Network Modeling Approaches and Applications to Unravelling Non-Alcoholic Fatty Liver Disease


Abstract
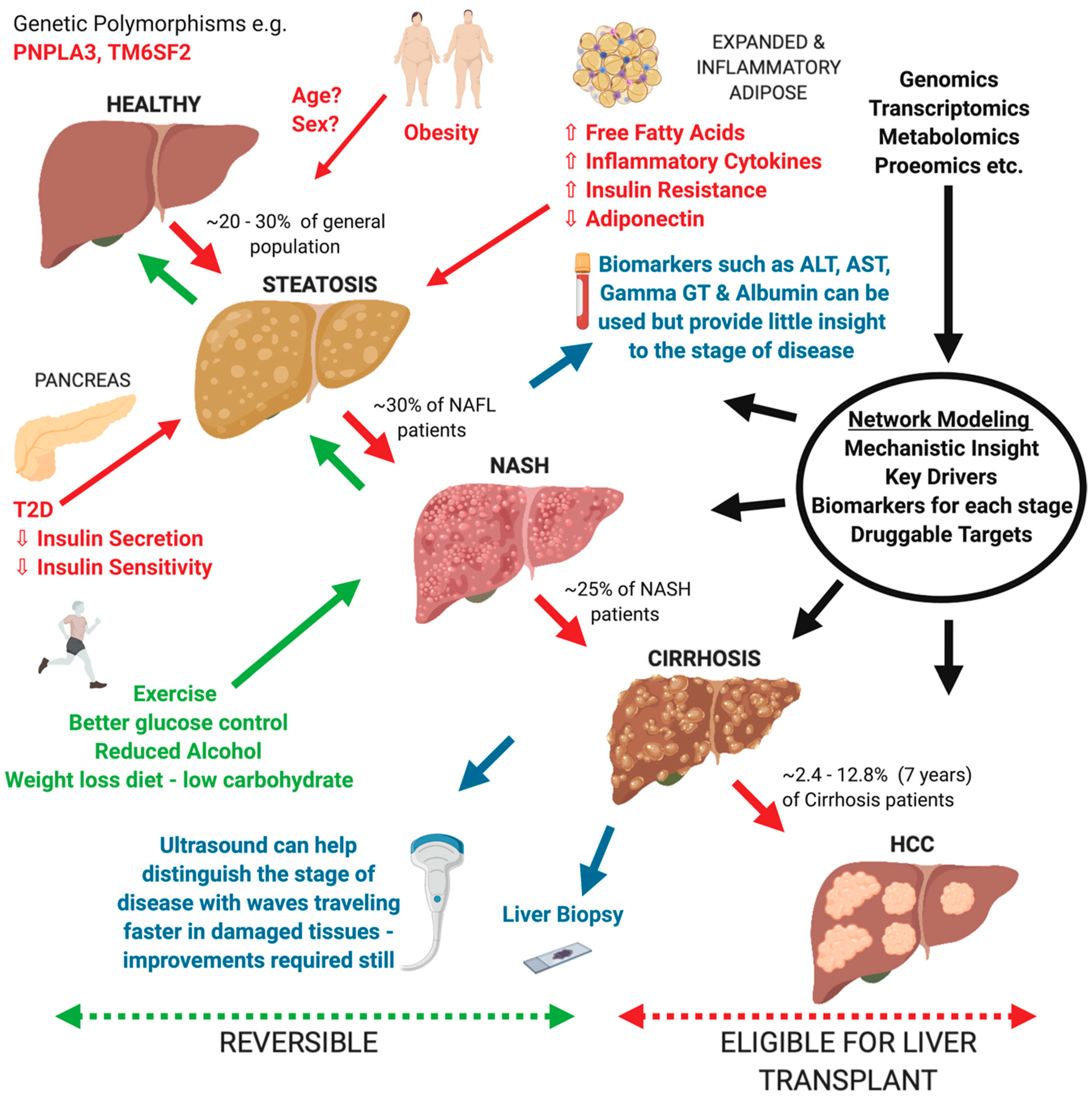
1. Introduction
2. Current Understanding of NAFLD and Remaining Gaps
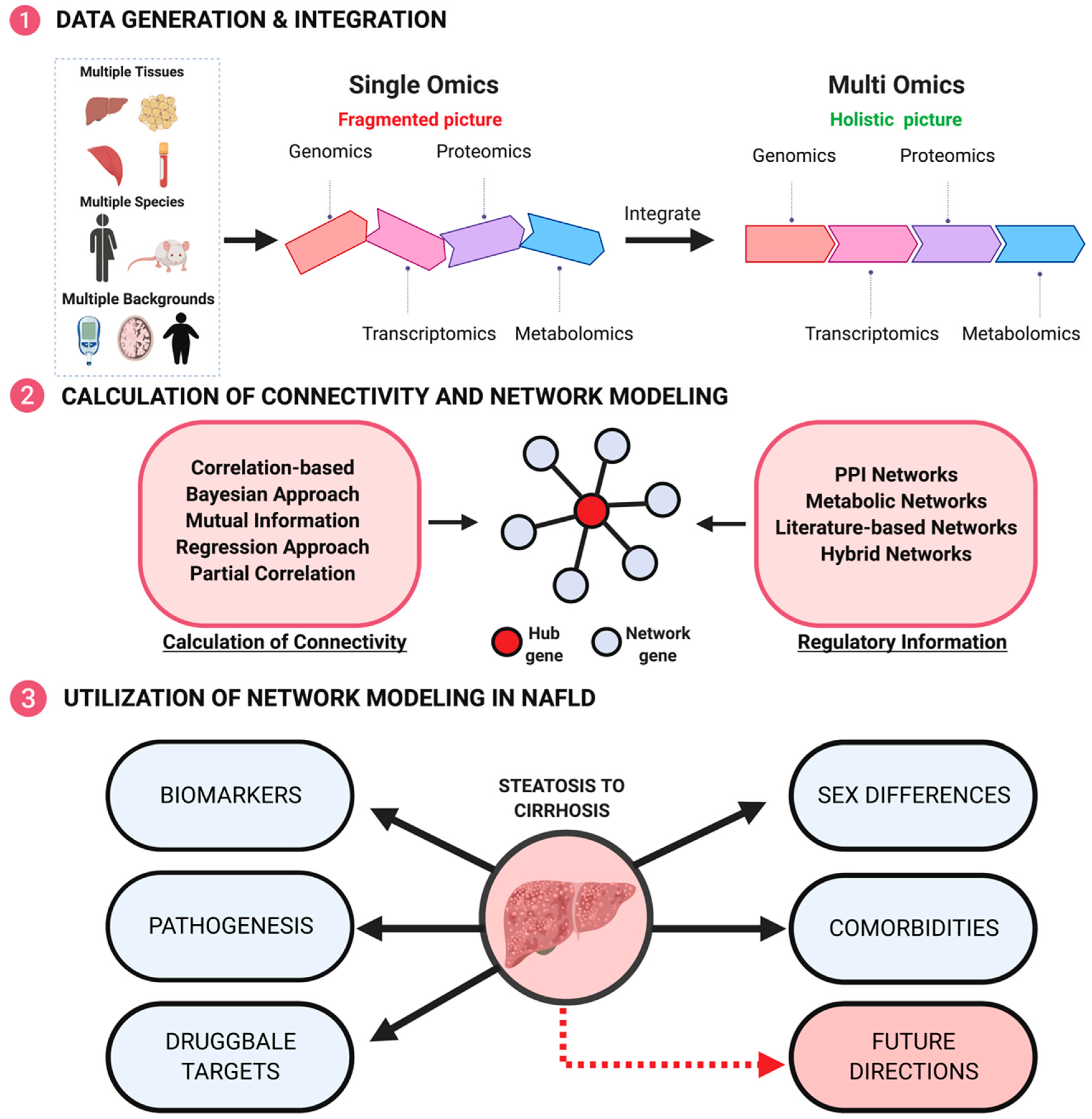
3. Importance of Omics Data in Offering Integrated Network Views of Complex Diseases
4. Commonly Used Network Models
4.1. Tissue and Single Cell GRNs
4.2. Protein-Protein Interaction (PPI) Networks
4.3. Metabolic Networks
4.4. Literature-based Pathways and Networks
4.5. Hybrid Networks
4.6. How to Determine What Type of Networks to Use
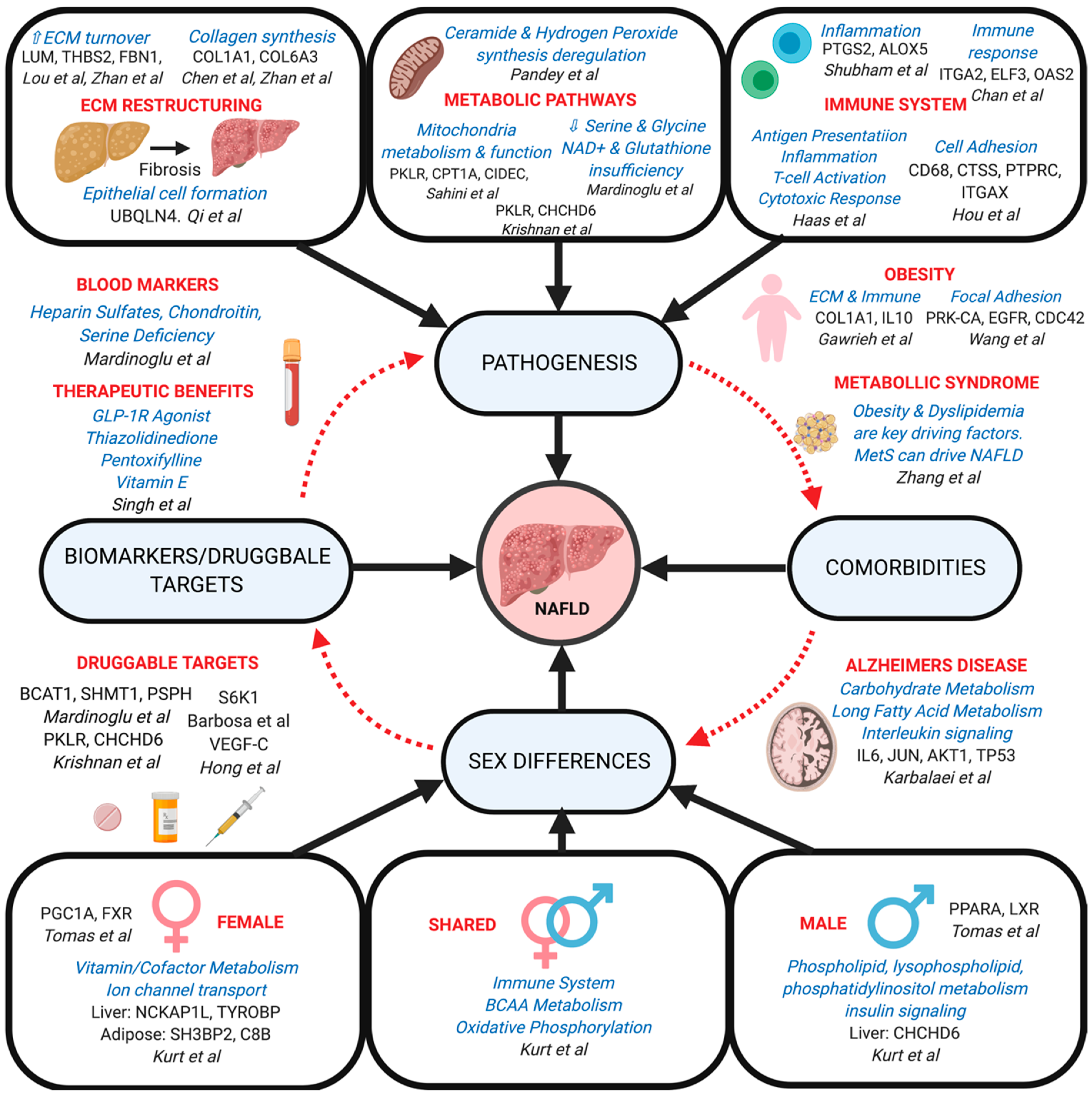
5. Use of Network models to understand the pathogenic mechanisms in NAFLD development and progression
5.1. Modeling of single tissue networks in NAFLD
5.1.1. ECM Structure in Liver Tissue
5.1.2. Metabolic Pathways in Liver and Adipose Tissues
5.1.3. Immune System in Liver and Adipose Tissues
5.1.4. Coordination of ECM, Metabolism, and Immune Pathways in Liver
5.2. Network Modeling of Multiple Tissues
5.3. Network Modeling of NAFLD Comorbidities
5.4. Network Modeling for Sex Differences in NAFLD
5.5. Use of Network Models as Predictive and Diagnostic Tools for NAFLD development
5.6. Network Insights into Drug Repositioning for NAFLD Treatment
5.7. NAFLD Insights Learned From Network Modeling so Far
6. Future Directions for Network Modeling in NAFLD
7. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef]
- McCullough, A.J. The clinical features, diagnosis and natural history of nonalcoholic fatty liver disease. Clin. Liver Dis. 2004, 8, 521–533. [Google Scholar] [CrossRef]
- Bottcher, K.; Pinzani, M. Pathophysiology of liver fibrosis and the methodological barriers to the development of anti-fibrogenic agents. Adv. Drug Deliv. Rev. 2017, 121, 3–8. [Google Scholar] [CrossRef]
- Charlton, M. Nonalcoholic fatty liver disease: A review of current understanding and future impact. Clin. Gastroenterol. Hepatol. 2004, 2, 1048–1058. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Targher, G.; Day, C.P. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 330–344. [Google Scholar] [CrossRef]
- Sookoian, S.; Pirola, C.J. Genetic predisposition in nonalcoholic fatty liver disease. Clin. Mol. Hepatol. 2017, 23, 1–12. [Google Scholar] [CrossRef]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef]
- Buch, S.; Stickel, F.; Trepo, E.; Way, M.; Herrmann, A.; Nischalke, H.D.; Brosch, M.; Rosendahl, J.; Berg, T.; Ridinger, M.; et al. A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat. Genet. 2015, 47, 1443–1448. [Google Scholar] [CrossRef]
- Horvath, S.; Erhart, W.; Brosch, M.; Ammerpohl, O.; von Schonfels, W.; Ahrens, M.; Heits, N.; Bell, J.T.; Tsai, P.C.; Spector, T.D.; et al. Obesity accelerates epigenetic aging of human liver. Proc. Natl. Acad. Sci. USA 2014, 111, 15538–15543. [Google Scholar] [CrossRef]
- Trovato, F.M.; Martines, G.F.; Brischetto, D.; Catalano, D.; Musumeci, G.; Trovato, G.M. Fatty liver disease and lifestyle in youngsters: Diet, food intake frequency, exercise, sleep shortage and fashion. Liver Int. 2016, 36, 427–433. [Google Scholar] [CrossRef]
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.H.; et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 1063–1075. [Google Scholar] [CrossRef]
- Bruce, K.D.; Cagampang, F.R.; Argenton, M.; Zhang, J.; Ethirajan, P.L.; Burdge, G.C.; Bateman, A.C.; Clough, G.F.; Poston, L.; Hanson, M.A.; et al. Maternal high-fat feeding primes steatohepatitis in adult mice offspring, involving mitochondrial dysfunction and altered lipogenesis gene expression. Hepatology 2009, 50, 1796–1808. [Google Scholar] [CrossRef]
- Portillo-Sanchez, P.; Bril, F.; Maximos, M.; Lomonaco, R.; Biernacki, D.; Orsak, B.; Subbarayan, S.; Webb, A.; Hecht, J.; Cusi, K. High Prevalence of Nonalcoholic Fatty Liver Disease in Patients With Type 2 Diabetes Mellitus and Normal Plasma Aminotransferase Levels. J. Clin. Endocrinol. Metab. 2015, 100, 2231–2238. [Google Scholar] [CrossRef]
- Singh, S.; Allen, A.M.; Wang, Z.; Prokop, L.J.; Murad, M.H.; Loomba, R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: A systematic review and meta-analysis of paired-biopsy studies. Clin. Gastroenterol. Hepatol. 2015, 13, 643–654. [Google Scholar] [CrossRef]
- Zhang, X.; Ji, X.; Wang, Q.; Li, J.Z. New insight into inter-organ crosstalk contributing to the pathogenesis of non-alcoholic fatty liver disease (NAFLD). Protein Cell 2018, 9, 164–177. [Google Scholar] [CrossRef]
- Stojsavljevic, S.; Gomercic Palcic, M.; Virovic Jukic, L.; Smircic Duvnjak, L.; Duvnjak, M. Adipokines and proinflammatory cytokines, the key mediators in the pathogenesis of nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 18070–18091. [Google Scholar] [CrossRef]
- Meng, X.; Li, S.; Li, Y.; Gan, R.-Y.; Li, H.-B. Gut Microbiota’s Relationship with Liver Disease and Role in Hepatoprotection by Dietary Natural Products and Probiotics. Nutrients 2018, 10, 1457. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, C. Adipose “talks” to distant organs to regulate insulin sensitivity and vascular function. Obesity (Silver Spring) 2010, 18, 2071–2076. [Google Scholar] [CrossRef]
- Khan, R.S.; Bril, F.; Cusi, K.; Newsome, P.N. Modulation of Insulin Resistance in Nonalcoholic Fatty Liver Disease. Hepatology 2019, 70, 711–724. [Google Scholar] [CrossRef]
- Loria, P.; Carulli, L.; Bertolotti, M.; Lonardo, A. Endocrine and liver interaction: The role of endocrine pathways in NASH. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 236–247. [Google Scholar] [CrossRef]
- Lombardi, R.; Fargion, S.; Fracanzani, A.L. Brain involvement in non-alcoholic fatty liver disease (NAFLD): A systematic review. Dig. Liver Dis. 2019, 51, 1214–1222. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.G.; Farrell, G.C. Epidemiology of non-alcoholic fatty liver disease in China. J. Hepatol. 2009, 50, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Ballestri, S.; Nascimbeni, F.; Baldelli, E.; Marrazzo, A.; Romagnoli, D.; Lonardo, A. NAFLD as a Sexual Dimorphic Disease: Role of Gender and Reproductive Status in the Development and Progression of Nonalcoholic Fatty Liver Disease and Inherent Cardiovascular Risk. Adv. Ther. 2017, 34, 1291–1326. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Nascimbeni, F.; Ballestri, S.; Fairweather, D.; Win, S.; Than, T.A.; Abdelmalek, M.F.; Suzuki, A. Sex Differences in Nonalcoholic Fatty Liver Disease: State of the Art and Identification of Research Gaps. Hepatology 2019, 70, 1457–1469. [Google Scholar] [CrossRef]
- Boyle, E.A.; Li, Y.I.; Pritchard, J.K. An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell 2017, 169, 1177–1186. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: The central role of nontriglyceride fatty acid metabolites. Hepatology 2010, 52, 774–788. [Google Scholar] [CrossRef]
- Cusi, K. Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: Pathophysiology and clinical implications. Gastroenterology 2012, 142, 711–725. [Google Scholar] [CrossRef]
- Hirsova, P.; Ibrahim, S.H.; Gores, G.J.; Malhi, H. Lipotoxic lethal and sublethal stress signaling in hepatocytes: Relevance to NASH pathogenesis. J. Lipid Res. 2016, 57, 1758–1770. [Google Scholar] [CrossRef]
- Lomonaco, R.; Ortiz-Lopez, C.; Orsak, B.; Webb, A.; Hardies, J.; Darland, C.; Finch, J.; Gastaldelli, A.; Harrison, S.; Tio, F.; et al. Effect of adipose tissue insulin resistance on metabolic parameters and liver histology in obese patients with nonalcoholic fatty liver disease. Hepatology 2012, 55, 1389–1397. [Google Scholar] [CrossRef]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin. Invest. 2005, 115, 1343–1351. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Campbell-Sargent, C.; Mirshahi, F.; Rizzo, W.B.; Contos, M.J.; Sterling, R.K.; Luketic, V.A.; Shiffman, M.L.; Clore, J.N. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001, 120, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Csak, T.; Ganz, M.; Pespisa, J.; Kodys, K.; Dolganiuc, A.; Szabo, G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology 2011, 54, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Kaufman, R.J. The role of ER stress in lipid metabolism and lipotoxicity. J. Lipid Res. 2016, 57, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Loomba, R.; Abraham, M.; Unalp, A.; Wilson, L.; Lavine, J.; Doo, E.; Bass, N.M.; Nonalcoholic Steatohepatitis Clinical Research. Association between diabetes, family history of diabetes, and risk of nonalcoholic steatohepatitis and fibrosis. Hepatology 2012, 56, 943–951. [Google Scholar] [CrossRef]
- Ioannou, G.N. The Role of Cholesterol in the Pathogenesis of NASH. Trends Endocrinol. Metab. 2016, 27, 84–95. [Google Scholar] [CrossRef]
- Trevaskis, J.L.; Griffin, P.S.; Wittmer, C.; Neuschwander-Tetri, B.A.; Brunt, E.M.; Dolman, C.S.; Erickson, M.R.; Napora, J.; Parkes, D.G.; Roth, J.D. Glucagon-like peptide-1 receptor agonism improves metabolic, biochemical, and histopathological indices of nonalcoholic steatohepatitis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G762–G772. [Google Scholar] [CrossRef]
- Boursier, J.; Mueller, O.; Barret, M.; Machado, M.; Fizanne, L.; Araujo-Perez, F.; Guy, C.D.; Seed, P.C.; Rawls, J.F.; David, L.A.; et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 2016, 63, 764–775. [Google Scholar] [CrossRef]
- Loomba, R.; Schork, N.; Chen, C.H.; Bettencourt, R.; Bhatt, A.; Ang, B.; Nguyen, P.; Hernandez, C.; Richards, L.; Salotti, J.; et al. Heritability of Hepatic Fibrosis and Steatosis Based on a Prospective Twin Study. Gastroenterology 2015, 149, 1784–1793. [Google Scholar] [CrossRef]
- Pirazzi, C.; Valenti, L.; Motta, B.M.; Pingitore, P.; Hedfalk, K.; Mancina, R.M.; Burza, M.A.; Indiveri, C.; Ferro, Y.; Montalcini, T.; et al. PNPLA3 has retinyl-palmitate lipase activity in human hepatic stellate cells. Hum. Mol. Genet. 2014, 23, 4077–4085. [Google Scholar] [CrossRef]
- Smagris, E.; BasuRay, S.; Li, J.; Huang, Y.; Lai, K.M.; Gromada, J.; Cohen, J.C.; Hobbs, H.H. Pnpla3I148M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology 2015, 61, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.H.; Tybjaerg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Cohen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.L.; Reeves, H.L.; Burt, A.D.; Tiniakos, D.; McPherson, S.; Leathart, J.B.; Allison, M.E.; Alexander, G.J.; Piguet, A.C.; Anty, R.; et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014, 5, 4309. [Google Scholar] [CrossRef]
- Hui, S.T.; Parks, B.W.; Org, E.; Norheim, F.; Che, N.; Pan, C.; Castellani, L.W.; Charugundla, S.; Dirks, D.L.; Psychogios, N.; et al. The genetic architecture of NAFLD among inbred strains of mice. Elife 2015, 4, e05607. [Google Scholar] [CrossRef] [PubMed]
- Hui, S.T.; Kurt, Z.; Tuominen, I.; Norheim, F.; Davis, R.C.; Pan, C.; Dirks, D.L.; Magyar, C.E.; French, S.W.; Chella Krishnan, K.; et al. The Genetic Architecture of Diet-Induced Hepatic Fibrosis in Mice. Hepatology 2018, 68, 2182–2196. [Google Scholar] [CrossRef] [PubMed]
- Arneson, D.; Shu, L.; Tsai, B.; Barrere-Cain, R.; Sun, C.; Yang, X. Multidimensional Integrative Genomics Approaches to Dissecting Cardiovascular Disease. Front. Cardiovasc. Med. 2017, 4, 8. [Google Scholar] [CrossRef]
- Meng, Q.; Makinen, V.P.; Luk, H.; Yang, X. Systems Biology Approaches and Applications in Obesity, Diabetes, and Cardiovascular Diseases. Curr. Cardiovasc. Risk Rep. 2013, 7, 73–83. [Google Scholar] [CrossRef]
- Zhao, Y.; Barrere-Cain, R.E.; Yang, X. Nutritional systems biology of type 2 diabetes. Genes Nutr. 2015, 10, 481. [Google Scholar] [CrossRef]
- Chai, L.E.; Loh, S.K.; Low, S.T.; Mohamad, M.S.; Deris, S.; Zakaria, Z. A review on the computational approaches for gene regulatory network construction. Comput. Biol. Med. 2014, 48, 55–65. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Song, W.M.; Zhang, B. Multiscale Embedded Gene Co-expression Network Analysis. PLoS Comput. Biol. 2015, 11, e1004574. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhou, D.; Qiu, W.; Shi, Y.; Yang, J.J.; Chen, S.; Wang, Q.; Pan, H. Application of Weighted Gene Co-expression Network Analysis for Data from Paired Design. Sci. Rep. 2018, 8, 622. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D. Introduction to Graphical Modelling; Springer: Berlin, Germany, 2012. [Google Scholar]
- Bernal, V.; Bischoff, R.; Guryev, V.; Grzegorczyk, M.; Horvatovich, P. Exact hypothesis testing for shrinkage based Gaussian Graphical Models. Bioinformatics 2019. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Guo, M.; Liu, X.; Wang, C.; Wang, L.; Zhang, Y. An improved Bayesian network method for reconstructing gene regulatory network based on candidate auto selection. BMC Genom. 2017, 18, 844. [Google Scholar] [CrossRef] [PubMed]
- Blencowe, M.; Arneson, D.; Ding, J.; Chen, Y.-W.; Saleem, Z.; Yang, X. Network modeling of single-cell omics data: Challenges, opportunities, and progresses. Emerg. Top. Life Sci. 2019, 3, 379–398. [Google Scholar] [CrossRef]
- Chen, S.; Mar, J.C. Evaluating methods of inferring gene regulatory networks highlights their lack of performance for single cell gene expression data. BMC Bioinform. 2018, 19, 232. [Google Scholar] [CrossRef]
- Chatr-Aryamontri, A.; Ceol, A.; Palazzi, L.M.; Nardelli, G.; Schneider, M.V.; Castagnoli, L.; Cesareni, G. MINT: The Molecular INTeraction database. Nucleic Acids Res. 2007, 35, D572–D574. [Google Scholar] [CrossRef]
- Peri, S.; Navarro, J.D.; Amanchy, R.; Kristiansen, T.Z.; Jonnalagadda, C.K.; Surendranath, V.; Niranjan, V.; Muthusamy, B.; Gandhi, T.K.; Gronborg, M.; et al. Development of human protein reference database as an initial platform for approaching systems biology in humans. Genome Res. 2003, 13, 2363–2371. [Google Scholar] [CrossRef]
- Stark, C.; Breitkreutz, B.J.; Reguly, T.; Boucher, L.; Breitkreutz, A.; Tyers, M. BioGRID: A general repository for interaction datasets. Nucleic Acids Res. 2006, 34, D535–D539. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Xenarios, I.; Fernandez, E.; Salwinski, L.; Duan, X.J.; Thompson, M.J.; Marcotte, E.M.; Eisenberg, D. DIP: The Database of Interacting Proteins: 2001 update. Nucleic Acids Res. 2001, 29, 239–241. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Kim, G.B.; Kim, W.J.; Kim, H.U.; Lee, S.Y. Current status and applications of genome-scale metabolic models. Genome Biol. 2019, 20, 121. [Google Scholar] [CrossRef] [PubMed]
- Agren, R.; Bordel, S.; Mardinoglu, A.; Pornputtapong, N.; Nookaew, I.; Nielsen, J. Reconstruction of genome-scale active metabolic networks for 69 human cell types and 16 cancer types using INIT. PLoS Comput. Biol. 2012, 8, e1002518. [Google Scholar] [CrossRef] [PubMed]
- Mardinoglu, A.; Agren, R.; Kampf, C.; Asplund, A.; Uhlen, M.; Nielsen, J. Genome-scale metabolic modelling of hepatocytes reveals serine deficiency in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014, 5, 3083. [Google Scholar] [CrossRef]
- Thiele, I.; Swainston, N.; Fleming, R.M.; Hoppe, A.; Sahoo, S.; Aurich, M.K.; Haraldsdottir, H.; Mo, M.L.; Rolfsson, O.; Stobbe, M.D. A community-driven global reconstruction of human metabolism. Nat. Biotechnol. 2013, 31, 419. [Google Scholar] [CrossRef]
- Gille, C.; Bölling, C.; Hoppe, A.; Bulik, S.; Hoffmann, S.; Hübner, K.; Karlstädt, A.; Ganeshan, R.; König, M.; Rother, K. HepatoNet1: A comprehensive metabolic reconstruction of the human hepatocyte for the analysis of liver physiology. Mol. Syst. Biol. 2010, 6, 411. [Google Scholar] [CrossRef]
- Kramer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Santos, A.; von Mering, C.; Jensen, L.J.; Bork, P.; Kuhn, M. STITCH 5: Augmenting protein-chemical interaction networks with tissue and affinity data. Nucleic Acids Res. 2016, 44, D380–D384. [Google Scholar] [CrossRef]
- Ekins, S.; Bugrim, A.; Brovold, L.; Kirillov, E.; Nikolsky, Y.; Rakhmatulin, E.; Sorokina, S.; Ryabov, A.; Serebryiskaya, T.; Melnikov, A.; et al. Algorithms for network analysis in systems-ADME/Tox using the MetaCore and MetaDrug platforms. Xenobiotica 2006, 36, 877–901. [Google Scholar] [CrossRef]
- Cirillo, E.; Parnell, L.D.; Evelo, C.T. A Review of Pathway-Based Analysis Tools That Visualize Genetic Variants. Front. Genet. 2017, 8, 174. [Google Scholar] [CrossRef]
- Pandey, V.; Hatzimanikatis, V. Investigating the deregulation of metabolic tasks via Minimum Network Enrichment Analysis (MiNEA) as applied to nonalcoholic fatty liver disease using mouse and human omics data. PLoS Comput. Biol. 2019, 15, e1006760. [Google Scholar] [CrossRef]
- Dweep, H.; Gretz, N. miRWalk2.0: A comprehensive atlas of microRNA-target interactions. Nat. Methods 2015, 12, 697. [Google Scholar] [CrossRef]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4. [Google Scholar] [CrossRef]
- Chen, Y.; Zhu, J.; Lum, P.Y.; Yang, X.; Pinto, S.; MacNeil, D.J.; Zhang, C.; Lamb, J.; Edwards, S.; Sieberts, S.K.; et al. Variations in DNA elucidate molecular networks that cause disease. Nature 2008, 452, 429–435. [Google Scholar] [CrossRef]
- Yang, X.; Deignan, J.L.; Qi, H.; Zhu, J.; Qian, S.; Zhong, J.; Torosyan, G.; Majid, S.; Falkard, B.; Kleinhanz, R.R.; et al. Validation of candidate causal genes for obesity that affect shared metabolic pathways and networks. Nat. Genet. 2009, 41, 415–423. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, B.; Molony, C.; Chudin, E.; Hao, K.; Zhu, J.; Gaedigk, A.; Suver, C.; Zhong, H.; Leeder, J.S.; et al. Systematic genetic and genomic analysis of cytochrome P450 enzyme activities in human liver. Genome Res. 2010, 20, 1020–1036. [Google Scholar] [CrossRef]
- Zhu, J.; Sova, P.; Xu, Q.; Dombek, K.M.; Xu, E.Y.; Vu, H.; Tu, Z.; Brem, R.B.; Bumgarner, R.E.; Schadt, E.E. Stitching together multiple data dimensions reveals interacting metabolomic and transcriptomic networks that modulate cell regulation. PLoS Biol. 2012, 10, e1001301. [Google Scholar] [CrossRef]
- Zhu, J.; Wiener, M.C.; Zhang, C.; Fridman, A.; Minch, E.; Lum, P.Y.; Sachs, J.R.; Schadt, E.E. Increasing the power to detect causal associations by combining genotypic and expression data in segregating populations. PLoS Comput. Biol. 2007, 3, e69. [Google Scholar] [CrossRef]
- Zhu, J.; Zhang, B.; Smith, E.N.; Drees, B.; Brem, R.B.; Kruglyak, L.; Bumgarner, R.E.; Schadt, E.E. Integrating large-scale functional genomic data to dissect the complexity of yeast regulatory networks. Nat. Genet. 2008, 40, 854–861. [Google Scholar] [CrossRef]
- Chan, K.H.; Huang, Y.T.; Meng, Q.; Wu, C.; Reiner, A.; Sobel, E.M.; Tinker, L.; Lusis, A.J.; Yang, X.; Liu, S. Shared molecular pathways and gene networks for cardiovascular disease and type 2 diabetes mellitus in women across diverse ethnicities. Circ. Cardiovasc. Genet. 2014, 7, 911–919. [Google Scholar] [CrossRef]
- Chella Krishnan, K.; Kurt, Z.; Barrere-Cain, R.; Sabir, S.; Das, A.; Floyd, R.; Vergnes, L.; Zhao, Y.; Che, N.; Charugundla, S.; et al. Integration of Multi-omics Data from Mouse Diversity Panel Highlights Mitochondrial Dysfunction in Non-alcoholic Fatty Liver Disease. Cell Syst. 2018, 6, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Huan, T.; Meng, Q.; Saleh, M.A.; Norlander, A.E.; Joehanes, R.; Zhu, J.; Chen, B.H.; Zhang, B.; Johnson, A.D.; Ying, S.; et al. Integrative network analysis reveals molecular mechanisms of blood pressure regulation. Mol. Syst. Biol. 2015, 11, 799. [Google Scholar] [CrossRef]
- Huan, T.; Zhang, B.; Wang, Z.; Joehanes, R.; Zhu, J.; Johnson, A.D.; Ying, S.; Munson, P.J.; Raghavachari, N.; Wang, R.; et al. A systems biology framework identifies molecular underpinnings of coronary heart disease. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1427–1434. [Google Scholar] [CrossRef] [PubMed]
- Makinen, V.P.; Civelek, M.; Meng, Q.; Zhang, B.; Zhu, J.; Levian, C.; Huan, T.; Segre, A.V.; Ghosh, S.; Vivar, J.; et al. Integrative genomics reveals novel molecular pathways and gene networks for coronary artery disease. PLoS Genet. 2014, 10, e1004502. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Ying, Z.; Noble, E.; Zhao, Y.; Agrawal, R.; Mikhail, A.; Zhuang, Y.; Tyagi, E.; Zhang, Q.; Lee, J.H.; et al. Systems Nutrigenomics Reveals Brain Gene Networks Linking Metabolic and Brain Disorders. EBioMedicine 2016, 7, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Zhuang, Y.; Ying, Z.; Agrawal, R.; Yang, X.; Gomez-Pinilla, F. Traumatic Brain Injury Induces Genome-Wide Transcriptomic, Methylomic, and Network Perturbations in Brain and Blood Predicting Neurological Disorders. EBioMedicine 2017, 16, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Shu, L.; Chan, K.H.K.; Zhang, G.; Huan, T.; Kurt, Z.; Zhao, Y.; Codoni, V.; Tregouet, D.A.; Cardiogenics, C.; Yang, J.; et al. Shared genetic regulatory networks for cardiovascular disease and type 2 diabetes in multiple populations of diverse ethnicities in the United States. PLoS Genet. 2017, 13, e1007040. [Google Scholar] [CrossRef] [PubMed]
- Shu, L.; Meng, Q.; Diamante, G.; Tsai, B.; Chen, Y.W.; Mikhail, A.; Luk, H.; Ritz, B.; Allard, P.; Yang, X. Prenatal Bisphenol A Exposure in Mice Induces Multitissue Multiomics Disruptions Linking to Cardiometabolic Disorders. Endocrinology 2019, 160, 409–429. [Google Scholar] [CrossRef]
- Zhao, Y.; Blencowe, M.; Shi, X.; Shu, L.; Levian, C.; Ahn, I.S.; Kim, S.K.; Huan, T.; Levy, D.; Yang, X. Integrative Genomics Analysis Unravels Tissue-Specific Pathways, Networks, and Key Regulators of Blood Pressure Regulation. Front. Cardiovasc. Med. 2019, 6, 21. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, J.; Freudenberg, J.M.; Meng, Q.; Rajpal, D.K.; Yang, X. Network-Based Identification and Prioritization of Key Regulators of Coronary Artery Disease Loci. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 928–941. [Google Scholar] [CrossRef]
- Zhao, Y.; Jhamb, D.; Shu, L.; Arneson, D.; Rajpal, D.K.; Yang, X. Multi-omics integration reveals molecular networks and regulators of psoriasis. BMC Syst. Biol. 2019, 13, 8. [Google Scholar] [CrossRef] [PubMed]
- Arneson, D.; Zhang, G.; Ying, Z.; Zhuang, Y.; Byun, H.R.; Ahn, I.S.; Gomez-Pinilla, F.; Yang, X. Single Cell Molecular Alterations Reveal Target Cells and Pathways of Concussive Brain Injury. Nat. Commun. 2019, 9, 3894. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, M.; Huynh, J.L.; Wang, K.; Yang, X.; Yoo, S.; McElwee, J.; Zhang, B.; Zhang, C.; Lamb, J.R.; Xie, T.; et al. Common dysregulation network in the human prefrontal cortex underlies two neurodegenerative diseases. Mol. Syst. Biol. 2014, 10, 743. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Byun, H.R.; Ying, Z.; Blencowe, M.; Zhao, Y.; Hong, J.; Shu, L.; Krishnan, K.C.K.; Gomez-Pinilla, F.; Yang, X. Differential Metabolic and Multi-tissue Transcriptomic Responses to Fructose Consumption among Genetically Diverse Mice. bioRxiv 2019, 439562. [Google Scholar] [CrossRef] [PubMed]
- Ying, Z.; Byun, H.R.; Meng, Q.; Noble, E.; Zhang, G.; Yang, X.; Gomez-Pinilla, F. Biglycan gene connects metabolic dysfunction with brain disorder. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2018, 1864, 3679–3687. [Google Scholar] [CrossRef]
- Maldonado, E.M.; Fisher, C.P.; Mazzatti, D.J.; Barber, A.L.; Tindall, M.J.; Plant, N.J.; Kierzek, A.M.; Moore, J.B. Multi-scale, whole-system models of liver metabolic adaptation to fat and sugar in non-alcoholic fatty liver disease. NPJ Syst. Biol. Appl. 2018, 4, 33. [Google Scholar] [CrossRef]
- Zhu, M.; Wang, Q.; Zhou, W.; Liu, T.; Yang, L.; Zheng, P.; Zhang, L.; Ji, G. Integrated analysis of hepatic mRNA and miRNA profiles identified molecular networks and potential biomarkers of NAFLD. Sci. Rep. 2018, 8, 7628. [Google Scholar] [CrossRef]
- Ma, H.; Xu, C.F.; Shen, Z.; Yu, C.H.; Li, Y.M. Application of Machine Learning Techniques for Clinical Predictive Modeling: A Cross-Sectional Study on Nonalcoholic Fatty Liver Disease in China. BioMed Res. Int. 2018, 2018, 4304376. [Google Scholar] [CrossRef]
- Shubham, K.; Vinay, L.; Vinod, P.K. Systems-level organization of non-alcoholic fatty liver disease progression network. Mol. Biosyst. 2017, 13, 1898–1911. [Google Scholar] [CrossRef]
- Mardinoglu, A.; Bjornson, E.; Zhang, C.; Klevstig, M.; Soderlund, S.; Stahlman, M.; Adiels, M.; Hakkarainen, A.; Lundbom, N.; Kilicarslan, M.; et al. Personal model-assisted identification of NAD(+) and glutathione metabolism as intervention target in NAFLD. Mol. Syst. Biol. 2017, 13, 916. [Google Scholar] [CrossRef]
- Hou, C.; Feng, W.; Wei, S.; Wang, Y.; Xu, X.; Wei, J.; Ma, Z.; Du, Y.; Guo, J.; He, Y.; et al. Bioinformatics Analysis of Key Differentially Expressed Genes in Nonalcoholic Fatty Liver Disease Mice Models. Gene Expr. 2018, 19, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Lou, Y.; Tian, G.Y.; Song, Y.; Liu, Y.L.; Chen, Y.D.; Shi, J.P.; Yang, J. Characterization of transcriptional modules related to fibrosing-NAFLD progression. Sci. Rep. 2017, 7, 4748. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.L.; Ming, Y.N.; Zhang, J.Y.; Chen, X.Y.; Zeng, M.D.; Mao, Y.M. Gene-metabolite network analysis in different nonalcoholic fatty liver disease phenotypes. Exp. Mol. Med. 2017, 49, e283. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Kuang, H.; Ansari, S.; Liu, T.; Gong, J.; Wang, S.; Zhao, X.Y.; Ji, Y.; Li, C.; Guo, L.; et al. Landscape of Intercellular Crosstalk in Healthy and NASH Liver Revealed by Single-Cell Secretome Gene Analysis. Mol. Cell 2019, 75, 644–660. [Google Scholar] [CrossRef]
- Sahini, N.; Borlak, J. Genomics of human fatty liver disease reveal mechanistically linked lipid droplet-associated gene regulations in bland steatosis and nonalcoholic steatohepatitis. Transl. Res. 2016, 177, 41–69. [Google Scholar] [CrossRef]
- Qi, S.; Wang, C.; Li, C.; Wang, P.; Liu, M. Candidate genes investigation for severe nonalcoholic fatty liver disease based on bioinformatics analysis. Medicine 2017, 96, e7743. [Google Scholar] [CrossRef]
- Chan, K.M.; Wu, T.H.; Wu, T.J.; Chou, H.S.; Yu, M.C.; Lee, W.C. Bioinformatics microarray analysis and identification of gene expression profiles associated with cirrhotic liver. Kaohsiung J. Med. Sci. 2016, 32, 165–176. [Google Scholar] [CrossRef]
- Chen, W.; Zhao, W.; Yang, A.; Xu, A.; Wang, H.; Cong, M.; Liu, T.; Wang, P.; You, H. Integrated analysis of microRNA and gene expression profiles reveals a functional regulatory module associated with liver fibrosis. Gene 2017, 636, 87–95. [Google Scholar] [CrossRef]
- Lee, S.; Zhang, C.; Liu, Z.; Klevstig, M.; Mukhopadhyay, B.; Bergentall, M.; Cinar, R.; Stahlman, M.; Sikanic, N.; Park, J.K.; et al. Network analyses identify liver-specific targets for treating liver diseases. Mol. Syst. Biol. 2017, 13, 938. [Google Scholar] [CrossRef]
- Karbalaei, R.; Allahyari, M.; Rezaei-Tavirani, M.; Asadzadeh-Aghdaei, H.; Zali, M.R. Protein-protein interaction analysis of Alzheimer’s disease and NAFLD based on systems biology methods unhide common ancestor pathways. Gastroenterol. Hepatol. Bed Bench 2018, 11, 27–33. [Google Scholar]
- Haas, J.T.; Vonghia, L.; Mogilenko, D.A.; Verrijken, A.; Molendi-Coste, O.; Fleury, S.; Deprince, A.; Nikitin, A.; Woitrain, E.; Ducrocq-Geoffroy, L.; et al. Transcriptional network analysis implicates altered hepatic immune function in NASH development and resolution. Nat. Metab. 2019, 1, 604–614. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Wang, X.; Zhuang, L. Gene expression profiling reveals key genes and pathways related to the development of non-alcoholic fatty liver disease. Ann. Hepatol. 2016, 15, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Gawrieh, S.; Baye, T.M.; Carless, M.; Wallace, J.; Komorowski, R.; Kleiner, D.E.; Andris, D.; Makladi, B.; Cole, R.; Charlton, M.; et al. Hepatic gene networks in morbidly obese patients with nonalcoholic fatty liver disease. Obes. Surg. 2010, 20, 1698–1709. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, T.; Zhang, C.; Tang, F.; Zhong, N.; Li, H.; Song, X.; Lin, H.; Liu, Y.; Xue, F. Identification of reciprocal causality between non-alcoholic fatty liver disease and metabolic syndrome by a simplified Bayesian network in a Chinese population. BMJ Open 2015, 5, e008204. [Google Scholar] [CrossRef]
- Kurt, Z.; Barrere-Cain, R.; LaGuardia, J.; Mehrabian, M.; Pan, C.; Hui, S.T.; Norheim, F.; Zhou, Z.; Hasin, Y.; Lusis, A.J.; et al. Tissue-specific pathways and networks underlying sexual dimorphism in non-alcoholic fatty liver disease. Biol. Sex Differ. 2018, 9, 46. [Google Scholar] [CrossRef]
- Hong, W.; Li, S.; Wu, L.; He, B.; Jiang, J.; Chen, Z. Prediction of VEGF-C as a Key Target of Pure Total Flavonoids From Citrus Against NAFLD in Mice via Network Pharmacology. Front. Pharmacol. 2019, 10, 582. [Google Scholar] [CrossRef]
- Barbosa, I.A.; Santos, E.M.; Paraíso, A.F.; Ferreira, P.V.; Oliveira, L.P.; Andrade, J.M.O.; Farias, L.C.; de Cravalho, B.M.; de Paula, A.M.B.; Guimarães, A.L.S.; et al. Liraglutide alters hepatic metabolism in high-fat fed obese mice: A bioinformatic prediction and functional analysis. Meta Gene 2019, 20, 100553. [Google Scholar] [CrossRef]
- Singh, S.; Khera, R.; Allen, A.M.; Murad, M.H.; Loomba, R. Comparative effectiveness of pharmacological interventions for nonalcoholic steatohepatitis: A systematic review and network meta-analysis. Hepatology 2015, 62, 1417–1432. [Google Scholar] [CrossRef]
- Krishnan, A.; Li, X.; Kao, W.-Y.; Viker, K.; Butters, K.; Masuoka, H.; Knudsen, B.; Gores, G.; Charlton, M. Lumican, an extracellular matrix proteoglycan, is a novel requisite for hepatic fibrosis. Lab. Investig. 2012, 92, 1712. [Google Scholar] [CrossRef]
- Nomoto, S.; Kanda, M.; Okamura, Y.; Nishikawa, Y.; Qiyong, L.; Fujii, T.; Sugimoto, H.; Takeda, S.; Nakao, A. Epidermal growth factor-containing fibulin-like extracellular matrix protein 1, EFEMP1, a novel tumor-suppressor gene detected in hepatocellular carcinoma using double combination array analysis. Ann. Surg. Oncol. 2010, 17, 923–932. [Google Scholar] [CrossRef]
- Vestentoft, P.S.; Jelnes, P.; Andersen, J.B.; Tran, T.A.T.; Jørgensen, T.; Rasmussen, M.; Bornholdt, J.; Grøvdal, L.M.; Jensen, C.H.; Vogel, L.K. Molecular constituents of the extracellular matrix in rat liver mounting a hepatic progenitor cell response for tissue repair. Fibrogenesis Tissue Repair 2013, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Agostini, J.; Benoist, S.; Seman, M.; Julié, C.; Imbeaud, S.; Letourneur, F.; Cagnard, N.; Rougier, P.; Brouquet, A.; Zucman-Rossi, J. Identification of molecular pathways involved in oxaliplatin-associated sinusoidal dilatation. J. Hepatol. 2012, 56, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]
- Zhan, Z.; Chen, Y.; Duan, Y.; Li, L.; Mew, K.; Hu, P.; Ren, H.; Peng, M. Identification of key genes, pathways and potential therapeutic agents for liver fibrosis using an integrated bioinformatics analysis. PeerJ 2019, 7, e6645. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Kerner, J.; Hoppel, C.L. Mitochondrial carnitine palmitoyltransferase 1a (CPT1a) is part of an outer membrane fatty acid transfer complex. J. Biol. Chem. 2011, 286, 25655–25662. [Google Scholar] [CrossRef] [PubMed]
- Jerby, L.; Shlomi, T.; Ruppin, E. Computational reconstruction of tissue-specific metabolic models: Application to human liver metabolism. Mol. Syst. Biol. 2010, 6, 401. [Google Scholar] [CrossRef]
- Kalhan, S.C.; Guo, L.; Edmison, J.; Dasarathy, S.; McCullough, A.J.; Hanson, R.W.; Milburn, M. Plasma metabolomic profile in nonalcoholic fatty liver disease. Metabolism 2011, 60, 404–413. [Google Scholar] [CrossRef]
- Bjornson, E.; Adiels, M.; Taskinen, M.R.; Boren, J. Kinetics of plasma triglycerides in abdominal obesity. Curr. Opin. Lipidol. 2017, 28, 11–18. [Google Scholar] [CrossRef]
- Kolak, M.; Westerbacka, J.; Velagapudi, V.R.; Wagsater, D.; Yetukuri, L.; Makkonen, J.; Rissanen, A.; Hakkinen, A.M.; Lindell, M.; Bergholm, R.; et al. Adipose tissue inflammation and increased ceramide content characterize subjects with high liver fat content independent of obesity. Diabetes 2007, 56, 1960–1968. [Google Scholar] [CrossRef]
- Blachnio-Zabielska, A.U.; Pulka, M.; Baranowski, M.; Nikolajuk, A.; Zabielski, P.; Gorska, M.; Gorski, J. Ceramide metabolism is affected by obesity and diabetes in human adipose tissue. J. Cell. Physiol. 2012, 227, 550–557. [Google Scholar] [CrossRef]
- Masoodi, M.; Kuda, O.; Rossmeisl, M.; Flachs, P.; Kopecky, J. Lipid signaling in adipose tissue: Connecting inflammation & metabolism. Biochim. Biophys. Acta 2015, 1851, 503–518. [Google Scholar] [CrossRef]
- Chakrabarti, S.K.; Wen, Y.; Dobrian, A.D.; Cole, B.K.; Ma, Q.; Pei, H.; Williams, M.D.; Bevard, M.H.; Vandenhoff, G.E.; Keller, S.R.; et al. Evidence for activation of inflammatory lipoxygenase pathways in visceral adipose tissue of obese Zucker rats. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E175–E187. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Clemente, M.; Claria, J.; Titos, E. The 5-lipoxygenase/leukotriene pathway in obesity, insulin resistance, and fatty liver disease. Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Lusis, A.J.; Seldin, M.M.; Allayee, H.; Bennett, B.J.; Civelek, M.; Davis, R.C.; Eskin, E.; Farber, C.R.; Hui, S.; Mehrabian, M. The Hybrid Mouse Diversity Panel: A resource for systems genetics analyses of metabolic and cardiovascular traits. J. Lipid Res. 2016, 57, 925–942. [Google Scholar] [CrossRef] [PubMed]
- Shu, L.; Blencowe, M.; Yang, X. Translating GWAS Findings to Novel Therapeutic Targets for Coronary Artery Disease. Front. Cardiovasc. Med. 2018, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- VanWagner, L.B. New insights into NAFLD and subclinical coronary atherosclerosis. J. Hepatol. 2018, 68, 890–892. [Google Scholar] [CrossRef] [PubMed]
- Paredes-Turrubiarte, G.; Gonzalez-Chavez, A.; Perez-Tamayo, R.; Salazar-Vazquez, B.Y.; Hernandez, V.S.; Garibay-Nieto, N.; Fragoso, J.M.; Escobedo, G. Severity of non-alcoholic fatty liver disease is associated with high systemic levels of tumor necrosis factor alpha and low serum interleukin 10 in morbidly obese patients. Clin. Exp. Med. 2016, 16, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Jorge, A.S.B.; Andrade, J.M.O.; Paraíso, A.F.; Jorge, G.C.B.; Silveira, C.M.; de Souza, L.R.; Santos, E.P.; Guimaraes, A.L.S.; Santos, S.H.S.; De-Paula, A.M.B. Body mass index and the visceral adipose tissue expression of IL-6 and TNF-alpha are associated with the morphological severity of non-alcoholic fatty liver disease in individuals with class III obesity. Obes. Res. Clin. Pract. 2018, 12, 1–8. [Google Scholar] [CrossRef]
- Cojocaru, I.M.; Cojocaru, M.; Miu, G.; Sapira, V. Study of interleukin-6 production in Alzheimer’s disease. Rom. J. Intern. Med. 2011, 49, 55–58. [Google Scholar]
- Sclip, A.; Tozzi, A.; Abaza, A.; Cardinetti, D.; Colombo, I.; Calabresi, P.; Salmona, M.; Welker, E.; Borsello, T. c-Jun N-terminal kinase has a key role in Alzheimer disease synaptic dysfunction in vivo. Cell Death Dis. 2014, 5, e1019. [Google Scholar] [CrossRef]
- Shu, L.; Zhao, Y.; Kurt, Z.; Byars, S.G.; Tukiainen, T.; Kettunen, J.; Orozco, L.D.; Pellegrini, M.; Lusis, A.J.; Ripatti, S.; et al. Mergeomics: Multidimensional data integration to identify pathogenic perturbations to biological systems. BMC Genom. 2016, 17, 874. [Google Scholar] [CrossRef] [PubMed]
- Arneson, D.; Bhattacharya, A.; Shu, L.; Makinen, V.P.; Yang, X. Mergeomics: A web server for identifying pathological pathways, networks, and key regulators via multidimensional data integration. BMC Genom. 2016, 17, 722. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, C.; Lee, S.; Kim, W.; Klevstig, M.; Harzandi, A.M.; Sikanic, N.; Arif, M.; Stahlman, M.; Nielsen, J.; et al. Pyruvate kinase L/R is a regulator of lipid metabolism and mitochondrial function. Metab. Eng. 2019, 52, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Almanza, D.; Gharaee-Kermani, M.; Zhilin-Roth, A.; Rodriguez-Nieves, J.A.; Colaneri, C.; Riley, T.; Macoska, J.A. Nonalcoholic Fatty Liver Disease Demonstrates a Pre-fibrotic and Premalignant Molecular Signature. Dig. Dis. Sci. 2019, 64, 1257–1269. [Google Scholar] [CrossRef] [PubMed]
- Cvitanovic Tomas, T.; Urlep, Z.; Moskon, M.; Mraz, M.; Rozman, D. LiverSex Computational Model: Sexual Aspects in Hepatic Metabolism and Abnormalities. Front. Physiol. 2018, 9, 360. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C.P.; Plant, N.J.; Moore, J.B.; Kierzek, A.M. QSSPN: Dynamic simulation of molecular interaction networks describing gene regulation, signalling and whole-cell metabolism in human cells. Bioinformatics 2013, 29, 3181–3190. [Google Scholar] [CrossRef]
- Boeckmans, J.; Natale, A.; Buyl, K.; Rogiers, V.; De Kock, J.; Vanhaecke, T.; Rodrigues, R.M. Comment to Letter to the editor: Human-based systems: Mechanistic NASH modelling just around the corner? Pharmacol. Res. 2018, 137, 282–283. [Google Scholar] [CrossRef]
- Cassidy, S.; Syed, B.A. Nonalcoholic steatohepatitis (NASH) drugs market. Nat. Rev. Drug Discov. 2016, 15, 745–746. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Application categories | Paper | Network Method | Omics Data | Tissue | Pathways | Key Drivers/ Findings |
|---|---|---|---|---|---|---|
| Prediction | Maldonado et al. [97] | ODE, GEM, and QSSPN Stochastic Simulation | Transcriptomics and Proteomics | Liver (human) | Lipid metabolism | No significant difference between glucose and fructose on lipogenesis. PPARα activation implicated in steatosis |
| Zhu et al. [98] | STRING and miRWalk | Transcriptomics | Liver (rat) | Lipid metabolism (steroid synthesis, fat digestion, bile secretion) Inflammatory/Immune response (NF-κB, cytokine signaling) | Abcg8, Cyp1a1, Cyp51, Hmgcr, Acat2, Cyp7a1, Cyp7b1, Cd36, Cd44, RT1-Da, RT1-Ba, RT1-Bb, RT1-Db1 | |
| Ma et al. [99] | Bayesian | N/A | N/A | N/A | Bayesian network provides screening and predictive value for NAFLD | |
| Pathogenesis | Pandey et al. [72] | MiNEA and GEM | Metabolomics, Proteomics and Transcriptomics | Liver (mouse and human) | Inflammatory/Immune response (ceramide synthesis, oxidative stress) Lipid metabolism (cholesterol synthesis) | Identified deregulation in metabolic networks of ceramide and hydrogen peroxide synthesis for NASH in both mice and humans |
| Mardinoglu et al. [65] | GEM | Metabolomics and Proteomics | Liver (human) | Lipid metabolism (peroxisome, steroid biosynthesis, FA biosynthesis) Protein metabolism (amino acid turnover) Inflammatory/Immune response Mitochondrial stress | Constructed a consensus GEM for hepatocytes termed iHepatocytes2322, which was used to identify serine deficiency in NASH and possible therapeutic targets PSPH, SHMT1 and BCAT1 | |
| Shubham et al. [100] | WGCNA and GEM | Transcriptomics | Visceral Adipose Tissue (human) | Lipid metabolismInflammatory/Immune response (hypoxia) ECM remodeling Protein metabolism | FOSL1, HIF1A, CHSY1, NAMPT, NAMP, NCOR2, SUV39H1, SUV420H1, CHD9, CAT, ALDH2, HADH, ETFA, ETFB, PPRC1, CYP2C8, ADH4, DAPK1 | |
| Mardinoglu et al. [101] | GEM | Metabolomics and Proteomics | Liver and Adipose (human) | Lipid metabolism Carbohydrate metabolism GSH biosynthesis NAD+ repletion | Dietary supplementation of GSH and NAD+ precursors are possible NAFLD treatment options | |
| Hou et al. [102] | STRING | Transcriptomics | Liver (mouse) | Lipid metabolism (steroid synthesis, FA activation) Cell cycle (PI3K–Akt signaling) Inflammatory/Immune response (phagocytosis) | Itgb2, Hck, Rac2, CD48 Ptprc, Jun, Cd68, Tyrobp, Ctss, Itgax, Hsd3b5, Cyp2c44 Cyp2c54, Cyp1a2, Cyp2c70, Ugt2b1, C8b, Egfr, Gyp7b1, Slco1a1 | |
| Lou et al. [103] | WGCNA | Transcriptomics | Liver (human) | Lipid metabolism Inflammatory/Immune response (fibrosis) Cell cycle (PI3K-Akt signaling) Cell adhesion ECM remodeling | LUM, THBS2, FBN1, EFEMP1, SELENBP1 | |
| Liu et al. [104] | STRING | Transcriptomics and Metabolomics | Liver and Blood (rat) | Lipid metabolism Inflammatory/Immune response (TNF, cytokine signaling, TLR signaling) Cell cycle (NF-κB, p53 signaling) ECM remodeling Mitochondrial stress | Jun, Ccl2, Ccl12, Icam1, Cxcl2, Cdkn1a, Serpine1, Rprm, Fabp4, Fabp5, Bcl2a1, Cxcl10, Olr1 | |
| Krishnan et al. [82] | WGCNA, MEGENA and Bayesian | Genomics and Transcriptomics | Liver and Adipose (mouse) | Lipid metabolism Cell cycle Inflammatory/Immune response (peroxisomal pathways) Insulin signaling Carbohydrate metabolism (TCA cycle) Apoptosis | Fasn, Pklr, Chchd6, Thrsp, Cd36, Acly, Hmgcr, Acaca, Acacb, Col1a2, Elovl6, Ptpn6, Echs1 | |
| Xiong et al. (Single cell) [105] | Ligand-Receptor Interaction (Fantom 5) | Transcriptomics and Proteomics | Liver (mouse) | Lipid metabolism Inflammatory/Immune response (cytokine signaling) Cell adhesion ECM remodeling | Hepatic stellate cells serve as a hub of intrahepatic cell signaling. Identification of novel Trem2+ NASH-associated macrophages (NAMs) linked to disease severity | |
| Sahini et al. [106] | STRING | Genomics and Transcriptomics | Liver (human) | Lipid metabolism (lipogenesis, lipid droplet growth) Inflammatory/Immune response Carbohydrate metabolism Mitochondrial stress | PLIN2, CIDEC, HILPDA, STAT1 | |
| Qi et al. [107] | PPI (HPRD) | Transcriptomics | Liver (human) | Inflammatory/Immune response (cytokine activity) Cell adhesion | UBQLN4, APP, SHBG, CTNNB1, COL1A1 | |
| Chan et al. [108] | MetaCore | Transcriptomics | Liver (human) | Lipid metabolism Carbohydrate metabolism Inflammatory/Immune response Cell cycle ECM remodeling (fibrosis) | Identified 87 “significant” genes (not hub genes) associated with cirrhosis | |
| Chen et al. [109] | miRWalk and STRING | Transcriptomics | Liver (human) | ECM remodeling (focal adhesion, fibrosis) Cell cycle (PI3K-Akt signaling) | COL6A1, COL6A2, COL6A3, PIK3R3, COL1A1, CCND2 | |
| Lee et al. [110] | Co-expression, PPI, TR and GEM | Metabolomics, Proteomics and Transcriptomics | Liver, Adipose, and Muscle (human) | Lipid metabolism (FASN enzyme activity) Inflammatory/Immune response Mitochondrial stress (transport) Cell Cycle | FASN, PKLR, PNPLA3, PCSK9 | |
| Comorbidities | Karbalaei et al. (AD) [111] | STRING (DisGeNet) | Genomics, Transcriptomics and Proteomics | Not specified (human) | Lipid metabolism Carbohydrate metabolism Insulin signaling Regulation of JAK-STAT Inflammatory/Immune response (IL signaling) | IL6, AKT1, TP53, TNF JUN, VEGFA, PPARG, MAPK3, IGF1, LEP |
| Haas et al. (Obesity) [112] | WGCNA | Transcriptomics | Liver (human) | Inflammatory/Immune response | CXCL9, CXCL10, and LYZ higher expression in patients with NASH than steatosis | |
| Wang et al. (Obesity) [113] | PPI (HPRD) | Transcriptomics | Liver (human) | Cell cycle Cell adhesion Protein metabolism Inflammatory/Immune response (phagocytosis) | PRKCA, EGFR, CDC42, VEGFA, CRK | |
| Gawrieh et al. (Obesity) [114] | Ingenuity Pathway Analysis (IPA) | Transcriptomics | Liver (human) | Lipid metabolism Cell cycle (development, movement) Apoptosis (ubiquitination) Inflammatory/Immune response (cell death) | COL1A1, IL10, DCN, IGFBP3, HSPA5, USP25, FABP4, PPFIBP1, ZAK, RGN, SMUG1, CYP4F22, CSN2 | |
| Zhang et al. (MetS) [115] | Bayesian | N/A | N/A | Inflammatory response (oxidative stress) Insulin signaling Lipid metabolism (dyslipidemia) | The effect of MetS on NAFLD is significantly greater than that of NAFLD on MetS | |
| Sex Differences | Kurt et al. [116] | WGCNA, MEGENA and Bayesian | Genomics and Transcriptomics | Liver and Adipose (mouse and human) | Lipid metabolism Protein metabolism Insulin signaling Inflammatory/Immune response Cell cycle Apoptosis ECM remodeling | AHSG, FASN, RBP4, SREBF1, ACOT2, DECR1, DHCR7, SQLE, INSIG1, ACSS2, BCKDHA, MCCC1, ECHS1, DCA8, MKI67, CCNA2, FBN1, COL1A2, CCDC80, RELB, IFNG, CXCL10, PTPRO, SH3BP2, TYROBP, C8B, CD36, CPT2, CHCHD6, GYS1, INPP5D, FCER1G, NCKAP1L, ANXA2, CIDEC, PNPLA3, TRIB1, CY7B1, HCK, FGL2, SLC2A3 |
| Drugs | Hong et al. (PTFC) [117] | WGCNA and STRING | Transcriptomics and Toxicogenomics | Liver (mouse) | Lipid metabolism Cell cycle (activation) Cell adhesion Inflammatory/Immune response (Toll-like receptor, cytokine and chemokine signaling) Apoptosis | VEGF-C and COL4A1 may play a regulatory role in NAFLD development and are possible targets of PTFC |
| Barbosa et al. (GLP-1 Receptor Agonist - Liraglutide) [118] | STRING and STITCH | Transcriptomics and Proteomics | Liver (mouse) | Insulin signaling Cell cycle Inflammatory/Immune response Lipid metabolism | AKT1, RPS6KB1/S6K1; Liraglutide decreases liver fat content and improves metabolic conditions | |
| Singh et al. (Vitamin E, Pentoxifylline, Obeticholic Acid and TZDs) [119] | Bayesian | N/A | N/A | N/A | Pentoxifylline and Obeticholic Acid improve fibrosis. Vitamin E, TZDs, and Obeticholic Acid improve ballooning degeneration in NASH patients |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blencowe, M.; Karunanayake, T.; Wier, J.; Hsu, N.; Yang, X. Network Modeling Approaches and Applications to Unravelling Non-Alcoholic Fatty Liver Disease. Genes 2019, 10, 966. https://doi.org/10.3390/genes10120966
Blencowe M, Karunanayake T, Wier J, Hsu N, Yang X. Network Modeling Approaches and Applications to Unravelling Non-Alcoholic Fatty Liver Disease. Genes. 2019; 10(12):966. https://doi.org/10.3390/genes10120966
Chicago/Turabian StyleBlencowe, Montgomery, Tilan Karunanayake, Julian Wier, Neil Hsu, and Xia Yang. 2019. "Network Modeling Approaches and Applications to Unravelling Non-Alcoholic Fatty Liver Disease" Genes 10, no. 12: 966. https://doi.org/10.3390/genes10120966
APA StyleBlencowe, M., Karunanayake, T., Wier, J., Hsu, N., & Yang, X. (2019). Network Modeling Approaches and Applications to Unravelling Non-Alcoholic Fatty Liver Disease. Genes, 10(12), 966. https://doi.org/10.3390/genes10120966