Genomic Diversity of Common Sequence Types of Listeria monocytogenes Isolated from Ready-to-Eat Products of Animal Origin in South Africa


Abstract
1. Introduction
2. Materials and Methods
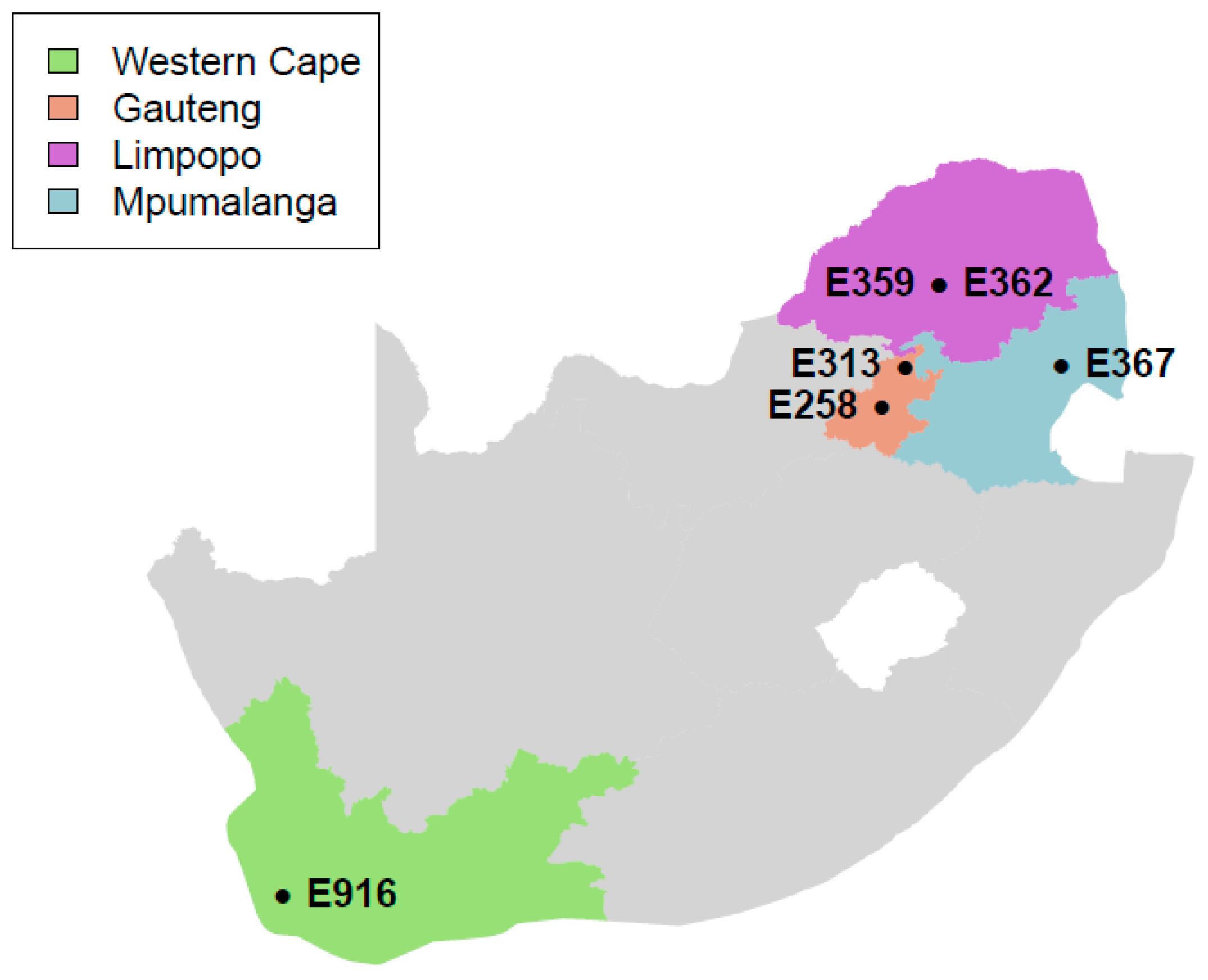
2.1. Sample Information
2.2. Microbiological Analysis
2.3. Genomic Deoxyribonucleic Acid (DNA) Extraction
2.4. Genome Sequencing and De Novo Assembly
2.5. Core Genome Determination
2.6. Core Genome Phylogenetic Analysis
2.7. Prophage Identification and Analysis
2.8. Determination of Plasmids
2.9. Average Nucleotide Identity Calculation
2.10. In Silico PCR-Serogroup and ST Prediction
2.11. Virulence Factors
2.12. Resistance Profiles
2.13. Data Availability
3. Results
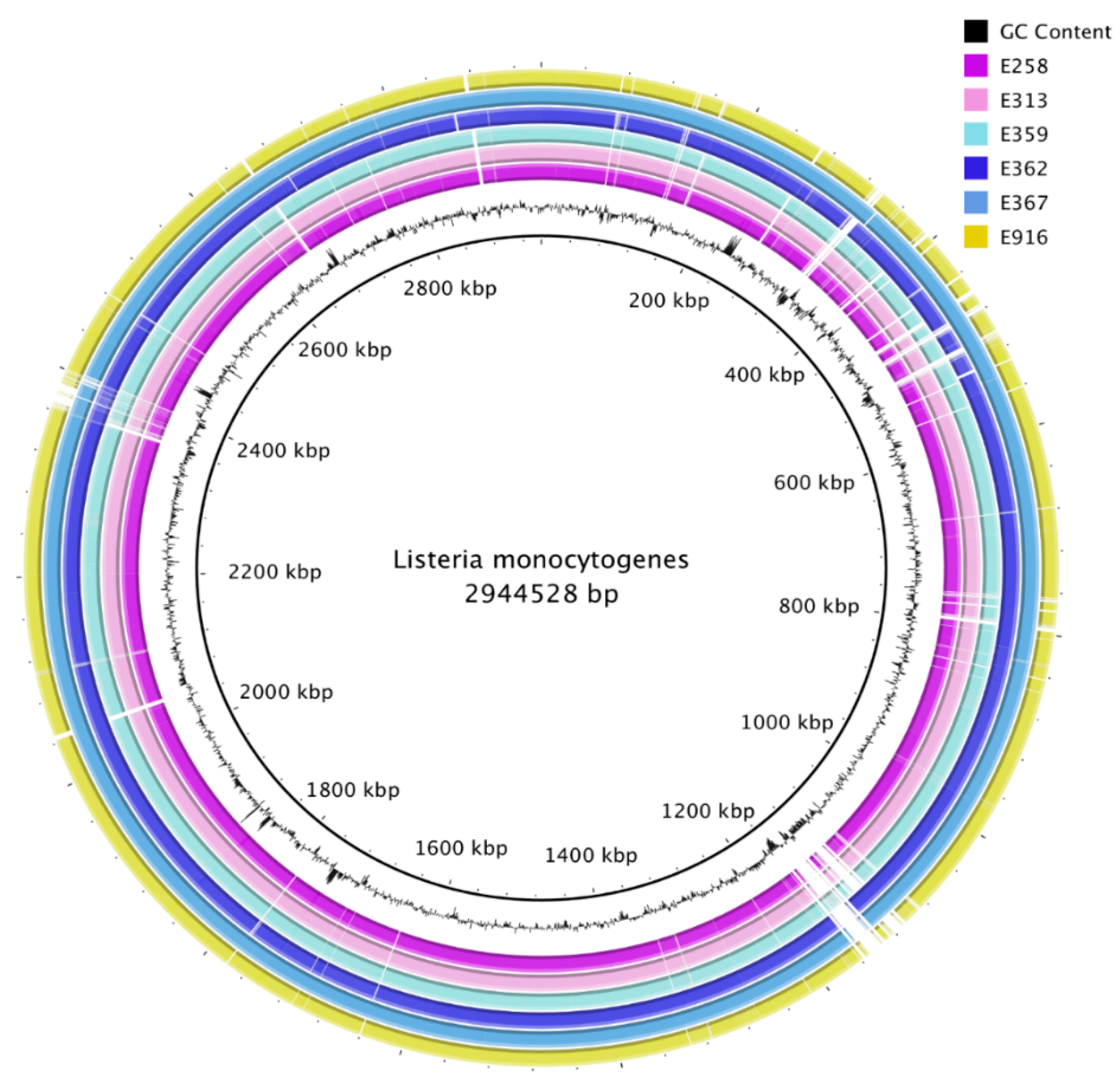
3.1. Genome Sequencing, Assembly, and Annotation
3.2. Plasmid Identification
3.3. Stress Survival Islet
3.4. Listeria Pathogenicity Islands
3.5. Multi-Locus Sequence Typing (MLST) and PCR-Serogroups
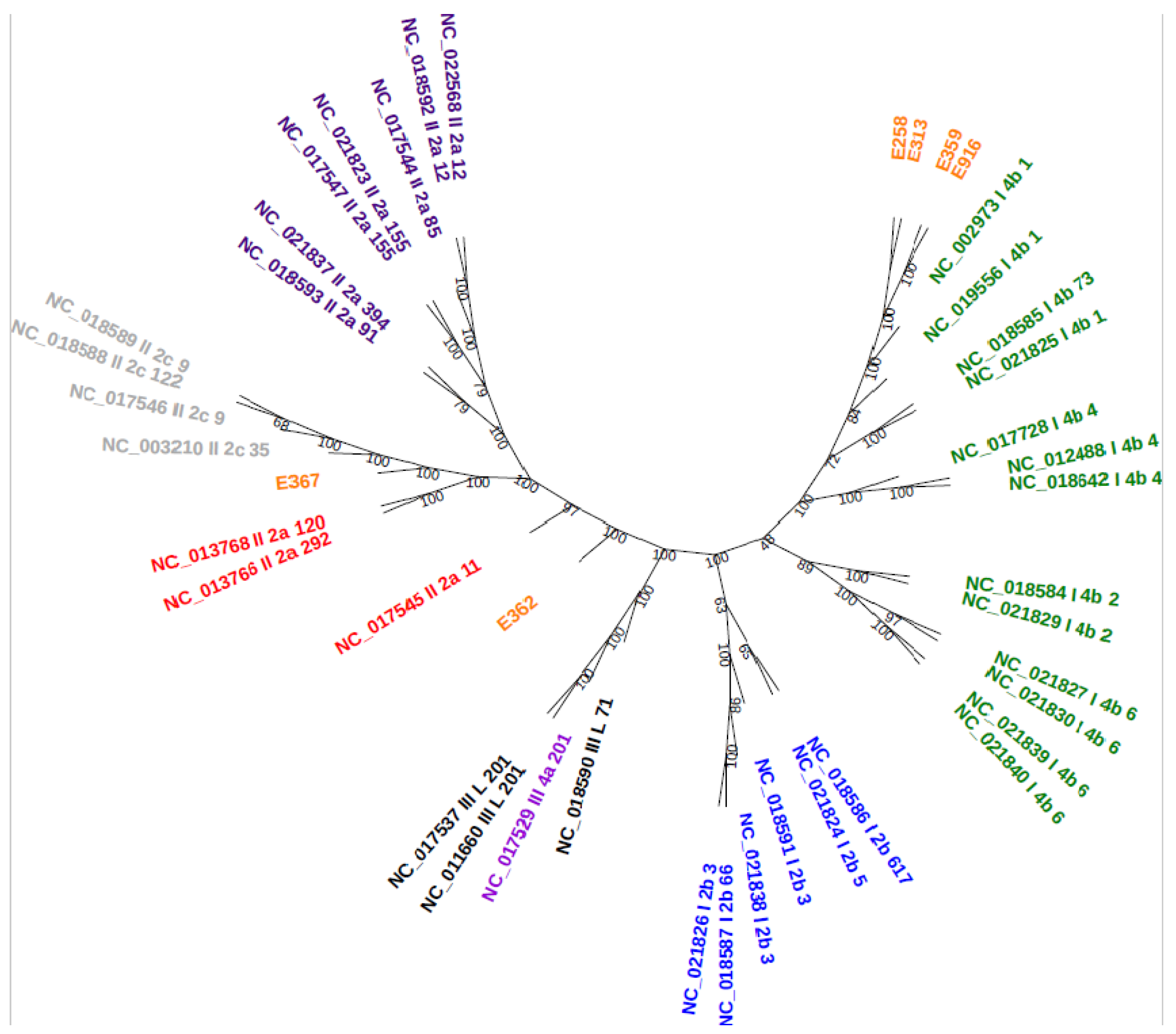
3.6. Core-Genome Phylogenetic Analysis
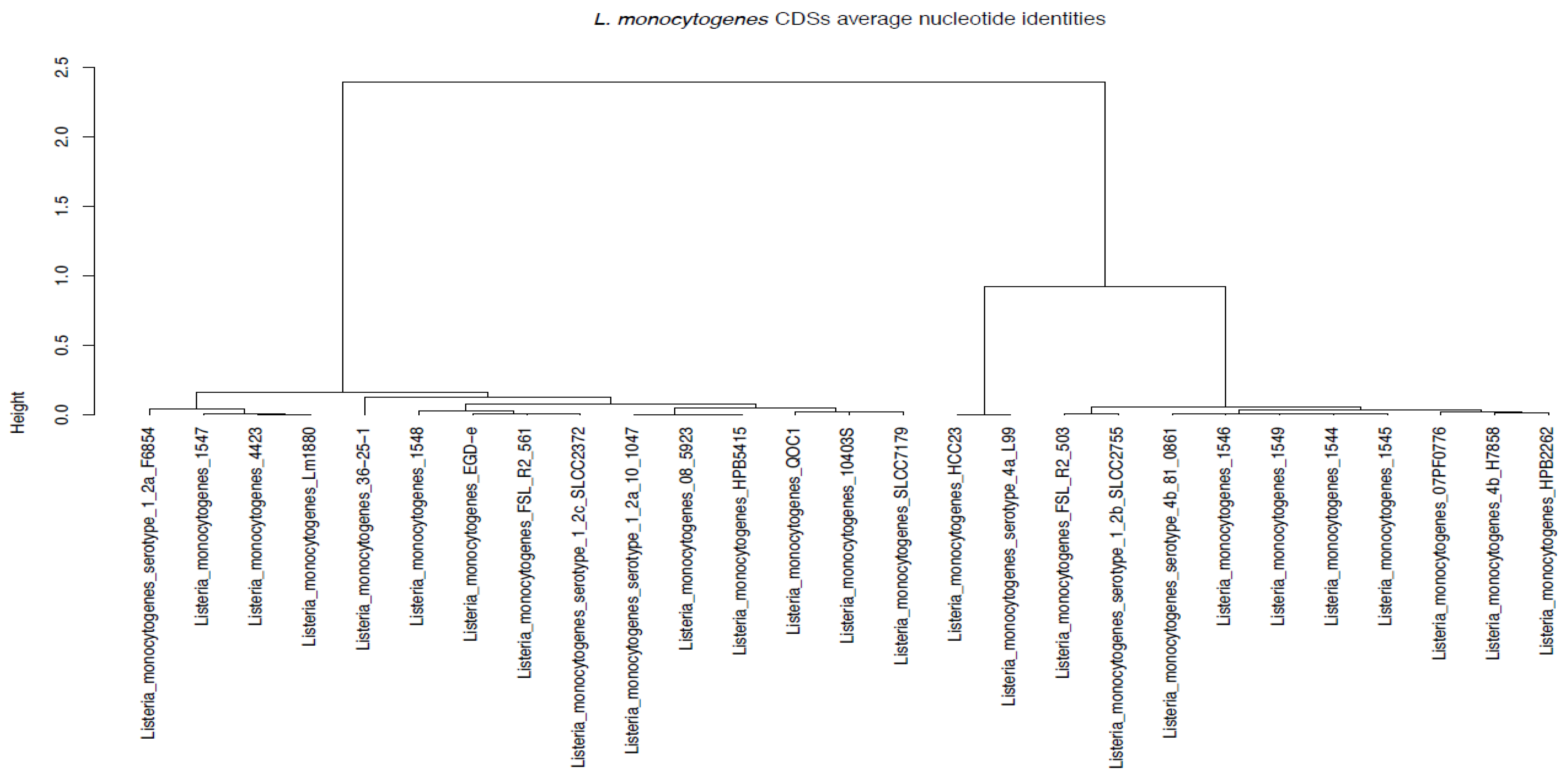
3.7. Average Nucleotide Identities
3.8. Virulence Factors
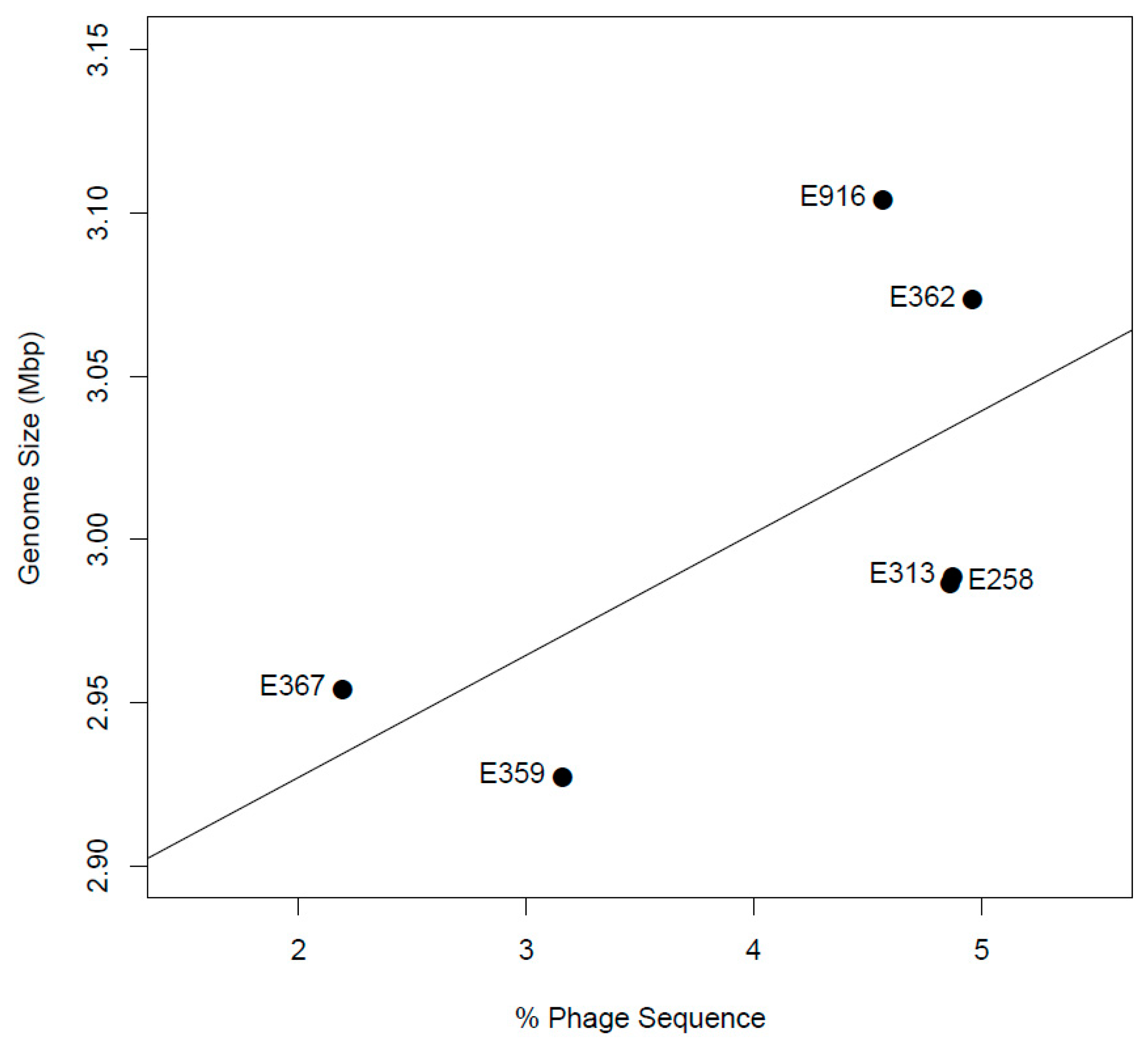
3.9. Prophage Identification and Analysis
3.10. Resistance Genes
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hilliard, A.; Leong, D.; Callaghan, A.O.; Culligan, E.P.; Morgan, C.A.; Delappe, N.; Hill, C.; Id, K.J.; Cormican, M.; Gahan, C.G.M. Genomic Characterization of Listeria monocytogenes Isolates Associated with Clinical Listeriosis and the Food Production Environment in Ireland. Genes 2018, 9, 171. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.; Begley, M.; Hill, C.; Gahan, C.G.M. A five-gene stress survival islet (SSI-1) that contributes to the growth of Listeria monocytogenes in suboptimal conditions. J. Appl. Microbiol. 2010, 109, 984–995. [Google Scholar] [CrossRef] [PubMed]
- Burall, L.S.; Grim, C.J.; Mammel, M.K.; Datta, A.R. Whole genome sequence analysis using jspecies tool establishes clonal relationships between Listeria monocytogenes strains from epidemiologically unrelated listeriosis outbreaks. PLoS ONE 2016, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Den Bakker, H.C.; Cummings, C.A.; Ferreira, V.; Vatta, P.; Orsi, R.H.; Degoricija, L.; Barker, M.; Petrauskene, O.; Furtado, M.R.; Wiedmann, M. Comparative genomics of the bacterial genus Listeria: Genome evolution is characterized by limited gene acquisition and limited gene loss. BMC Genomics 2010, 11, 688. [Google Scholar] [CrossRef] [PubMed]
- Haase, J.K.; Didelot, X.; Lecuit, M.; Korkeala, H.; Achtman, M.; Leclercq, A.; Grant, K.; Wiedmann, M.; Apfalter, P. The ubiquitous nature of Listeria monocytogenes clones: A large-scale Multilocus Sequence Typing study. Environ. Microbiol. 2014, 16, 405–416. [Google Scholar] [CrossRef]
- Laksanalamai, P.; Huang, B.; Sabo, J.; Burall, L.S.; Zhao, S.; Bates, J.; Datta, A.R. Genomic characterization of novel Listeria monocytogenes serotype 4b variant strains. PLoS ONE 2014, 9, e89024. [Google Scholar] [CrossRef]
- Hyden, P.; Pietzka, A.; Lennkh, A.; Murer, A.; Springer, B.; Blaschitz, M.; Indra, A.; Huhulescu, S.; Allerberger, F.; Ruppitsch, W.; et al. Whole genome sequence-based serogrouping of Listeria monocytogenes isolates. J. Biotechnol. 2016, 235, 181–186. [Google Scholar] [CrossRef]
- Liu, D. Identification, subtyping and virulence determination of Listeria monocytogenes, an important foodborne pathogen. J. Med. Microbiol. 2006, 55, 645–659. [Google Scholar] [CrossRef]
- Franz, E.; Gras, L.M.; Dallman, T. Significance of whole genome sequencing for surveillance, source attribution and microbial risk assessment of foodborne pathogens. Curr. Opin. Food Sci. 2016, 8, 74–79. [Google Scholar] [CrossRef]
- Schmid, D.; Allerberger, F.; Huhulescu, S.; Pietzka, A.; Amar, C.; Kleta, S.; Prager, R.; Preußel, K.; Aichinger, E. Whole genome sequencing as a tool to investigate a cluster of seven cases of listeriosis in Austria and Germany, 2011–2013. Clin. Microbiol. Infect. 2014, 20, 431–436. [Google Scholar] [CrossRef]
- Jackson, B.R.; Tarr, C.; Strain, E.; Jackson, K.A.; Conrad, A.; Carleton, H.; Katz, L.S.; Stroika, S.; Gould, L.H.; Mody, R.K.; et al. Implementation of Nationwide Real-time Whole-genome Sequencing to Enhance Listeriosis Outbreak Detection and Investigation. Clin. Infect. Dis. 2016, 63, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Gilmour, M.W.; Graham, M.; Van Domselaar, G.; Tyler, S.; Kent, H.; Trout-Yakel, K.M.; Larios, O.; Allen, V.; Lee, B.; Nadon, C. High-throughput genome sequencing of two Listeria monocytogenes clinical isolates during a large foodborne outbreak. BMC Genomics 2010, 11, 120. [Google Scholar] [CrossRef] [PubMed]
- Jensen, A. Factors Influencing Persistence and Virulence of Listeria monocytogenes. Ph.D. Thesis, Technical University of Denmark, Copenhagen, Denmark, 2007. [Google Scholar]
- Smith, A.M.; Naicker, P.; Bamford, C.; Shuping, L.; McCarthy, K.M.; Sooka, A.; Smouse, S.L.; Tau, N.; Keddy, K.H. Genome Sequences for a Cluster of Human Isolates of Listeria monocytogenes Identified in South Africa in 2015. Genome Announc. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Tau, N.P.; Smouse, S.L.; Allam, M.; Ismail, A.; Ramalwa, N.R.; Disenyeng, B.; Ngomane, M.; Thomas, J. Outbreak of Listeria monocytogenes in South Africa, 2017–2018: Laboratory Activities and Experiences Associated with Whole-Genome Sequencing Analysis of Isolates. Foodborne Pathog. Dis. 2019, 16, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Leong, D.; Alvarez-Ordóñez, A.; Jooste, P.; Jordan, K. Listeria monocytogenes in food: Control by monitoring the food processing environment. Afr. J. Microbiol. Res. 2016, 10, 1–14. [Google Scholar]
- Naidoo, K. The Microbial Ecology of Biltong in South Africa during Production and at Point-of-Sale. Ph.D. Thesis, University of the Witwatersrand, Johannesburg, South Afrika, 2014. [Google Scholar]
- Matle, I.; Mbatha, K.R.; Lentsoane, O.; Magwedere, K.; Morey, L.; Madoroba, E. Occurrence, serotypes, and characteristics of Listeria monocytogenes in meat and meat products in South Africa between 2014 and 2016. J. Food Saf. 2019, 39, e12629. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Alikhan, N.F.; Petty, N.K.; Ben Zakour, N.L.; Beatson, S.A. BLAST Ring Image Generator (BRIG): Simple prokaryote genome comparisons. BMC Genomics 2011, 12, 402. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed]
- Moura, A.; Tourdjman, M.; Leclercq, A.; Hamelin, E.; Laurent, E.; Fredriksen, N.; Van Cauteren, D.; Bracq-dieye, H.; Thouvenot, P.; Vales, G.; et al. Real-time whole-genome sequencing for surveillance of Listeria monocytogenes, France. Emerg. Infect. Dis. 2017, 23, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Lipman, D.J. Protein database searches for multiple alignments. Proc. Natl. Acad. Sci. USA 2019, 87, 5509–5513. [Google Scholar] [CrossRef] [PubMed]
- Enright, A.J.; Van Dongen, S.; Ouzounis, C.A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30, 1575–1584. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, 242–245. [Google Scholar] [CrossRef]
- Bosi, E.; Donati, B.; Galardini, M.; Brunetti, S.; Sagot, M.F.; Lió, P.; Crescenzi, P.; Fani, R.; Fondi, M. MeDuSa: A multi-draft based scaffolder. Bioinformatics 2015, 31, 2443–2451. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, 16–21. [Google Scholar] [CrossRef]
- Carattoli, A.; Miriagou, V.; Bertini, A.; Loli, A.; Colinon, C.; Villa, L.; Whichard, J.M.; Rossolini, G.M. Replicon typing of plasmids encoding resistance to newer β-lactams. Emerg. Infect. Dis. 2006, 12, 1145–1148. [Google Scholar] [CrossRef]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Jordan, I.K.; Rishishwar, L. stringMLST: A fast k-mer based tool for multilocus sequence typing. Bioinformatics 2017, 33, 119–121. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zheng, D.; Liu, B.; Yang, J.; Jin, Q. VFDB 2016: Hierarchical and refined dataset for big data analysis—10 years on. Nucleic Acids Res. 2016, 44, D694–D697. [Google Scholar] [CrossRef] [PubMed]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef] [PubMed]
- Pal, C.; Bengtsson-Palme, J.; Rensing, C.; Kristiansson, E.; Larsson, D.G.J. BacMet: Antibacterial biocide and metal resistance genes database. Nucleic Acids Res. 2014, 42, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Lakin, S.M.; Dean, C.; Noyes, N.R.; Dettenwanger, A.; Ross, A.S.; Doster, E.; Rovira, P.; Abdo, Z.; Jones, K.L.; Ruiz, J.; et al. MEGARes: An antimicrobial resistance database for high throughput sequencing. Nucleic Acids Res. 2017, 45, D574–D580. [Google Scholar] [CrossRef]
- Harter, E.; Wagner, E.M.; Zaiser, A.; Halecker, S.; Wagner, M.; Rychli, K. Stress Survival Islet 2, Predominantly Present in Listeria monocytogenes Strains of Sequence Type 121, Is Involved in the Alkaline and Oxidative Stress Responses. Appl. Environ. Microbiol. 2017, 83. [Google Scholar] [CrossRef]
- Muhterem-Uyar, M.; Ciolacu, L.; Wagner, K.; Wagner, M.; Schmitz-Esser, S.; Stessl, B. New Aspects on Listeria monocytogenes ST5-ECVI Predominance in a Heavily Contaminated Cheese Processing Environment. Front. Microbiol. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- McNair, K.; Bailey, B.A.; Edwards, R.A. PHACTS, a computational approach to classifying the lifestyle of phages. Bioinformatics 2012, 28, 614–618. [Google Scholar] [CrossRef]
- Kuenne, C.; Billion, A.; Mraheil, M.A.; Strittmatter, A.; Daniel, R.; Goesmann, A.; Barbuddhe, S.; Hain, T.; Chakraborty, T. Reassessment of the Listeria monocytogenes pan-genome reveals dynamic integration hotspots and mobile genetic elements as major components of the accessory genome. BMC Genomics 2013, 14, 1–19. [Google Scholar] [CrossRef]
- Schmitz-Esser, S.; Müller, A.; Stessl, B.; Wagner, M. Genomes of sequence type 121 Listeria monocytogenes strains harbor highly conserved plasmids and prophages. Front. Microbiol. 2015, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Ragon, M.; Wirth, T.; Hollandt, F.; Lavenir, R.; Lecuit, M.; Monnier, A.L.; Brisse, S. A New Perspective on Listeria monocytogenes Evolution. PLoS Pathog. 2008, 4, e1000146. [Google Scholar] [CrossRef] [PubMed]
- Chenal-Francisque, V.; Lopez, J.; Cantinelli, T.; Caro, V.; Tran, C.; Leclercq, A.; Lecuit, M.; Brisse, S. World wide distribution of major clones of listeria monocytogenes. Emerg. Infect. Dis. 2011, 17, 1110–1112. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, A.; Zhu, R.; Lan, R.; Jin, D.; Cui, Z.; Wang, Y.; Li, Z.; Wang, Y.; Xu, J.; et al. Genetic diversity and molecular typing of Listeria monocytogenes in China. BMC Microbiol. 2012, 12, 119. [Google Scholar] [CrossRef] [PubMed]
- Fox, E.M.; Allnutt, T.; Bradbury, M.I.; Fanning, S.; Chandry, P.S. Comparative Genomics of the Listeria monocytogenes ST204 Subgroup. Front. Microbiol. 2016, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Suokko, A.; Savijoki, K.; Malinen, E.; Palva, A.; Varmanen, P. Characterization of a mobile clpL gene from Lactobacillus rhamnosus. Appl. Environ. Microbiol. 2005, 71, 2061–2069. [Google Scholar] [CrossRef]
- Klumpp, J.; Loessner, M.J. Listeria phages: Genomes, evolution, and application. J. Bacteriophage 2013, 3, e26861. [Google Scholar] [CrossRef]
- Denes, T.; Vongkamjan, K.; Ackermann, H.; Switt, I.M.; Wiedmann, M. Comparative Genomic and Morphological Analyses of Listeria Phages Isolated from Farm Environments. Appl. Environ. Microbiol. 2014, 80, 4616–4625. [Google Scholar] [CrossRef]
- Casey, A.; Jordan, K.; Neve, H.; Coffey, A.; Mcauliffe, O. A tail of two phages: Genomic and functional analysis of Listeria monocytogenes phages vB_LmoS_188 and vB_LmoS_293 reveal the receptor-binding proteins involved in host specificity. Front. Microbiol. 2015, 6, 1–14. [Google Scholar] [CrossRef]
- Verghese, B.; Lok, M.; Wen, J.; Alessandria, V.; Chen, Y.; Kathariou, S.; Knabel, S. comK Prophage junction fragments as markers for Listeria monocytogenes Genotypes unique to individual meat and poultry processing plants and a model for rapid niche-specific adaptation, biofilm formation, and persistence. Appl. Environ. Microbiol. 2011, 77, 5064. [Google Scholar] [CrossRef]
- Rabinovich, L.; Sigal, N.; Borovok, I.; Nir-Paz, R.; Herskovits, A.A. Prophage excision activates Listeria competence genes that promote phagosomal escape and virulence. Cell 2012, 150, 792–802. [Google Scholar] [CrossRef] [PubMed]
- Nightingale, K.K.; Schukken, Y.H.; Nightingale, C.R.; Fortes, E.D.; Ho, A.J.; Her, Z.; Grohn, Y.T.; Mcdonough, P.L.; Wiedmann, M. Ecology and Transmission of Listeria monocytogenes infecting ruminants and in the farm environment. Appl. Environ. Microbiol. 2004, 70, 4458–4467. [Google Scholar] [CrossRef] [PubMed]
- Cotter, P.D.; Draper, L.A.; Lawton, E.M.; Daly, K.M.; Groeger, D.S.; Casey, P.G.; Ross, R.P.; Hill, C. Listeriolysin S, a novel peptide haemolysin associated with a subset of lineage I Listeria monocytogenes. PLoS Pathog. 2008, 4, e1000144. [Google Scholar] [CrossRef] [PubMed]
- Chenal-francisque, V.; Maury, M.M.; Lavina, M.; Touchon, M.; Leclercq, A.; Lecuit, M. Clonogrouping, A Rapid Multiplex PCR Method for Identification of Major Clones of Listeria monocytogenes. J. Clin. Microbiol. 2015, 53, 3355–3358. [Google Scholar] [CrossRef] [PubMed]
- Ducey, T.F.; Page, B.; Usgaard, T.; Borucki, M.K.; Pupedis, K.; Ward, T.J. A Single-Nucleotide-Polymorphism-Based Multilocus Genotyping Assay for Subtyping Lineage I Isolates of Listeria monocytogenes. Appl. Environ. Microbiol. 2006, 73, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Rychli, K.; Wagner, E.M.; Ciolacu, L.; Zaiser, A.; Tasara, T.; Wagner, M.; Schmitz-Esser, S. Comparative genomics of human and non- human Listeria monocytogenes sequence type 121 strains. PLoS ONE 2017, 12, e0176857. [Google Scholar] [CrossRef] [PubMed]
- Mata, M.T.; Baquero, F.; Pe, J.C. A multidrug efflux transporter in Listeria monocytogenes. FEMS Microbiol. Lett. 2000, 187, 185–188. [Google Scholar] [CrossRef]
- Mereghetti, L.; Lanotte, P.; Savoye-Marczuk, V.; Marquet-Van Der Mee, N.; Audurier, A.; Quentin, R. Combined ribotyping and random multiprimer DNA analysis to probe the population structure of Listeria monocytogenes. Appl. Environ. Microbiol. 2002, 68, 2849–2857. [Google Scholar] [CrossRef]
- Elhanafi, D.; Utta, V.; Kathariou, S. Genetic characterization of plasmid-associated benzalkonium chloride resistance determinants in a Listeria monocytogenes train from the 1998–1999 outbreak. Appl. Environ. Microbiol. 2010, 76, 8231–8238. [Google Scholar] [CrossRef]
- Müller, A.; Rychli, K.; Muhterem-Uyar, M.; Zaiser, A.; Stessl, B.; Guinane, C.M.; Cotter, P.D.; Wagner, M.; Schmitz-Esser, S. Tn6188—A Novel Transposon in Listeria monocytogenes Responsible for Tolerance to Benzalkonium Chloride. PLoS ONE 2013, 8, e76835. [Google Scholar] [CrossRef]
- Meier, A.B.; Guldimann, C.; Markkula, A.; Pöntinen, A. Comparative Phenotypic and Genotypic Analysis of Swiss and Finnish Listeria monocytogenes Isolates with Respect to Benzalkonium Chloride Resistance. Front. Microbiol. 2017, 8, 57. [Google Scholar] [CrossRef] [PubMed]
- O’Grady, J.; Sedano-Balbás, S.; Maher, M.; Smith, T.; Barry, T. Rapid real-time PCR detection of Listeria monocytogenes in enriched food samples based on the ssrA gene, a novel diagnostic target. Food Microbiol. 2008, 25, 75–84. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | * MLST-ST | Lineage | Serogroup | * NCBI RefSeq Accession Number |
|---|---|---|---|---|
| EGD-e | 35 | II | 2c | NC_003210 |
| 07PF0776 | 4 | I | 4b | NC_017728 |
| 08-5578 | 292 | II | 2a | NC_013766 |
| 08-5923 | 120 | II | 2a | NC_013768 |
| 10403S | 85 | II | 2a | NC_017544 |
| ATCC 19117 | 2 | I | 4b | NC_018584 |
| C1-387 | 155 | II | 2a | NC_021823 |
| Clip81459 | 4 | I | 4b | NC_012488 |
| F2365 | 1 | I | 4b | NC_002973 |
| FInlAnd 1998 | 155 | II | 2a | NC_017547 |
| FSL R2-561 | 9 | II | 2c | NC_017546 |
| HCC23 | 201 | III | L | NC_011660 |
| J0161 | 11 | II | 2a | NC_017545 |
| J1-220 | 6 | I | 4b | NC_021830 |
| J1776 | 6 | I | 4b | NC_021839 |
| J1816 | 2 | I | 4b | NC_021829 |
| J1817 | 6 | I | 4b | NC_021827 |
| J1926 | 6 | I | 4b | NC_021840 |
| J2-031 | 394 | II | 2a | NC_021837 |
| J2-064 | 5 | I | 2b | NC_021824 |
| J2-1091 | 1 | I | 4b | NC_021825 |
| L312 | 4 | I | 4b | NC_018642 |
| L99 | 201 | III | 4a | NC_017529 |
| LL195 | 1 | I | 4b | NC_019556 |
| M7 | 201 | III | L | NC_017537 |
| N1-011A | 3 | I | 2b | NC_021826 |
| R2-502 | 3 | I | 2b | NC_021838 |
| SLCC2372 | 122 | II | 2c | NC_018588 |
| SLCC2376 | 71 | III | L | NC_018590 |
| SLCC2378 | 73 | I | 4b | NC_018585 |
| SLCC2479 | 9 | II | 2c | NC_018589 |
| SLCC2482 | 3 | I | 2b | NC_018591 |
| SLCC2540 | 617 | I | 2b | NC_018586 |
| SLCC2755 | 66 | I | 2b | NC_018587 |
| SLCC5850 | 12 | II | 2a | NC_018592 |
| SLCC7179 | 91 | II | 2a | NC_018593 |
| EGD-e | 12 | II | 2a | NC_022568 |
| Isolates Characteristics | L. monocytogenes Strain | |||||
|---|---|---|---|---|---|---|
| E258 | E313 | E359 | E362 | E367 | E916 | |
| Source | Biltong | Biltong | Biltong | Biltong | Biltong | Polony |
| Year isolated | 2016 | 2015 | 2016 | 2015 | 2016 | 2015 |
| Province (food establishment) | 1 (butchery) | 1 (butchery) | 2 (retail outlet) | 2 (retail outlet) | 3 (retail outlet) | 4 (retail outlet) |
| Genome length (bp) * | 2,994,232 | 2,997,211 | 3,020,685 | 3,101,293 | 2,994,400 | 3,107,420 |
| Number of contigs | 38 | 48 | 127 | 44 | 42 | 39 |
| G+C Content (%) | 37.88 | 37.90 | 37.84 | 37.82 | 37.98 | 37.70 |
| N50 | 411,134 | 411,134 | 63,798 | 480,372 | 437,780 | 521,621 |
| No. of plasmids | 0 | 0 | 0 | 0 | 0 | 0 |
| Number of Proteins | 2983 | 2985 | 2967 | 3070 | 2955 | 3110 |
| Listeria pathogenicity islands (LIPI-1) | + | + | + | + | + | + |
| Listeria pathogenicity islands (LIPI-3) | + | + | + | - | - | + |
| Stress Survival Islet (SSI-1) | - | + | - | - | - | - |
| Stress Survival Islet (SSI-2) | - | - | - | + | + | - |
| Strain | Source | MLST | Lineage | PCR-Serogroup |
|---|---|---|---|---|
| E258 | Biltong | 1 | I | IVb |
| E313 | Biltong | 1 | I | IVb |
| E359 | Biltong | 876 | I | IVb |
| E362 | Biltong | 121 | II | IIa |
| E367 | Biltong | 204 | II | IIa |
| E916 | Polony | 1 | I | IVb |
| Strain | Scaffold Length (bp) | Prophage Number | Status | Size (Kb) | Number of Proteins | Position | Most Common Phage |
|---|---|---|---|---|---|---|---|
| E258 | 2,986,597 | 1 | Questionable | 10.7 | 17 | 39,802–50,530 | PHAGE_Lister_A118_NC_003216(5) |
| 2 | Intact | 53.3 | 78 | 2,287,899–2,341,226 | PHAGE_Lister_vB_LmoS_188_NC_028871(31) | ||
| 3 | Incomplete | 33 | 29 | 2,554,382–2,587,413 | PHAGE_Lister_vB_LmoS_188_NC_028871(13) | ||
| 4 | Intact | 48.1 | 62 | 2,938,128–2,986,260 | PHAGE_Lister_vB_LmoS_293_NC_028929(42) | ||
| E313 | 2,988,893 | 1 | Intact | 41.3 | 62 | 338–41,646 | PHAGE_Lister_vB_LmoS_293_NC_028929(42) |
| 2 | Incomplete | 42.1 | 29 | 390,171–432,271 | PHAGE_Lister_vB_LmoS_188_NC_028871(13) | ||
| 3 | Intact | 51.4 | 74 | 645,517–697,015 | PHAGE_Lister_vB_LmoS_188_NC_028871(28) | ||
| 4 | Questionable | 10.7 | 17 | 2,935,515–2,946,243 | PHAGE_Lister_A118_NC_003216(5) | ||
| E359 | 2,927,179 | 1 | Intact | 37.1 | 57 | 261,259–298,366 | PHAGE_Lister_vB_LmoS_188_NC_028871(31) |
| 2 | Intact | 44.6 | 70 | 565,277–609,901 | PHAGE_Lister_vB_LmoS_293_NC_028929(46) | ||
| 3 | Questionable | 10.7 | 17 | 2,778,084–2,788,812 | PHAGE_Lister_A118_NC_003216(5) | ||
| E362 | 3,073,586 | 1 | Incomplete | 10.7 | 17 | 416,400–427,126 | PHAGE_Lister_A118_NC_003216(6) |
| 2 | Intact | 32.2 | 45 | 1,028,426–1,060,665 | PHAGE_Lister_A006_NC_009815(7) | ||
| 3 | Intact | 40.6 | 55 | 1,630,887–1,671,526 | PHAGE_Lister_LP_101_NC_024387(40) | ||
| 4 | Incomplete | 26.2 | 18 | 2,743,121–2,769,384 | PHAGE_Lister_A500_NC_009810(9) | ||
| 5 | Incomplete | 15.1 | 22 | 2,803,865–2,819,005 | PHAGE_Lister_A118_NC_003216(10) | ||
| 6 | Incomplete | 27.4 | 39 | 3,010,996–3,038,428 | PHAGE_Lister_A118_NC_003216(29) | ||
| E367 | 2,954,073 | 1 | Questionable | 10.7 | 17 | 40,491–51,217 | PHAGE_Lister_A118_NC_003216(5) |
| 2 | Questionable | 42.8 | 64 | 568,244–611,055 | PHAGE_Lister_A500_NC_009810(35) | ||
| 3 | Incomplete | 11.2 | 18 | 1,359,390–1,370,672 | PHAGE_Psychr_pOW20_A_NC_020841(1) | ||
| E916 | 3,104,227 | 1 | Questionable | 10.7 | 17 | 99,555–110,283 | PHAGE_Lister_A118_NC_003216(5) |
| 2 | Intact | 40.7 | 63 | 646,498–687,254 | PHAGE_Lister_LP_030_2_NC_021539(50) | ||
| 3 | Intact | 41.4 | 58 | 822,319–863,785 | PHAGE_Lister_vB_LmoS_188_NC_028871(46) | ||
| 4 | Intact | 48.8 | 76 | 1,932,379–1,981,196 | PHAGE_Lister_B054_NC_009813(67) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matle, I.; Pierneef, R.; Mbatha, K.R.; Magwedere, K.; Madoroba, E. Genomic Diversity of Common Sequence Types of Listeria monocytogenes Isolated from Ready-to-Eat Products of Animal Origin in South Africa. Genes 2019, 10, 1007. https://doi.org/10.3390/genes10121007
Matle I, Pierneef R, Mbatha KR, Magwedere K, Madoroba E. Genomic Diversity of Common Sequence Types of Listeria monocytogenes Isolated from Ready-to-Eat Products of Animal Origin in South Africa. Genes. 2019; 10(12):1007. https://doi.org/10.3390/genes10121007
Chicago/Turabian StyleMatle, Itumeleng, Rian Pierneef, Khanyisile R. Mbatha, Kudakwashe Magwedere, and Evelyn Madoroba. 2019. "Genomic Diversity of Common Sequence Types of Listeria monocytogenes Isolated from Ready-to-Eat Products of Animal Origin in South Africa" Genes 10, no. 12: 1007. https://doi.org/10.3390/genes10121007
APA StyleMatle, I., Pierneef, R., Mbatha, K. R., Magwedere, K., & Madoroba, E. (2019). Genomic Diversity of Common Sequence Types of Listeria monocytogenes Isolated from Ready-to-Eat Products of Animal Origin in South Africa. Genes, 10(12), 1007. https://doi.org/10.3390/genes10121007