | 1811 | A-vs.-E | 274 |
| profile1_0 | 1811 | M-vs.-A | 27 |
| profile1_0 | 1811 | M-vs.-E | 574 |
| profile6 | 3702 | A-vs.-E | 214 |
| profile6 | 3702 | M-vs.-A | 18 |
| profile6 | 3702 | M-vs.-E | 323 |
| Gene | Description | Group |
|---|---|---|
| IGF2 | insulin-like growth factor 2 | M-E; A-E; profile0_1 |
| TMEM8C | transmembrane protein 8c | M-E; A-E; profile0_1 |
| MUSTN1 | musculoskeletal, embryonic nuclear protein I | M-E; A-E; profile6 |
| CACNA1S | calcium voltage-gated channel subunit alpha1 S | M-E; A-E; profile6 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, X.; Jia, C.; Chu, M.; Fu, D.; Lei, Q.; Ding, X.; Wu, X.; Guo, X.; Pei, J.; Bao, P.; et al. Transcriptome and DNA Methylation Analyses of the Molecular Mechanisms Underlying with Longissimus dorsi Muscles at Different Stages of Development in the Polled Yak. Genes 2019, 10, 970. https://doi.org/10.3390/genes10120970
Ma X, Jia C, Chu M, Fu D, Lei Q, Ding X, Wu X, Guo X, Pei J, Bao P, et al. Transcriptome and DNA Methylation Analyses of the Molecular Mechanisms Underlying with Longissimus dorsi Muscles at Different Stages of Development in the Polled Yak. Genes. 2019; 10(12):970. https://doi.org/10.3390/genes10120970
Chicago/Turabian StyleMa, Xiaoming, Congjun Jia, Min Chu, Donghai Fu, Qinhui Lei, Xuezhi Ding, Xiaoyun Wu, Xian Guo, Jie Pei, Pengjia Bao, and et al. 2019. "Transcriptome and DNA Methylation Analyses of the Molecular Mechanisms Underlying with Longissimus dorsi Muscles at Different Stages of Development in the Polled Yak" Genes 10, no. 12: 970. https://doi.org/10.3390/genes10120970
APA StyleMa, X., Jia, C., Chu, M., Fu, D., Lei, Q., Ding, X., Wu, X., Guo, X., Pei, J., Bao, P., Yan, P., & Liang, C. (2019). Transcriptome and DNA Methylation Analyses of the Molecular Mechanisms Underlying with Longissimus dorsi Muscles at Different Stages of Development in the Polled Yak. Genes, 10(12), 970. https://doi.org/10.3390/genes10120970