The Fusion of Lipid and DNA Nanotechnology

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Liposomes
2.1. Liposome Structure
2.2. Lipid Chemistry
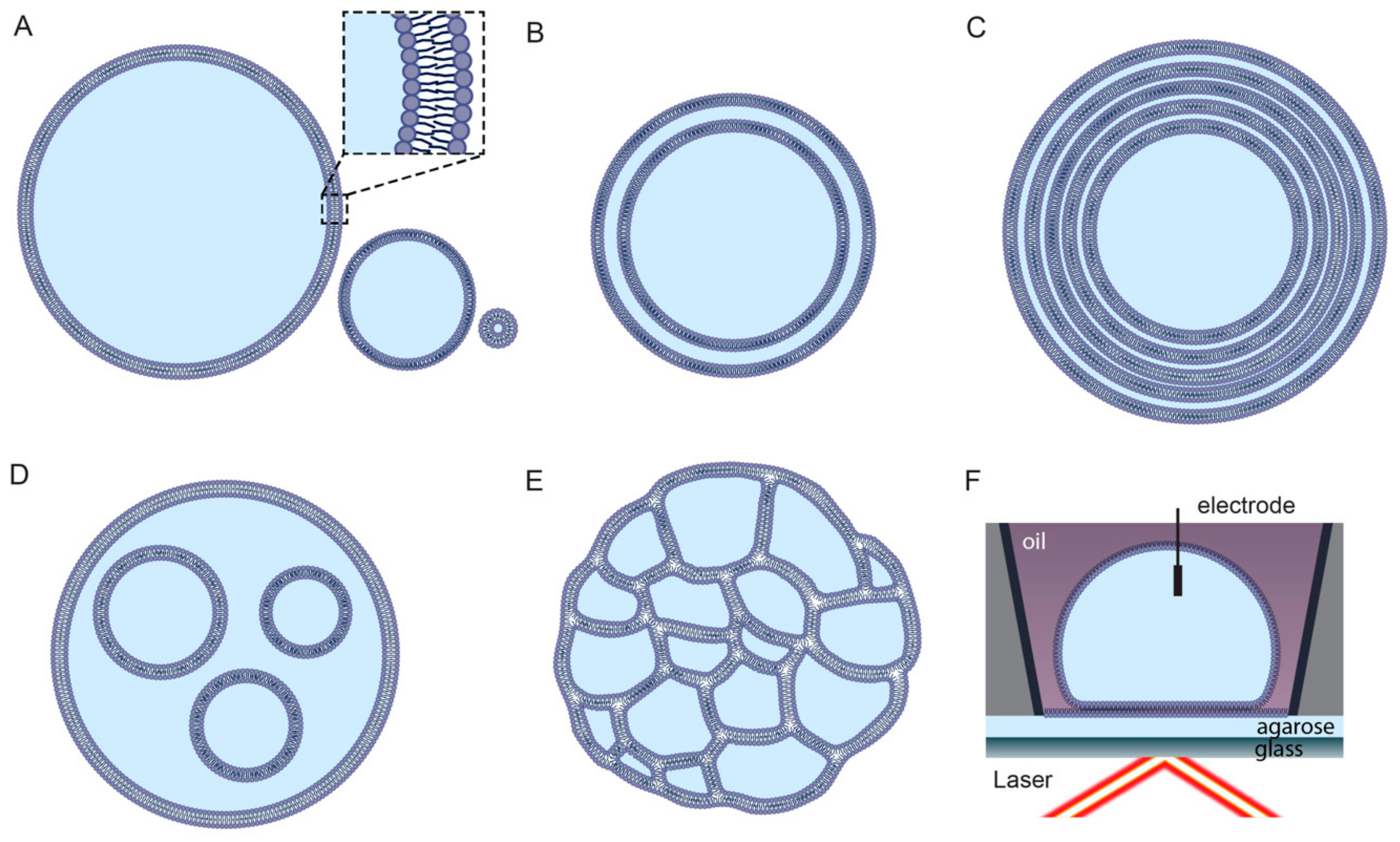
2.3. Preparation of Synthetic Liposomes
2.4. Limitations of Liposome Technology
3. DNA Nanotechnology
3.1. DNA Nanostructures
3.2. Switchable DNA Nanomachines
3.3. Limitations of DNA Nanotechnology
4. Hydrophobic DNA Modification for Membrane Attachment
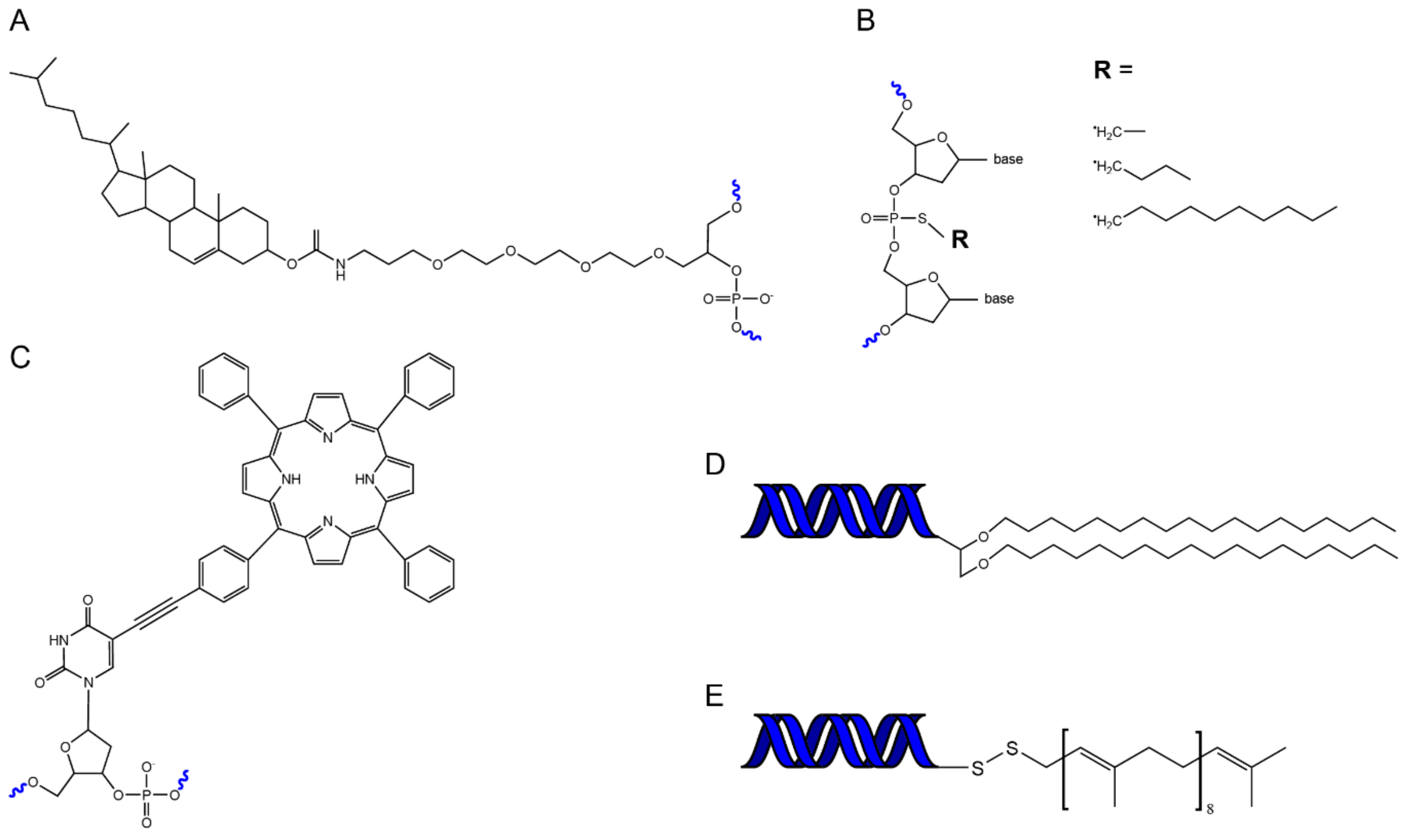
4.1. Chemistry of Hydrophobic DNA Modifications
4.2. Assembly Methods for Lipid-DNA Nanostructures
4.3. Properties of Different Lipid Modifiations
4.4. Bridging DNA and Lipid Nanotechnology
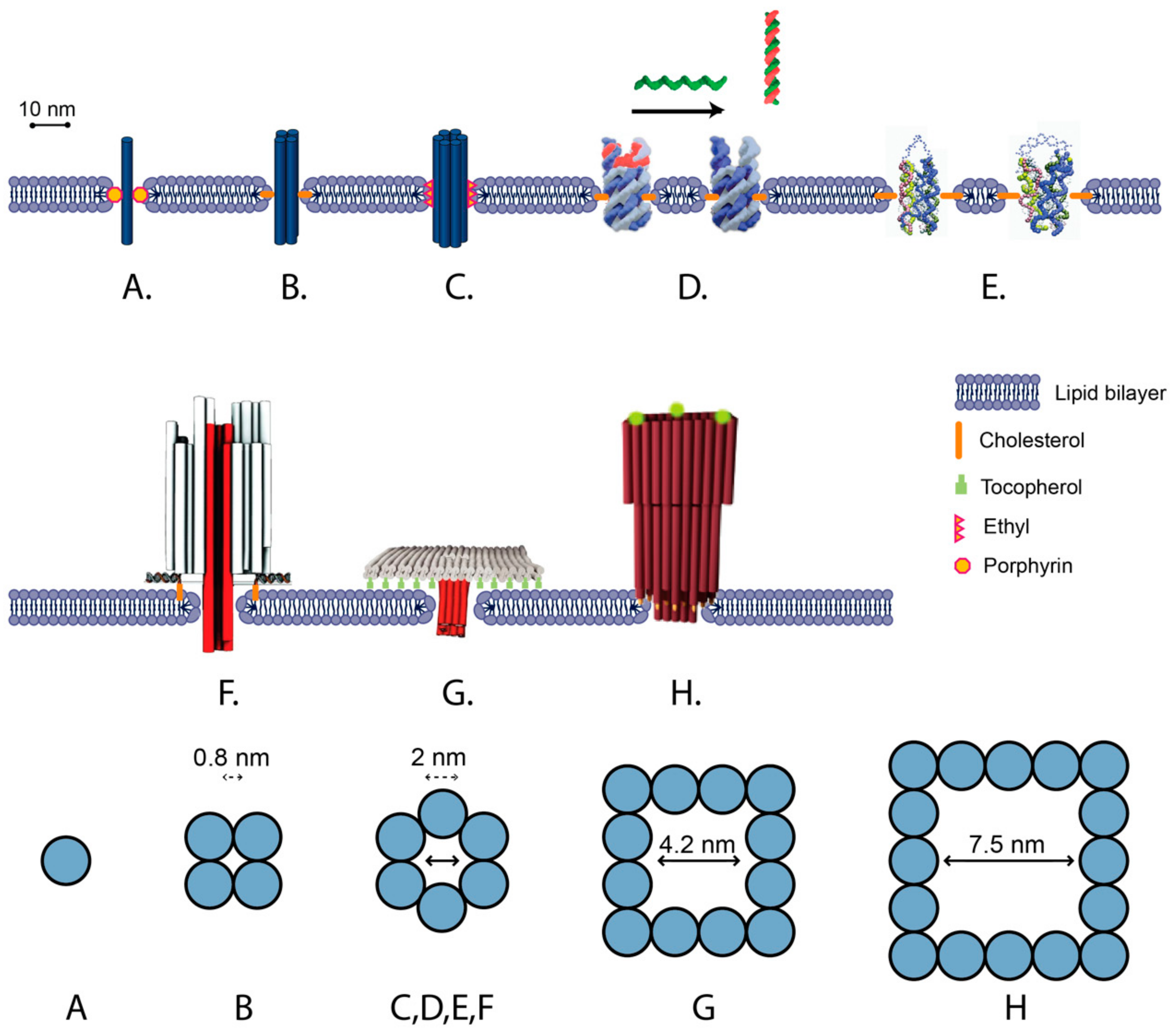
5. Membrane-Spanning DNA Nanostructures
5.1. Towards DNA Nanostructure Transmembrane Signaling
5.2. Unscaffolded Membrane-Spanning DNA Nanostructures
5.3. Scaffolded Membrane-Spanning DNA Nanostructures
5.4. Switchable DNA Lipid Channels
5.5. Challenges and Future Directions of Membrane-Spanning DNA Nanostructures
6. Membrane-Shaping DNA Nanostructures
6.1. Bio-Inspired Membrane Shaping
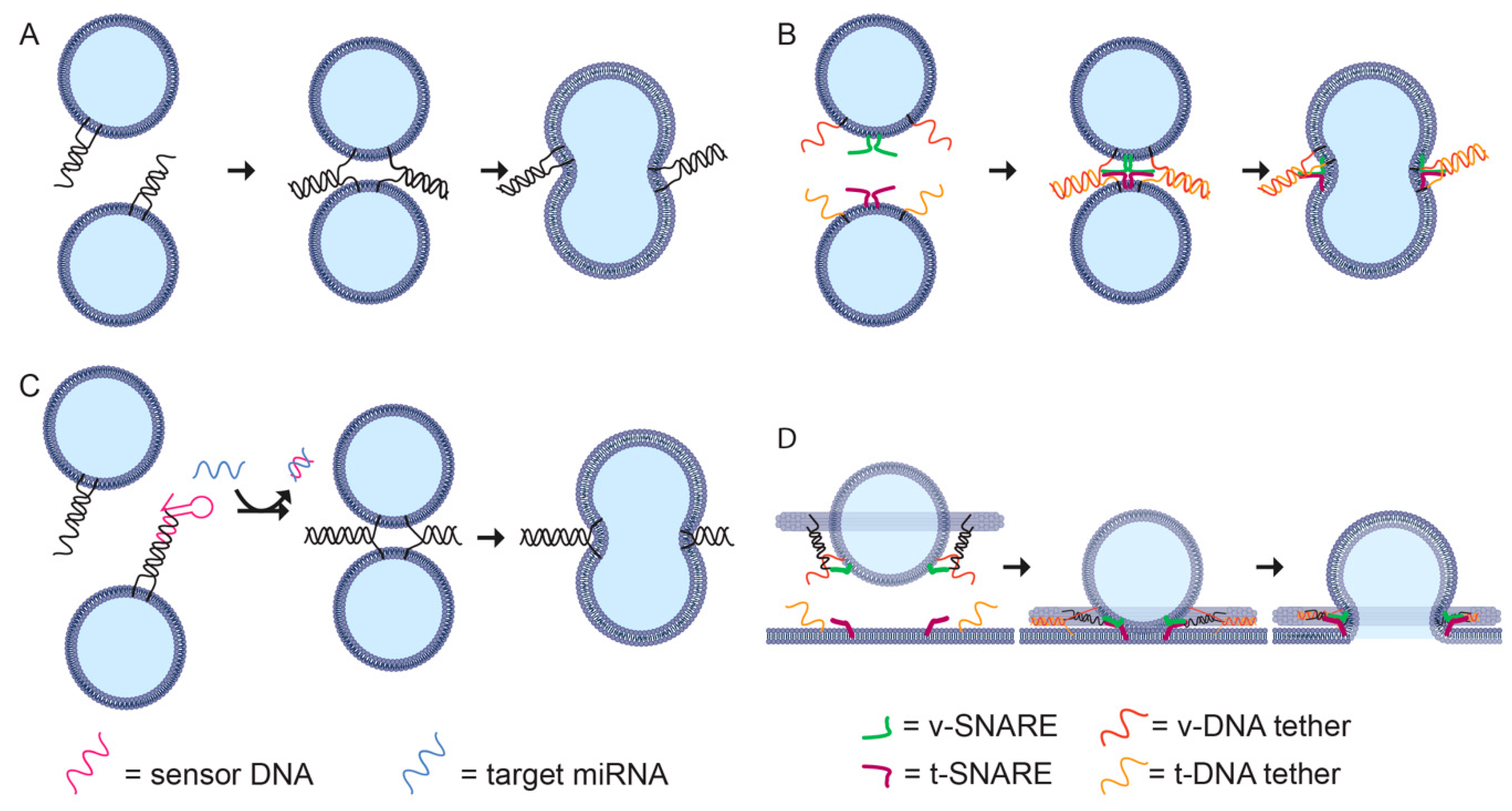
6.2. Deforming Liposomes
6.3. Directing Liposome Formation
6.4. Controlling Fusion
6.5. Functionalising and Characterising Membrane Surfaces
7. Applications and Future Directions
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Diekmann, Y.; Pereira-Leal, J.B. Evolution of intracellular compartmentalization. Biochem. J. 2013, 449, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Szostak, J.W.; Bartel, D.P.; Luisi, P.L. Synthesizing life. Nature 2001, 409, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, S.F. Multicellularity: The Evolution of Differentiation. In Developmental Biology, 6th ed.; Sinauer Associates: Sunderland, MA, USA, 2000. [Google Scholar]
- Yin, H.; Flynn, A.D. Drugging Membrane Protein Interactions. Annu. Rev. Biomed. Eng. 2016, 18, 51–76. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Overington, J.P.; Al-Lazikani, B.; Hopkins, A.L. How many drug targets are there? Nat. Rev. Drug Discov. 2006, 5, 993–996. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Araki, Y.; Yamamoto, T.; Nakaya, T. Trafficking of Alzheimer’s disease-related membrane proteins and its participation in disease pathogenesis. J. Biochem. 2006, 139, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Gadsby, D.C.; Vergani, P.; Csanády, L. The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature 2006, 440, 477–483. [Google Scholar] [CrossRef]
- Sezgin, E.; Levental, I.; Mayor, S.; Eggeling, C. The mystery of membrane organization: Composition, regulation and roles of lipid rafts. Nat. Rev. Mol. Cell Biol. 2017, 18, 361–374. [Google Scholar] [CrossRef]
- He, K.; Marsland Iii, R.; Upadhyayula, S.; Song, E.; Dang, S.; Capraro, B.R.; Wang, W.; Skillern, W.; Gaudin, R.; Ma, M.; et al. Dynamics of phosphoinositide conversion in clathrin-mediated endocytic traffic. Nature 2017, 552, 410–414. [Google Scholar] [CrossRef]
- Daraee, H.; Etemadi, A.; Kouhi, M.; Alimirzalu, S.; Akbarzadeh, A. Application of liposomes in medicine and drug delivery. Artif. Cells Nanomed. Biotechnol. 2016, 44, 381–391. [Google Scholar] [CrossRef]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal formulations in clinical use: An updated review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Booth, M.J.; Schild, V.R.; Downs, F.G.; Bayley, H. Functional aqueous droplet networks. Mol. Biosyst. 2017, 13, 1658–1691. [Google Scholar] [CrossRef] [PubMed]
- Derr, N.D.; Goodman, B.S.; Jungmann, R.; Leschziner, A.E.; Shih, W.M.; Reck-Peterson, S.L. Tug-of-War in Motor Protein Ensembles Revealed with a Programmable DNA Origami Scaffold. Science 2012, 338, 662–665. [Google Scholar] [CrossRef]
- Wickham, S.F.J.; Bath, J.; Katsuda, Y.; Endo, M.; Hidaka, K.; Sugiyama, H.; Turberfield, A.J. A DNA-based molecular motor that can navigate a network of tracks. Nat. Nanotechnol. 2012, 7, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, J.B.; Liu, L.; Bank Kodal, A.L.; Madsen, M.; Li, Q.; Song, J.; Woehrstein, J.B.; Wickham, S.F.J.; Strauss, M.T.; Schueder, F.; et al. Routing of individual polymers in designed patterns. Nat. Nanotechnol. 2015, 10, 892–898. [Google Scholar] [CrossRef]
- Douglas, S.M.; Bachelet, I.; Church, G.M. A logic-gated nanorobot for targeted transport of molecular payloads. Science 2012, 335, 831–834. [Google Scholar] [CrossRef] [PubMed]
- Ye, D.; Zuo, X.; Fan, C. DNA Nanotechnology-Enabled Interfacial Engineering for Biosensor Development. Annu. Rev. Anal. Chem. 2018, 11, 171–195. [Google Scholar] [CrossRef]
- Langecker, M.; Arnaut, V.; Martin, T.G.; List, J.; Renner, S.; Mayer, M.; Dietz, H.; Simmel, F.C. Synthetic lipid membrane channels formed by designed DNA nanostructures. Science 2012, 338, 932–936. [Google Scholar] [CrossRef]
- Zhang, Z.; Yang, Y.; Pincet, F.; Llaguno, M.C.; Lin, C. Placing and shaping liposomes with reconfigurable DNA nanocages. Nat. Chem. 2017, 9, 653. [Google Scholar] [CrossRef]
- Xu, W.; Nathwani, B.; Lin, C.; Wang, J.; Karatekin, E.; Pincet, F.; Shih, W.; Rothman, J.E. A programmable DNA origami platform to organize SNAREs for membrane fusion. J. Am. Chem. Soc. 2016, 138, 4439–4447. [Google Scholar] [CrossRef]
- Fisher, P.D.E.; Shen, Q.; Akpinar, B.; Davis, L.K.; Chung, K.K.H.; Baddeley, D.; Šarić, A.; Melia, T.J.; Hoogenboom, B.W.; Lin, C.; et al. A Programmable DNA Origami Platform for Organizing Intrinsically Disordered Nucleoporins within Nanopore Confinement. ACS Nano 2018, 12, 1508–1518. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Endo, M.; Yang, Y.; Sugiyama, H. Dynamic Assembly/Disassembly Processes of Photoresponsive DNA Origami Nanostructures Directly Visualized on a Lipid Membrane Surface. J. Am. Chem. Soc. 2014, 136, 1714–1717. [Google Scholar] [CrossRef] [PubMed]
- Perrault, S.D.; Shih, W.M. Virus-Inspired Membrane Encapsulation of DNA Nanostructures To Achieve In Vivo Stability. ACS Nano 2014, 8, 5132–5140. [Google Scholar] [CrossRef] [PubMed]
- Bangham, A.; Standish, M.M.; Watkins, J.C. Diffusion of univalent ions across the lamellae of swollen phospholipids. J. Mol. Biol. 1965, 13, 238–252, IN26–IN27. [Google Scholar] [CrossRef]
- Storm, G.; Crommelin, D.J. Liposomes: Quo vadis? Pharm. Sci. Technol. Today 1998, 1, 19–31. [Google Scholar] [CrossRef]
- Ulrich, A.S. Biophysical aspects of using liposomes as delivery vehicles. Biosci. Rep. 2002, 22, 129–150. [Google Scholar] [CrossRef]
- Woodle, M.C.; Papahadjopoulos, D. Liposome preparation and size characterization. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1989; Volume 171, pp. 193–217. ISBN 0076-6879. [Google Scholar]
- Mantripragada, S. A lipid based depot (DepoFoam® technology) for sustained release drug delivery. Prog. Lipid Res. 2002, 41, 392–406. [Google Scholar] [CrossRef]
- Van Swaay, D.; DeMello, A. Microfluidic methods for forming liposomes. Lab Chip 2013, 13, 752–767. [Google Scholar] [CrossRef]
- Leptihn, S.; Castell, O.K.; Cronin, B.; Lee, E.-H.; Gross, L.C.M.; Marshall, D.P.; Thompson, J.R.; Holden, M.; Wallace, M.I. Constructing droplet interface bilayers from the contact of aqueous droplets in oil. Nat. Protoc. 2013, 8, 1048–1057. [Google Scholar] [CrossRef]
- Rosholm, K.R.; Baker, M.A.B.; Ridone, P.; Nakayama, Y.; Rohde, P.R.; Cuello, L.G.; Lee, L.K.; Martinac, B. Activation of the mechanosensitive ion channel MscL by mechanical stimulation of supported Droplet-Hydrogel bilayers. Sci. Rep. 2017, 7, 45180. [Google Scholar] [CrossRef]
- Jaggers, O.B.; Ridone, P.; Martinac, B.; Baker, M.A.B. Fluorescence microscopy of piezo1 in droplet hydrogel bilayers. Channels 2019, 13, 102–109. [Google Scholar] [CrossRef]
- Kuo, T.-C.; Tseng, Y.J. LipidPedia: A comprehensive lipid knowledgebase. Bioinformatics 2018, 34, 2982–2987. [Google Scholar] [CrossRef]
- Fahy, E.; Subramaniam, S.; Brown, H.A.; Glass, C.K.; Merrill, A.H., Jr.; Murphy, R.C.; Raetz, C.R.; Russell, D.W.; Seyama, Y.; Shaw, W. A comprehensive classification system for lipids. Eur. J. Lipid Sci. Technol. 2005, 107, 337–364. [Google Scholar] [CrossRef]
- Fahy, E.; Subramaniam, S.; Murphy, R.C.; Nishijima, M.; Raetz, C.R.; Shimizu, T.; Spener, F.; van Meer, G.; Wakelam, M.J.; Dennis, E.A. Update of the LIPID MAPS comprehensive classification system for lipids. J. Lipid Res. 2009, 50, S9–S14. [Google Scholar] [CrossRef]
- Simons, K.; Vaz, W.L.C. Model Systems, Lipid Rafts, and Cell Membranes. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 269–295. [Google Scholar] [CrossRef]
- Kates, M.; Pugh, E.; Ferrante, G. Regulation of membrane fluidity by lipid desaturases. In Membrane Fluidity; Springer: Berlin, Germany, 1984; pp. 379–395. [Google Scholar]
- Murata, N.; Los, D.A. Membrane fluidity and temperature perception. Plant Physiol. 1997, 115, 875. [Google Scholar] [CrossRef]
- Seu, K.J.; Cambrea, L.R.; Everly, R.M.; Hovis, J.S. Influence of lipid chemistry on membrane fluidity: Tail and headgroup interactions. Biophys. J. 2006, 91, 3727–3735. [Google Scholar] [CrossRef]
- Oh, Y.; Kim, J.; Yethiraj, A.; Sung, B.J. Swing motion as a diffusion mechanism of lipid bilayers in a gel phase. Phys. Rev. E 2016, 93, 012409. [Google Scholar] [CrossRef]
- Magarkar, A.; Dhawan, V.; Kallinteri, P.; Viitala, T.; Elmowafy, M.; Róg, T.; Bunker, A. Cholesterol level affects surface charge of lipid membranes in saline solution. Sci. Rep. 2014, 4, 5005. [Google Scholar] [CrossRef]
- Akbarzadeh, A.; Rezaei-Sadabady, R.; Davaran, S.; Joo, S.W.; Zarghami, N.; Hanifehpour, Y.; Samiei, M.; Kouhi, M.; Nejati-Koshki, K. Liposome: Classification, preparation, and applications. Nanoscale Res. Lett. 2013, 8, 102. [Google Scholar] [CrossRef]
- Bangham, A.; De Gier, J.; Greville, G. Osmotic properties and water permeability of phospholipid liquid crystals. Chem. Phys. Lipids 1967, 1, 225–246. [Google Scholar] [CrossRef]
- Kim, S.; Jacobs, R.E.; White, S.H. Preparation of multilamellar vesicles of defined size-distribution by solvent-spherule evaporation. Biochim. Biophys. Acta BBA Biomembr. 1985, 812, 793–801. [Google Scholar] [CrossRef]
- Payne, N.I.; Ambrose, C.V.; Timmins, P.; Ward, M.D.; Ridgway, F. Proliposomes: A novel solution to an old problem. J. Pharm. Sci. 1986, 75, 325–329. [Google Scholar] [CrossRef]
- Hope, M.; Bally, M.; Webb, G.; Cullis, P. Production of large unilamellar vesicles by a rapid extrusion procedure. Characterization of size distribution, trapped volume and ability to maintain a membrane potential. Biochim. Biophys. Acta BBA Biomembr. 1985, 812, 55–65. [Google Scholar] [CrossRef]
- Zumbuehl, O.; Weder, H.G. Liposomes of controllable size in the range of 40 to 180 nm by defined dialysis of lipid/detergent mixed micelles. Biochim. Biophys. Acta BBA Biomembr. 1981, 640, 252–262. [Google Scholar] [CrossRef]
- Deamer, D.; Bangham, A. Large volume liposomes by an ether vaporization method. Biochim. Biophys. Acta BBA Nucleic Acids Protein Synth. 1976, 443, 629–634. [Google Scholar] [CrossRef]
- Szoka, F.; Papahadjopoulos, D. Procedure for preparation of liposomes with large internal aqueous space and high capture by reverse-phase evaporation. Proc. Natl. Acad. Sci. USA 1978, 75, 4194–4198. [Google Scholar] [CrossRef]
- Mueller, P.; Chien, T.; Rudy, B. Formation and properties of cell-size lipid bilayer vesicles. Biophys. J. 1983, 44, 375–381. [Google Scholar] [CrossRef]
- Angelova, M.I.; Dimitrov, D.S. Liposome electroformation. Faraday Discuss. Chem. Soc. 1986, 81, 303–311. [Google Scholar] [CrossRef]
- Shimanouchi, T.; Umakoshi, H.; Kuboi, R. Kinetic study on giant vesicle formation with electroformation method. Langmuir 2009, 25, 4835–4840. [Google Scholar] [CrossRef]
- Jahn, A.; Vreeland, W.N.; Gaitan, M.; Locascio, L.E. Controlled vesicle self-assembly in microfluidic channels with hydrodynamic focusing. J. Am. Chem. Soc. 2004, 126, 2674–2675. [Google Scholar] [CrossRef]
- Funakoshi, K.; Suzuki, H.; Takeuchi, S. Formation of giant lipid vesiclelike compartments from a planar lipid membrane by a pulsed jet flow. J. Am. Chem. Soc. 2007, 129, 12608–12609. [Google Scholar] [CrossRef]
- Seeman, N.C.; Sleiman, H.F. DNA nanotechnology. Nat. Rev. Mater. 2018, 3, 17068. [Google Scholar] [CrossRef]
- Tinland, B.; Pluen, A.; Sturm, J.; Weill, G. Persistence Length of Single-Stranded DNA. Macromolecules 1997, 30, 5763–5765. [Google Scholar] [CrossRef]
- Bednar, J.; Furrer, P.; Katritch, V.; Stasiak, A.; Dubochet, J.; Stasiak, A. Determination of DNA Persistence Length by Cryo-electron Microscopy. Separation of the Static and Dynamic Contributions to the Apparent Persistence Length of DNA. J. Mol. Biol. 1995, 254, 579–594. [Google Scholar] [CrossRef]
- Caruthers, M.H. Gene synthesis machines: DNA chemistry and its uses. Science 1985, 230, 281–285. [Google Scholar] [CrossRef]
- Douglas, S.M.; Marblestone, A.H.; Teerapittayanon, S.; Vazquez, A.; Church, G.M.; Shih, W.M. Rapid prototyping of 3D DNA-origami shapes with caDNAno. Nucleic Acids Res. 2009, 37, 5001–5006. [Google Scholar] [CrossRef]
- Bandy, T.J.; Brewer, A.; Burns, J.R.; Marth, G.; Nguyen, T.; Stulz, E. DNA as supramolecular scaffold for functional molecules: Progress in DNA nanotechnology. Chem. Soc. Rev. 2011, 40, 138–148. [Google Scholar] [CrossRef]
- Seeman, N.C. Nucleic acid junctions and lattices. J. Theor. Biol. 1982, 99, 237–247. [Google Scholar] [CrossRef]
- Rothemund, P.W. Folding DNA to create nanoscale shapes and patterns. Nature 2006, 440, 297. [Google Scholar] [CrossRef]
- Douglas, S.M.; Dietz, H.; Liedl, T.; Högberg, B.; Graf, F.; Shih, W.M. Self-assembly of DNA into nanoscale three-dimensional shapes. Nature 2009, 459, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Ong, L.L.; Shih, W.M.; Yin, P. Three-Dimensional Structures Self-Assembled from DNA Bricks. Science 2012, 338, 1177–1183. [Google Scholar] [CrossRef] [PubMed]
- Wei, B.; Dai, M.; Yin, P. Complex shapes self–assembled from single–stranded DNA tiles. Nature 2012, 485, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Birktoft, J.J.; Chen, Y.; Wang, T.; Sha, R.; Constantinou, P.E.; Ginell, S.L.; Mao, C.; Seeman, N.C. From molecular to macroscopic via the rational design of a self-assembled 3D DNA crystal. Nature 2009, 461, 74–77. [Google Scholar] [CrossRef]
- Ke, Y.; Ong, L.L.; Sun, W.; Song, J.; Dong, M.; Shih, W.M.; Yin, P. DNA brick crystals with prescribed depths. Nat. Chem. 2014, 6, 994–1002. [Google Scholar] [CrossRef]
- Douglas, S.M.; Chou, J.J.; Shih, W.M. DNA-nanotube-induced alignment of membrane proteins for NMR structure determination. Proc. Natl. Acad. Sci. USA 2007, 104, 6644–6648. [Google Scholar] [CrossRef]
- Zhang, F.; Jiang, S.; Wu, S.; Li, Y.; Mao, C.; Liu, Y.; Yan, H. Complex wireframe DNA origami nanostructures with multi-arm junction vertices. Nat. Nanotechnol. 2015, 10, 779–784. [Google Scholar] [CrossRef]
- Han, D.; Pal, S.; Nangreave, J.; Deng, Z.; Liu, Y.; Yan, H. DNA origami with complex curvatures in three-dimensional space. Science 2011, 332, 342–346. [Google Scholar] [CrossRef]
- Li, S.; Jiang, Q.; Liu, S.; Zhang, Y.; Tian, Y.; Song, C.; Wang, J.; Zou, Y.; Anderson, G.J.; Han, J.-Y.; et al. A DNA nanorobot functions as a cancer therapeutic in response to a molecular trigger in vivo. Nat. Biotechnol. 2018, 36, 258–264. [Google Scholar] [CrossRef]
- Schreiber, R.; Do, J.; Roller, E.-M.; Zhang, T.; Schüller, V.J.; Nickels, P.C.; Feldmann, J.; Liedl, T. Hierarchical assembly of metal nanoparticles, quantum dots and organic dyes using DNA origami scaffolds. Nat. Nanotechnol. 2014, 9, 74–78. [Google Scholar] [CrossRef]
- Maune, H.T.; Han, S.-P.; Barish, R.D.; Bockrath, M.; Goddard, W.A.; Rothemund, P.W.K.; Winfree, E. Self-assembly of carbon nanotubes into two-dimensional geometries using DNA origami templates. Nat. Nanotechnol. 2010, 5, 61–66. [Google Scholar] [CrossRef]
- Lermusiaux, L.; Funston, A.M. Plasmonic isomers via DNA-based self-assembly of gold nanoparticles. Nanoscale 2018, 10, 19557–19567. [Google Scholar] [CrossRef]
- Bath, J.; Turberfield, A.J. DNA nanomachines. Nat. Nanotechnol. 2007, 2, 275–284. [Google Scholar] [CrossRef]
- Qian, L.; Winfree, E. Scaling up digital circuit computation with DNA strand displacement cascades. Science 2011, 332, 1196–1201. [Google Scholar] [CrossRef]
- Zhang, D.Y.; Seelig, G. Dynamic DNA nanotechnology using strand-displacement reactions. Nat. Chem. 2011, 3, 103–113. [Google Scholar] [CrossRef]
- Singh, J.K.D.; Luu, M.T.; Abbas, A.; Wickham, S.F. Switchable DNA-origami nanostructures that respond to their environment and their applications. Biophys. Rev. 2018, 10, 1283–1293. [Google Scholar] [CrossRef]
- Yurke, B.; Turberfield, A.J.; Mills, A.P., Jr.; Simmel, F.C.; Neumann, J.L. A DNA-fuelled molecular machine made of DNA. Nature 2000, 406, 605. [Google Scholar] [CrossRef]
- Gehring, K.; Leroy, J.-L.; Guéron, M. A tetrameric DNA structure with protonated cytosine-cytosine base pairs. Nature 1993, 363, 561. [Google Scholar] [CrossRef]
- Sannohe, Y.; Endo, M.; Katsuda, Y.; Hidaka, K.; Sugiyama, H. Visualization of Dynamic Conformational Switching of the G-Quadruplex in a DNA Nanostructure. J. Am. Chem. Soc. 2010, 132, 16311–16313. [Google Scholar] [CrossRef]
- Zhou, M.; Liang, X.; Mochizuki, T.; Asanuma, H. A light-driven DNA nanomachine for the efficient photoswitching of RNA digestion. Angew. Chem. Int. Ed. 2010, 49, 2167–2170. [Google Scholar] [CrossRef]
- Kopperger, E.; List, J.; Madhira, S.; Rothfischer, F.; Lamb, D.C.; Simmel, F.C. A self-assembled nanoscale robotic arm controlled by electric fields. Science 2018, 359, 296–301. [Google Scholar] [CrossRef]
- Lauback, S.; Mattioli, K.R.; Marras, A.E.; Armstrong, M.; Rudibaugh, T.P.; Sooryakumar, R.; Castro, C.E. Real-time magnetic actuation of DNA nanodevices via modular integration with stiff micro-levers. Nat. Commun. 2018, 9, 1446. [Google Scholar] [CrossRef]
- Iwaki, M.; Wickham, S.F.; Ikezaki, K.; Yanagida, T.; Shih, W.M. A programmable DNA origami nanospring that reveals force-induced adjacent binding of myosin VI heads. Nat. Commun. 2016, 7, 13715. [Google Scholar] [CrossRef]
- Huo, S.; Li, H.; Boersma, A.J.; Herrmann, A. DNA Nanotechnology Enters Cell Membranes. Adv. Sci. 2019, 6, 1900043. [Google Scholar] [CrossRef]
- Liu, K.; Zheng, L.; Liu, Q.; de Vries, J.W.; Gerasimov, J.Y.; Herrmann, A. Nucleic acid chemistry in the organic phase: From functionalized oligonucleotides to DNA side chain polymers. J. Am. Chem. Soc. 2014, 136, 14255–14262. [Google Scholar] [CrossRef]
- Hurley, J.H.; Hanson, P.I. Membrane budding and scission by the ESCRT machinery: It’s all in the neck. Nat. Rev. Mol. Cell Biol. 2010, 11, 556. [Google Scholar] [CrossRef]
- Loew, M.; Springer, R.; Scolari, S.; Altenbrunn, F.; Seitz, O.; Liebscher, J.; Huster, D.; Herrmann, A.; Arbuzova, A. Lipid domain specific recruitment of lipophilic nucleic acids: A key for switchable functionalization of membranes. J. Am. Chem. Soc. 2010, 132, 16066–16072. [Google Scholar] [CrossRef]
- Burns, J.R.; Seifert, A.; Fertig, N.; Howorka, S. A biomimetic DNA-based channel for the ligand-controlled transport of charged molecular cargo across a biological membrane. Nat. Nanotechnol. 2016, 11, 152–156. [Google Scholar] [CrossRef]
- Krishnan, S.; Ziegler, D.; Arnaut, V.; Martin, T.G.; Kapsner, K.; Henneberg, K.; Bausch, A.R.; Dietz, H.; Simmel, F.C. Molecular transport through large-diameter DNA nanopores. Nat. Commun. 2016, 7, 12787. [Google Scholar] [CrossRef]
- Burns, J.R.; Göpfrich, K.; Wood, J.W.; Thacker, V.V.; Stulz, E.; Keyser, U.F.; Howorka, S. Lipid-Bilayer-Spanning DNA Nanopores with a Bifunctional Porphyrin Anchor. Angew. Chem. Int. Ed. 2013, 52, 12069–12072. [Google Scholar] [CrossRef]
- Flavier, K.M.; Boxer, S.G. Vesicle Fusion Mediated by Solanesol-Anchored DNA. Biophys. J. 2017, 113, 1260–1268. [Google Scholar] [CrossRef]
- Khmelinskaia, A.; Franquelim, H.G.; Petrov, E.P.; Schwille, P. Effect of anchor positioning on binding and diffusion of elongated 3D DNA nanostructures on lipid membranes. J. Phys. Appl. Phys. 2016, 49, 194001. [Google Scholar] [CrossRef]
- Akbari, E.; Mollica, M.Y.; Lucas, C.R.; Bushman, S.M.; Patton, R.A.; Shahhosseini, M.; Song, J.W.; Castro, C.E. Engineering Cell Surface Function with DNA Origami. Adv. Mater. 2017, 29, 1703632. [Google Scholar] [CrossRef]
- Franquelim, H.G.; Khmelinskaia, A.; Sobczak, J.-P.; Dietz, H.; Schwille, P. Membrane sculpting by curved DNA origami scaffolds. Nat. Commun. 2018, 9, 811. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, J.; Shigematsu, H.; Xu, W.; Shih, W.M.; Rothman, J.E.; Lin, C. Self-assembly of size-controlled liposomes on DNA nanotemplates. Nat. Chem. 2016, 8, 476. [Google Scholar] [CrossRef]
- Grome, M.W.; Zhang, Z.; Pincet, F.; Lin, C. Vesicle Tubulation with Self-Assembling DNA Nanosprings. Angew. Chem. Int. Ed. 2018, 57, 5330–5334. [Google Scholar] [CrossRef]
- Khmelinskaia, A.; Mücksch, J.; Petrov, E.P.; Franquelim, H.G.; Schwille, P. Control of Membrane Binding and Diffusion of Cholesteryl-Modified DNA Origami Nanostructures by DNA Spacers. Langmuir 2018, 34, 14921–14931. [Google Scholar] [CrossRef]
- Iric, K.; Subramanian, M.; Oertel, J.; Agarwal, N.P.; Matthies, M.; Periole, X.; Sakmar, T.P.; Huber, T.; Fahmy, K.; Schmidt, T.L. DNA-encircled lipid bilayers. Nanoscale 2018, 10, 18463–18467. [Google Scholar] [CrossRef]
- Göpfrich, K.; Zettl, T.; Meijering, A.E.C.; Hernández-Ainsa, S.; Kocabey, S.; Liedl, T.; Keyser, U.F. DNA-Tile Structures Induce Ionic Currents through Lipid Membranes. Nano Lett. 2015, 15, 3134–3138. [Google Scholar] [CrossRef]
- Burns, J.R.; Stulz, E.; Howorka, S. Self-assembled DNA nanopores that span lipid bilayers. Nano Lett. 2013, 13, 2351–2356. [Google Scholar] [CrossRef]
- Mendoza, O.; Calmet, P.; Alves, I.; Lecomte, S.; Raoux, M.; Cullin, C.; Elezgaray, J. A tensegrity driven DNA nanopore. Nanoscale 2017, 9, 9762–9769. [Google Scholar] [CrossRef]
- Göpfrich, K.; Li, C.-Y.; Ricci, M.; Bhamidimarri, S.P.; Yoo, J.; Gyenes, B.; Ohmann, A.; Winterhalter, M.; Aksimentiev, A.; Keyser, U.F. Large-conductance transmembrane porin made from DNA origami. ACS Nano 2016, 10, 8207–8214. [Google Scholar] [CrossRef]
- Kohl, P.; Noble, D.; Hunter, P.J.; Kohl, P.; Sachs, F. Mechanoelectric feedback in cardiac cells. Philos. Trans. R. Soc. Lond. 2001, 359, 1173–1185. [Google Scholar] [CrossRef]
- Clarke, J.; Wu, H.-C.; Jayasinghe, L.; Patel, A.; Reid, S.; Bayley, H. Continuous base identification for single-molecule nanopore DNA sequencing. Nat. Nanotechnol. 2009, 4, 265–270. [Google Scholar] [CrossRef]
- Liu, D.; Wang, D.; Zhang, Y. DNA nanochannels. F1000Research 2017, 6. [Google Scholar] [CrossRef]
- Göpfrich, K.; Li, C.-Y.; Mames, I.; Bhamidimarri, S.P.; Ricci, M.; Yoo, J.; Mames, A.; Ohmann, A.; Winterhalter, M.; Stulz, E. Ion channels made from a single membrane-spanning DNA duplex. Nano Lett. 2016, 16, 4665–4669. [Google Scholar] [CrossRef]
- Joshi, H.; Maiti, P.K. Structure and electrical properties of DNA nanotubes embedded in lipid bilayer membranes. Nucleic Acids Res. 2018, 46, 2234–2242. [Google Scholar] [CrossRef]
- Seifert, A.; Göpfrich, K.; Burns, J.R.; Fertig, N.; Keyser, U.F.; Howorka, S. Bilayer-spanning DNA nanopores with voltage-switching between open and closed state. ACS Nano 2015, 9, 1117–1126. [Google Scholar] [CrossRef]
- Krasilnikov, O.; Sabirov, R.; Ternovsky, V.; Merzliak, P.; Muratkhodjaev, J. A simple method for the determination of the pore radius of ion channels in planar lipid bilayer membranes. FEMS Microbiol. Immunol. 1992, 5, 93–100. [Google Scholar] [CrossRef]
- Diederichs, T.; Pugh, G.; Dorey, A.; Xing, Y.; Burns, J.R.; Nguyen, Q.H.; Tornow, M.; Tampé, R.; Howorka, S. Synthetic protein-conductive membrane nanopores built with DNA. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef]
- Maingi, V.; Lelimousin, M.; Howorka, S.; Sansom, M.S.P. Gating-like Motions and Wall Porosity in a DNA Nanopore Scaffold Revealed by Molecular Simulations. ACS Nano 2015, 9, 11209–11217. [Google Scholar] [CrossRef]
- Arnott, P.M.; Howorka, S. A Temperature-Gated Nanovalve Self-Assembled from DNA to Control Molecular Transport across Membranes. ACS Nano 2019, 13, 3334–3340. [Google Scholar] [CrossRef]
- Langecker, M.; Arnaut, V.; List, J.; Simmel, F.C. DNA nanostructures interacting with lipid bilayer membranes. Acc. Chem. Res. 2014, 47, 1807–1815. [Google Scholar] [CrossRef]
- Marrink, S.J.; De Vries, A.H.; Mark, A.E. Coarse grained model for semiquantitative lipid simulations. J. Phys. Chem. B 2004, 108, 750–760. [Google Scholar] [CrossRef]
- Bruininks, B.M.; Souza, P.C.; Marrink, S.J. A practical view of the martini force field. In Biomolecular Simulations; Springer: Berlin, Germany, 2019; pp. 105–127. [Google Scholar]
- Bennett, W.D.; Tieleman, D.P. Molecular simulation of rapid translocation of cholesterol, diacylglycerol, and ceramide in model raft and nonraft membranes. J. Lipid Res. 2012, 53, 421–429. [Google Scholar] [CrossRef]
- Marrink, S.J.; Corradi, V.; Souza, P.C.T.; Ingólfsson, H.I.; Tieleman, D.P.; Sansom, M.S.P. Computational Modeling of Realistic Cell Membranes. Chem. Rev. 2019, 119, 6184–6226. [Google Scholar] [CrossRef]
- Jarsch, I.K.; Daste, F.; Gallop, J.L. Membrane curvature in cell biology: An integration of molecular mechanisms. J. Cell Biol. 2016, 214, 375–387. [Google Scholar] [CrossRef]
- McMahon, H.T.; Boucrot, E. Membrane curvature at a glance. J. Cell Sci. 2015, 128, 1065–1070. [Google Scholar] [CrossRef]
- Mim, C.; Cui, H.; Gawronski-Salerno, J.A.; Frost, A.; Lyman, E.; Voth, G.A.; Unger, V.M. Structural basis of membrane bending by the N-BAR protein endophilin. Cell 2012, 149, 137–145. [Google Scholar] [CrossRef]
- Stanishneva-Konovalova, T.; Derkacheva, N.; Polevova, S.; Sokolova, O. The role of BAR domain proteins in the regulation of membrane dynamics. Acta Naturae 2016, 8, 60–69. [Google Scholar] [CrossRef]
- Ferguson, S.M.; De Camilli, P. Dynamin, a membrane-remodelling GTPase. Nat. Rev. Mol. Cell Biol. 2012, 13, 75. [Google Scholar] [CrossRef]
- Li, F.; Pincet, F.; Perez, E.; Eng, W.S.; Melia, T.J.; Rothman, J.E.; Tareste, D. Energetics and dynamics of SNAREpin folding across lipid bilayers. Nat. Struct. Mol. Biol. 2007, 14, 890–896. [Google Scholar] [CrossRef]
- Zimmerberg, J.; Kozlov, M.M. How proteins produce cellular membrane curvature. Nat. Rev. Mol. Cell Biol. 2006, 7, 9. [Google Scholar] [CrossRef]
- Xu, W.; Wang, J.; Rothman, J.E.; Pincet, F. Accelerating SNARE-Mediated Membrane Fusion by DNA–Lipid Tethers. Angew. Chem. Int. Ed. 2015, 54, 14388–14392. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, M.; Hogle, J.M.; Shih, W.M.; Wagner, G.; Nasr, M.L. DNA-corralled nanodiscs for the structural and functional characterization of membrane proteins and viral entry. J. Am. Chem. Soc. 2018, 140, 10639–10643. [Google Scholar] [CrossRef]
- Sun, L.; Gao, Y.; Wang, Y.; Wei, Q.; Shi, J.; Chen, N.; Li, D.; Fan, C. Guiding protein delivery into live cells using DNA-programmed membrane fusion. Chem. Sci. 2018, 9, 5967–5975. [Google Scholar] [CrossRef]
- Jumeaux, C.; Wahlsten, O.; Block, S.; Kim, E.; Chandrawati, R.; Howes, P.D.; Höök, F.; Stevens, M.M. MicroRNA Detection by DNA-Mediated Liposome Fusion. ChemBioChem 2018, 19, 434–438. [Google Scholar] [CrossRef]
- Czogalla, A.; Kauert, D.J.; Franquelim, H.G.; Uzunova, V.; Zhang, Y.; Seidel, R.; Schwille, P. Amphipathic DNA Origami Nanoparticles to Scaffold and Deform Lipid Membrane Vesicles. Angew. Chem. Int. Ed. 2015, 54, 6501–6505. [Google Scholar] [CrossRef]
- Stengel, G.; Zahn, R.; Höök, F. DNA-Induced Programmable Fusion of Phospholipid Vesicles. J. Am. Chem. Soc. 2007, 129, 9584–9585. [Google Scholar] [CrossRef]
- Journot, C.M.A.; Ramakrishna, V.; Wallace, M.I.; Turberfield, A.J. Modifying Membrane Morphology and Interactions with DNA Origami Clathrin-Mimic Networks. ACS Nano 2019, 13, 9973–9979. [Google Scholar] [CrossRef]
- Manca, F.; Pincet, F.; Truskinovsky, L.; Rothman, J.E.; Foret, L.; Caruel, M. SNARE machinery is optimized for ultrafast fusion. Proc. Natl. Acad. Sci. USA 2019, 116, 2435–2442. [Google Scholar] [CrossRef]
- Johnson-Buck, A.; Jiang, S.; Yan, H.; Walter, N.G. DNA-cholesterol barges as programmable membrane-exploring agents. ACS Nano 2014, 8, 5641–5649. [Google Scholar] [CrossRef]
- Sato, Y.; Endo, M.; Morita, M.; Takinoue, M.; Sugiyama, H.; Murata, S.; Nomura, S.-I.M.; Suzuki, Y. Environment-Dependent Self-Assembly of DNA Origami Lattices on Phase-Separated Lipid Membranes. Adv. Mater. Interfaces 2018, 5, 1800437. [Google Scholar] [CrossRef]
- Czogalla, A.; Petrov, E.P.; Kauert, D.J.; Uzunova, V.; Zhang, Y.; Seidel, R.; Schwille, P. Switchable domain partitioning and diffusion of DNA origami rods on membranes. Faraday Discuss. 2012, 161, 31–43. [Google Scholar] [CrossRef]
- Czogalla, A.; Kauert, D.J.; Seidel, R.; Schwille, P.; Petrov, E.P. DNA origami nanoneedles on freestanding lipid membranes as a tool to observe isotropic-nematic transition in two dimensions. Nano Lett. 2015, 15, 649–655. [Google Scholar] [CrossRef]
- Bian, X.; Zhang, Z.; Xiong, Q.; Camilli, P.D.; Lin, C. A programmable DNA-origami platform for studying lipid transfer between bilayers. Nat. Chem. Biol. 2019, 15, 830–837. [Google Scholar] [CrossRef]
- Dou, Y.; Hynynen, K.; Allen, C. To heat or not to heat: Challenges with clinical translation of thermosensitive liposomes. J. Control. Release 2017, 249, 63–73. [Google Scholar] [CrossRef]
- Lajunen, T.; Nurmi, R.; Kontturi, L.; Viitala, L.; Yliperttula, M.; Murtomäki, L.; Urtti, A. Light activated liposomes: Functionality and prospects in ocular drug delivery. J. Control. Release 2016, 244, 157–166. [Google Scholar] [CrossRef]
- Mastrobattista, E.; Schoen, P.; Wilschut, J.; Crommelin, D.J.; Storm, G. Targeting influenza virosomes to ovarian carcinoma cells. FEBS Lett. 2001, 509, 71–76. [Google Scholar] [CrossRef]
- Fretz, M.M.; Mastrobattista, E.; Koning, G.A.; Jiskoot, W.; Storm, G. Strategies for cytosolic delivery of liposomal macromolecules. Int. J. Pharm. 2005, 298, 305–309. [Google Scholar] [CrossRef]
- Burns, J.R.; Al-Juffali, N.; Janes, S.M.; Howorka, S. Membrane-spanning DNA nanopores with cytotoxic effect. Angew. Chem.Int. Ed. 2014, 53, 12466–12470. [Google Scholar]
- Gilbert, R.J.C. Pore-forming toxins. Cell. Mol. Life Sci. CMLS 2002, 59, 832–844. [Google Scholar] [CrossRef]
- Guo, X.L.; Yuan, D.D.; Song, T.; Li, X.M. DNA nanopore functionalized with aptamer and cell-penetrating peptide for tumor cell recognition. Anal. Bioanal. Chem. 2017, 409, 3789–3797. [Google Scholar] [CrossRef]
- Bayley, H.; Martin, C.R. Resistive-pulse sensing from microbes to molecules. Chem. Rev. 2000, 100, 2575–2594. [Google Scholar] [CrossRef]
- Ishikawa, D.; Suzuki, Y.; Kurokawa, C.; Ohara, M.; Tsuchiya, M.; Morita, M.; Yanagisawa, M.; Endo, M.; Kawano, R.; Takinoue, M. DNA Origami Nanoplate-Based Emulsion with Nanopore Function. Angew. Chem. Int. Ed. 2019, 58, 15299–15303. [Google Scholar] [CrossRef]
- Seo, J.; Kim, S.; Park, H.H.; Choi, D.Y.; Nam, J.-M. Nano-bio-computing lipid nanotablet. Sci. Adv. 2019, 5, eaau2124. [Google Scholar] [CrossRef]
- Fabry-Wood, A.; Fetrow, M.E.; Oloyede, A.; Yang, K.-A.; Stojanovic, M.N.; Stefanovic, D.; Graves, S.W.; Carroll, N.J.; Lakin, M.R. Microcompartments for Protection and Isolation of Nanoscale DNA Computing Elements. ACS Appl. Mater. Interfaces 2019, 11, 11262–11269. [Google Scholar] [CrossRef]
- Spruijt, E.; Tusk, S.E.; Bayley, H. DNA scaffolds support stable and uniform peptide nanopores. Nat. Nanotechnol. 2018, 13, 739–745. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Darley, E.; Singh, J.K.D.; Surace, N.A.; Wickham, S.F.J.; Baker, M.A.B. The Fusion of Lipid and DNA Nanotechnology. Genes 2019, 10, 1001. https://doi.org/10.3390/genes10121001
Darley E, Singh JKD, Surace NA, Wickham SFJ, Baker MAB. The Fusion of Lipid and DNA Nanotechnology. Genes. 2019; 10(12):1001. https://doi.org/10.3390/genes10121001
Chicago/Turabian StyleDarley, Es, Jasleen Kaur Daljit Singh, Natalie A. Surace, Shelley F. J. Wickham, and Matthew A. B. Baker. 2019. "The Fusion of Lipid and DNA Nanotechnology" Genes 10, no. 12: 1001. https://doi.org/10.3390/genes10121001
APA StyleDarley, E., Singh, J. K. D., Surace, N. A., Wickham, S. F. J., & Baker, M. A. B. (2019). The Fusion of Lipid and DNA Nanotechnology. Genes, 10(12), 1001. https://doi.org/10.3390/genes10121001