Enriching Human Interactome with Functional Mutations to Detect High-Impact Network Modules Underlying Complex Diseases


Abstract
1. Introduction
2. Materials and Methods
2.1. Human Interactome Construction
2.2. GWAS Data Collection and Processing
2.3. Functional Annotation of nsSNV with the SNP-IN Tool
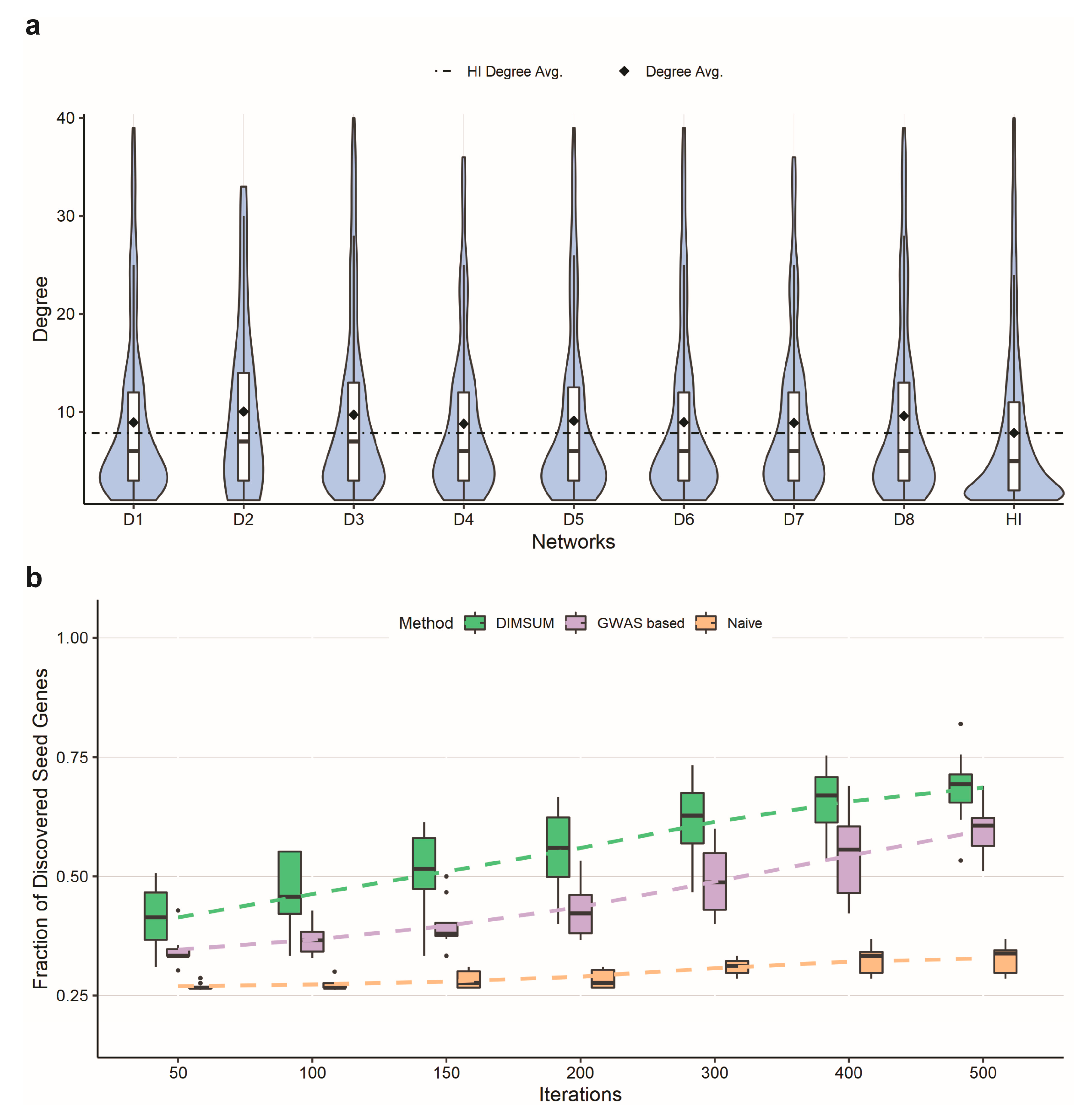
2.4. Network Annotation and Network Propagation
2.5. Sub-Network Extraction
- Assume that a seed gene set {, , …, } induces a subnetwork with initial size . For all immediate interacting partners of the seed gene set that are not in the subnetwork, define an impact score:where is the updated p-value after the network propagation and is the total number of mutations that disrupt the PPIs between the candidate gene i and every gene j in the module gene set. Thus, the impact score combines the disease relevance and potential disruption to existing subnetwork caused by the candidate gene.
- All the immediate interaction neighbors of the seed gene set, not included in the subnetwork, are ranked according to their impact score.
- Select the gene with the largest impact score. If there are multiple candidates with the same impact score, randomly pick one gene to break the tie. Add the gene with the biggest impact to the set of seed genes and increase the size of the induced subnetwork by 1: = + 1.
2.6. Validation and GO Analysis
3. Results
3.1. Seed Genes Generated from GWAS Datasets
3.2. Functional Predictions from the SNP-IN Tool
3.3. Network Annotation and Network Propagation
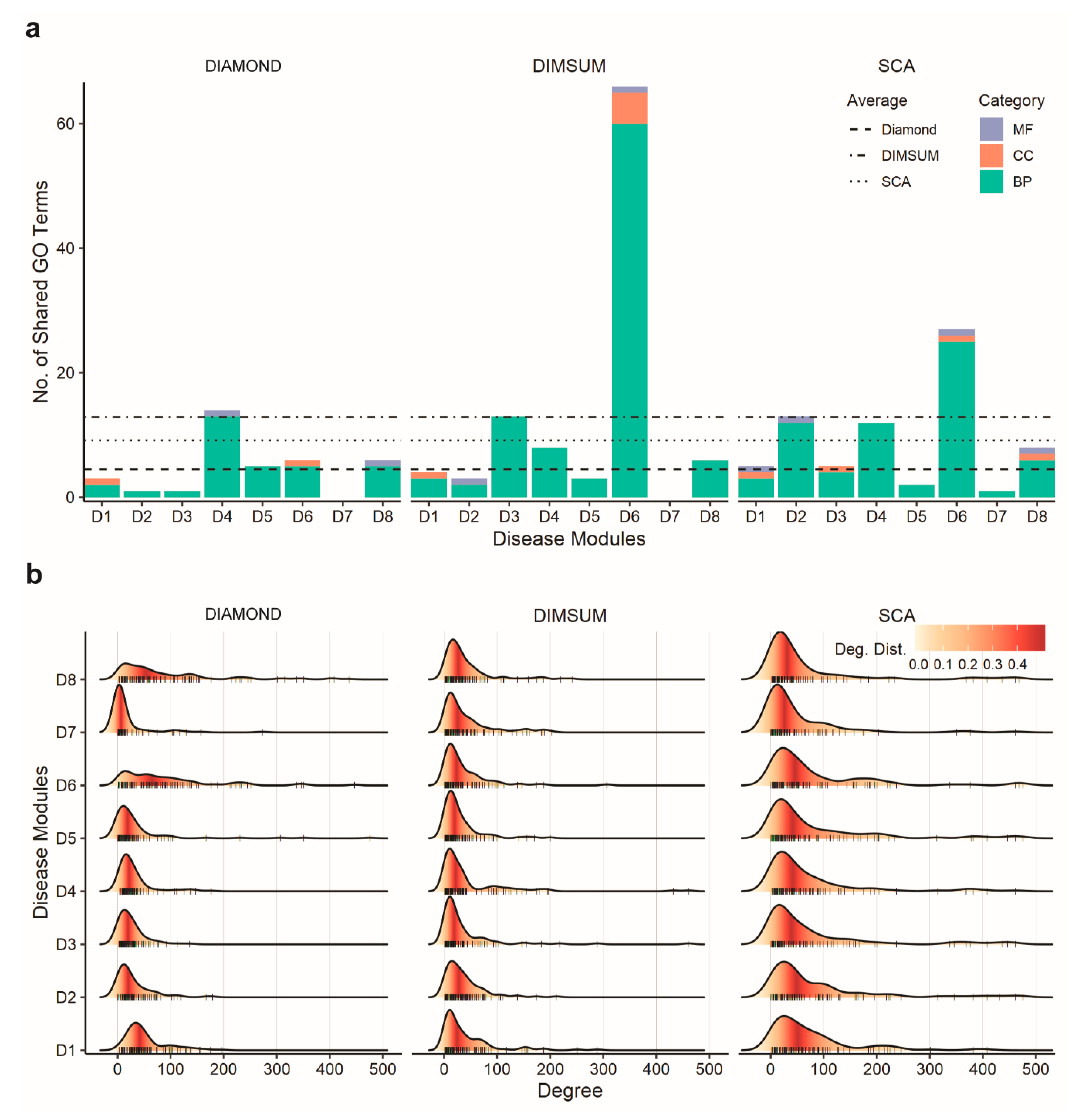
3.4. Comparison Against DIAMOnD and SCA
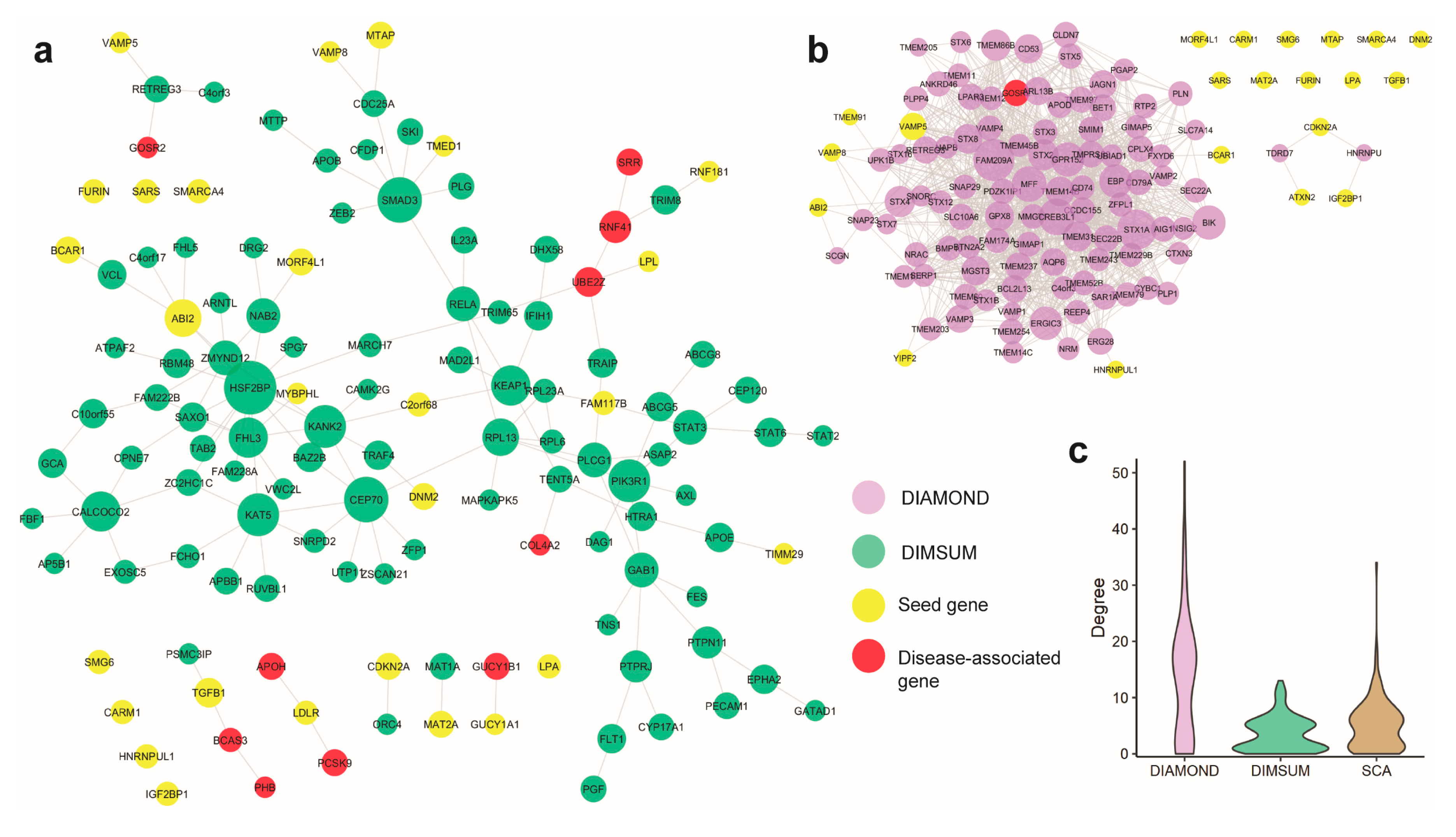
3.5. Case Study 1: Coronary Artery Disease
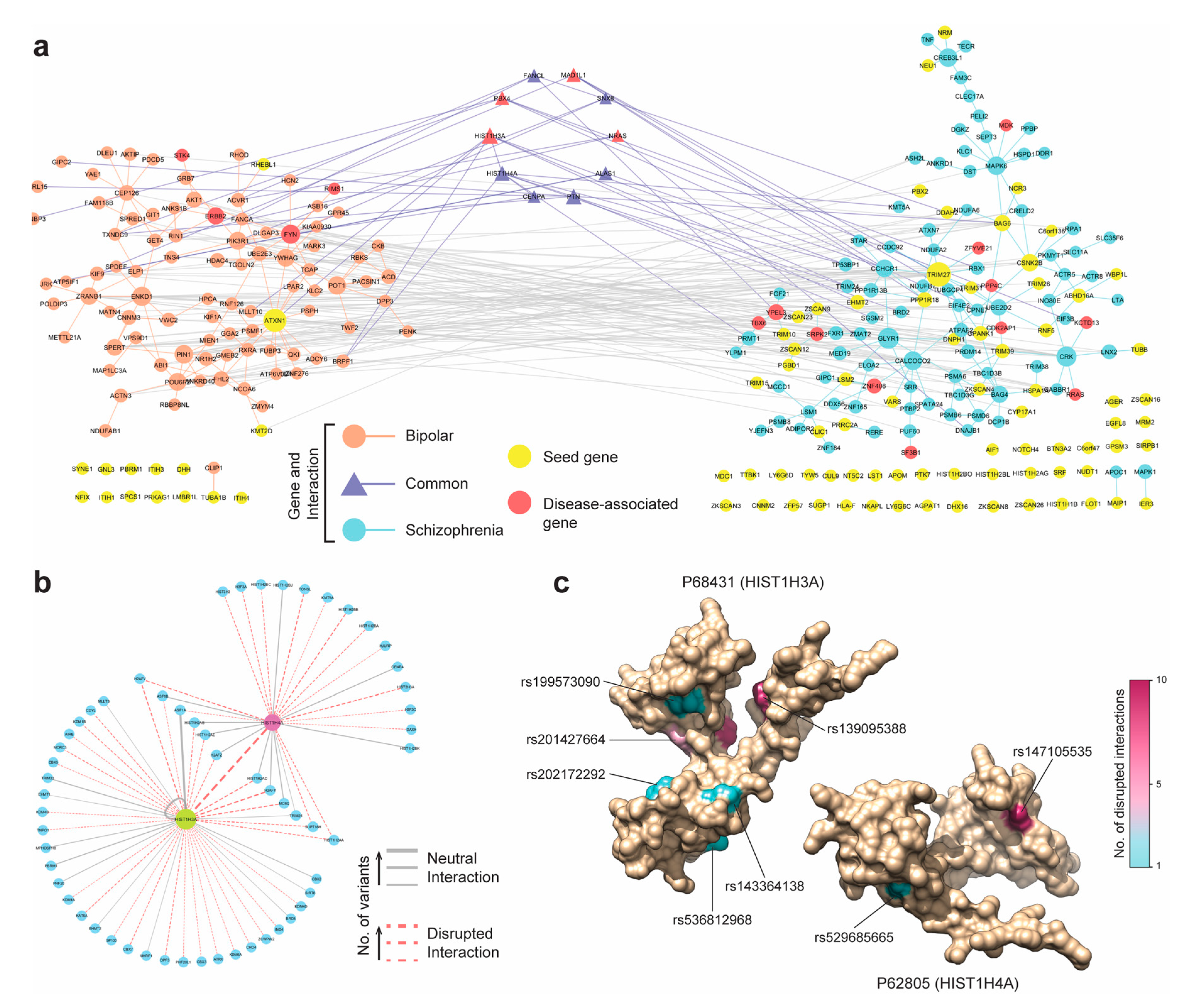
3.6. Case Study 2: Schizophrenia and Bipolar Disorder
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Anderson, D.E. Ceinical characteristics of the genetic variety of cutaneous melanoma in man. Cancer 1971, 28, 721–725. [Google Scholar] [CrossRef]
- Metzker, M.L. Sequencing technologies—The next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Ozsolak, F.; Milos, P.M. RNA sequencing: Advances, challenges and opportunities. Nat. Rev. Genet. 2011, 12, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Kolodziejczyk, A.A.; Kim, J.K.; Svensson, V.; Marioni, J.C.; Teichmann, S.A. The technology and biology of single-cell RNA sequencing. Mol. Cell 2015, 58, 610–620. [Google Scholar] [CrossRef]
- Zeggini, E. Next-generation association studies for complex traits. Nat. Genet. 2011, 43, 287–288. [Google Scholar] [CrossRef]
- Cui, H.; Dhroso, A.; Johnson, N.; Korkin, D. The variation game: Cracking complex genetic disorders with NGS and omics data. Methods 2015, 79, 18–31. [Google Scholar] [CrossRef]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- McCarthy, M.I.; Abecasis, G.R.; Cardon, L.R.; Goldstein, D.B.; Little, J.; Ioannidis, J.P.; Hirschhorn, J.N. Genome-wide association studies for complex traits: Consensus, uncertainty and challenges. Nat. Rev. Genet. 2008, 9, 356–369. [Google Scholar] [CrossRef]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef]
- Gonzalez-Perez, A.; Mustonen, V.; Reva, B.; Ritchie, G.R.; Creixell, P.; Karchin, R.; Vazquez, M.; Fink, J.L.; Kassahn, K.S.; Pearson, J.V. Computational approaches to identify functional genetic variants in cancer genomes. Nat. Methods 2013, 10, 723–729. [Google Scholar]
- Alexander, R.P.; Fang, G.; Rozowsky, J.; Snyder, M.; Gerstein, M.B. Annotating non-coding regions of the genome. Nat. Rev. Genet. 2010, 11, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Han, J.G.; Shyu, C.-R.; Korkin, D. Determining effects of non-synonymous SNPs on protein-protein interactions using supervised and semi-supervised learning. PLoS Comput. Biol. 2014, 10, e1003592. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Q.; Simonis, N.; Li, Q.R.; Charloteaux, B.; Heuze, F.; Klitgord, N.; Tam, S.; Yu, H.; Venkatesan, K.; Mou, D. Edgetic perturbation models of human inherited disorders. Mol. Syst. Biol. 2009, 5, 321. [Google Scholar] [CrossRef] [PubMed]
- Sahni, N.; Yi, S.; Zhong, Q.; Jailkhani, N.; Charloteaux, B.; Cusick, M.E.; Vidal, M. Edgotype: A fundamental link between genotype and phenotype. Curr. Opin. Genet. Dev. 2013, 23, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Zhao, N.; Korkin, D. Multilayer view of pathogenic SNVs in human interactome through in silico edgetic profiling. J. Mol. Biol. 2018, 430, 2974–2992. [Google Scholar] [CrossRef] [PubMed]
- Ideker, T.; Sharan, R. Protein networks in disease. Genome Res. 2008, 18, 644–652. [Google Scholar] [CrossRef]
- Cui, H.; Korkin, D. Effect-specific analysis of pathogenic SNVs in human interactome: Leveraging edge-based network robustness. In Proceedings of the 2016 38th Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC), Orlando, FL, USA, 16–20 August 2016. [Google Scholar]
- Wang, X.; Wei, X.; Thijssen, B.; Das, J.; Lipkin, S.M.; Yu, H. Three-dimensional reconstruction of protein networks provides insight into human genetic disease. Nat. Biotechnol. 2012, 30, 159–164. [Google Scholar] [CrossRef]
- Barabási, A.-L.; Gulbahce, N.; Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 2011, 12, 56–6854. [Google Scholar] [CrossRef]
- Rolland, T.; Taşan, M.; Charloteaux, B.; Pevzner, S.J.; Zhong, Q.; Sahni, N.; Yi, S.; Lemmens, I.; Fontanillo, C.; Mosca, R. A proteome-scale map of the human interactome network. Cell 2014, 159, 1212–1226. [Google Scholar] [CrossRef]
- Luck, K.; Kim, D.K.; Lambourne, L.; Spirohn, K.; Begg, B.E.; Bian, W.; Brignall, R.; Cafarelli, T.; Campos-Laborie, F.J.; Charloteaux, B. A reference map of the human protein interactome. bioRxiv 2019. [Google Scholar] [CrossRef]
- Cowen, L.; Ideker, T.; Raphael, B.J.; Sharan, R. Network propagation: A universal amplifier of genetic associations. Nat. Rev. Genet. 2017, 18, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Csermely, P.; Korcsmáros, T.; Kiss, H.J.; London, G.; Nussinov, R. Structure and dynamics of molecular networks: A novel paradigm of drug discovery: A comprehensive review. Pharmacol. Ther. 2013, 138, 333–408. [Google Scholar] [CrossRef] [PubMed]
- Choobdar, S.; Ahsen, M.E.; Crawford, J.; Tomasoni, M.; Fang, T.; Lamparter, D.; Lin, J.; Hescott, B.; Hu, X.; Mercer, J. Assessment of network module identification across complex diseases. Nat. Methods 2019, 16, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Girvan, M.; Newman, M.E. Community structure in social and biological networks. Proc. Natl. Acad. Sci. USA 2002, 99, 7821–7826. [Google Scholar] [CrossRef] [PubMed]
- Ghiassian, S.D.; Menche, J.; Barabási, A.-L. A DIseAse MOdule Detection (DIAMOnD) algorithm derived from a systematic analysis of connectivity patterns of disease proteins in the human interactome. PLoS Comput. Biol. 2015, 11, e1004120. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.; Moutari, S.; Dehmer, M.; Emmert-Streib, F. Comparison of module detection algorithms in protein networks and investigation of the biological meaning of predicted modules. BMC Bioinform. 2016, 17, 129. [Google Scholar] [CrossRef]
- Vlaic, S.; Conrad, T.; Tokarski-Schnelle, C.; Gustafsson, M.; Dahmen, U.; Guthke, R.; Schuster, S. ModuleDiscoverer: Identification of regulatory modules in protein-protein interaction networks. Sci. Rep. 2018, 8, 433. [Google Scholar] [CrossRef]
- Zhang, D.; Cui, H.; Korkin, D.; Wu, Z. Incorporation of protein binding effects into likelihood ratio test for exome sequencing data. In BMC Proceedings; BioMed Central: London, UK, 2016. [Google Scholar]
- Mitra, K.; Carvunis, A.-R.; Ramesh, S.K.; Ideker, T. Integrative approaches for finding modular structure in biological networks. Nat. Rev. Genet. 2013, 14, 719–732. [Google Scholar] [CrossRef]
- Das, J.; Yu, H. HINT: High-quality protein interactomes and their applications in understanding human disease. BMC Syst. Biol. 2012, 6, 92. [Google Scholar] [CrossRef]
- Lamparter, D.; Marbach, D.; Rueedi, R.; Kutalik, Z.; Bergmann, S. Fast and rigorous computation of gene and pathway scores from SNP-based summary statistics. PLoS Comput. Biol. 2016, 12, e1004714. [Google Scholar] [CrossRef]
- Wang, L.; Jia, P.; Wolfinger, R.D.; Chen, X.; Grayson, B.L.; Aune, T.M.; Zhao, Z. An efficient hierarchical generalized linear mixed model for pathway analysis of genome-wide association studies. Bioinformatics 2011, 27, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Consortium, G.P. An integrated map of genetic variation from 1092 human genomes. Nature 2012, 491, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Benedix, A.; Becker, C.M.; de Groot, B.L.; Caflisch, A.; Böckmann, R.A. Predicting free energy changes using structural ensembles. Nat. Methods 2009, 6, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Kamisetty, H.; Ramanathan, A.; Bailey-Kellogg, C.; Langmead, C.J. Accounting for conformational entropy in predicting binding free energies of protein-protein interactions. Proteins Struct. Funct. Bioinform. 2011, 79, 444–462. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Sussman, J.L.; Lin, D.; Jiang, J.; Manning, N.O.; Prilusky, J.; Ritter, O.; Abola, E.E. Protein Data Bank (PDB): Database of three-dimensional structural information of biological macromolecules. Acta Crystallogr. Sect. D Biol. Crystallogr. 1998, 54, 1078–1084. [Google Scholar] [CrossRef]
- Mosca, R.; Ceol, A.; Stein, A.; Olivella, R.; Aloy, P. 3did: A catalog of domain-based interactions of known three-dimensional structure. Nucleic Acids Res. 2013, 42, D374–D379. [Google Scholar] [CrossRef]
- Russell, R.B.; Alber, F.; Aloy, P.; Davis, F.P.; Korkin, D.; Pichaud, M.; Topf, M.; Sali, A. A structural perspective on protein–protein interactions. Curr. Opin. Struct. Biol. 2004, 14, 313–324. [Google Scholar] [CrossRef]
- Mosca, R.; Céol, A.; Aloy, P. Interactome3D: Adding structural details to protein networks. Nat. Methods 2013, 10, 47–53. [Google Scholar] [CrossRef]
- Vanunu, O.; Magger, O.; Ruppin, E.; Shlomi, T.; Sharan, R. Associating genes and protein complexes with disease via network propagation. PLoS Comput. Biol. 2010, 6, e1000641. [Google Scholar] [CrossRef]
- Wang, R.-S.; Loscalzo, J. Network-based disease module discovery by a novel seed connector algorithm with pathobiological implications. J. Mol. Biol. 2018, 430, 2939–2950. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Ball, E.V.; Mort, M.; Phillips, A.D.; Shiel, J.A.; Thomas, N.S.; Abeysinghe, S.; Krawczak, M.; Cooper, D.N. Human gene mutation database (HGMD®): 2003 update. Hum. Mutat. 2003, 21, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Hamosh, A.; Scott, A.F.; Amberger, J.S.; Bocchini, C.A.; McKusick, V.A. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005, 33 (Suppl. 1), D514–D517. [Google Scholar] [CrossRef] [PubMed]
- Rivals, I.; Personnaz, L.; Taing, L.; Potier, M.-C. Enrichment or depletion of a GO category within a class of genes: Which test? Bioinformatics 2006, 23, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Sahni, N.; Yi, S.; Taipale, M.; Bass, J.I.F.; Coulombe-Huntington, J.; Yang, F.; Peng, J.; Weile, J.; Karras, G.I.; Wang, Y. Widespread macromolecular interaction perturbations in human genetic disorders. Cell 2015, 161, 647–660. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Besenbacher, S.; Mailund, T.; Schierup, M.H. Identifying disease associated genes by network propagation. In BMC Systems Biology; BioMed Central: London, UK, 2014. [Google Scholar]
- Cao, M.; Zhang, H.; Park, J.; Daniels, N.M.; Crovella, M.E.; Cowen, L.J.; Hescott, B. Going the distance for protein function prediction: A new distance metric for protein interaction networks. PLoS ONE 2013, 8, e76339. [Google Scholar] [CrossRef]
- Craddock, N.; Sklar, P. Genetics of bipolar disorder. Lancet 2013, 381, 1654–1662. [Google Scholar] [CrossRef]
- Belmaker, R. Bipolar disorder. N. Engl. J. Med. 2004, 351, 476–486. [Google Scholar] [CrossRef]
- Fazel, S.; Gulati, G.; Linsell, L.; Geddes, J.R.; Grann, M. Schizophrenia and violence: Systematic review and meta-analysis. PLoS Med. 2009, 6, e1000120. [Google Scholar] [CrossRef]
- Lichtenstein, P.; Yip, B.H.; Björk, C.; Pawitan, Y.; Cannon, T.D.; Sullivan, P.F.; Hultman, C.M. Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: A population-based study. Lancet 2009, 373, 234–239. [Google Scholar] [CrossRef]
- Purcell, S.M.; Wray, N.R.; Stone, J.L.; Visscher, P.M.; O’Donovan, M.C.; Sullivan, P.F.; Sklar, P. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009, 460, 748–752. [Google Scholar] [PubMed]
- Ripke, S.; Sanders, A.R.; Kendler, K.S.; Levinson, D.F.; Sklar, P.; Holmans, P.A.; Lin, D.-Y.; Duan, J.; Ophoff, R.A.; Andreassen, O.A. Genome-wide association study identifies five new schizophrenia loci. Nat. Genet. 2011, 43, 969–976. [Google Scholar]
- Sklar, P.; Ripke, S.; Scott, L.J.; Andreassen, O.A.; Cichon, S.; Craddock, N.; Edenberg, H.J.; Nurnberger, J.I., Jr.; Rietschel, M.; Blackwood, D. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat. Genet. 2011, 43, 977–983. [Google Scholar]
- Stahl, E.A.; Breen, G.; Forstner, A.J.; McQuillin, A.; Ripke, S.; Trubetskoy, V.; Mattheisen, M.; Wang, Y.; Coleman, J.R.; Gaspar, H.A. Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat. Genet. 2019, 51, 793–803. [Google Scholar] [CrossRef]
- Pinto, D.; Delaby, E.; Merico, D.; Barbosa, M.; Merikangas, A.; Klei, L.; Thiruvahindrapuram, B.; Xu, X.; Ziman, R.; Wang, Z. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet. 2014, 94, 677–694. [Google Scholar] [CrossRef]
- Pirooznia, M.; Wang, T.; Avramopoulos, D.; Potash, J.B.; Zandi, P.P.; Goes, F.S. High-throughput sequencing of the synaptome in major depressive disorder. Mol. Psychiatry 2016, 21, 650–655. [Google Scholar] [CrossRef]
- Fabbri, C.; Serretti, A. Pharmacogenetics of major depressive disorder: Top genes and pathways toward clinical applications. Curr. Psychiatry Rep. 2015, 17, 50. [Google Scholar] [CrossRef]
- Castillo, P.E.; Schoch, S.; Schmitz, F.; Südhof, T.C.; Malenka, R.C. RIM1α is required for presynaptic long-term potentiation. Nature 2002, 415, 327–330. [Google Scholar] [CrossRef]
- Sisodiya, S.M.; Thompson, P.J.; Need, A.; Harris, S.E.; Weale, M.E.; Wilkie, S.E.; Michaelides, M.; Free, S.L.; Walley, N.; Gumbs, C. Genetic enhancement of cognition in a kindred with cone–rod dystrophy due to RIMS1 mutation. J. Med. Genet. 2007, 44, 373–380. [Google Scholar] [CrossRef]
- Stessman, H.A.; Xiong, B.; Coe, B.P.; Wang, T.; Hoekzema, K.; Fenckova, M.; Kvarnung, M.; Gerdts, J.; Trinh, S.; Cosemans, N. Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat. Genet. 2017, 49, 515–526. [Google Scholar] [CrossRef]
- Radulescu, E.; Jaffe, A.E.; Straub, R.E.; Chen, Q.; Shin, J.H.; Hyde, T.M.; Kleinman, J.E.; Weinberger, D.R. Identification and prioritization of gene sets associated with schizophrenia risk by co-expression network analysis in human brain. Mol. Psychiatry 2018. [Google Scholar] [CrossRef] [PubMed]
- Winkler, C.; Yao, S. The midkine family of growth factors: Diverse roles in nervous system formation and maintenance. Br. J. Pharmacol. 2014, 171, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, T. Midkine: A promising molecule for drug development to treat diseases of the central nervous system. Curr. Pharm. Des. 2011, 17, 410–423. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.; Keleshian, V.; Klein, S.; Rapoport, S. Epigenetic modifications in frontal cortex from Alzheimer’s disease and bipolar disorder patients. Transl. Psychiatry 2012, 2, e132. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.P.; Rosen, C.; Kartan, S.; Guidotti, A.; Costa, E.; Grayson, D.R.; Chase, K. Valproic acid and chromatin remodeling in schizophrenia and bipolar disorder: Preliminary results from a clinical population. Schizophr. Res. 2006, 88, 227–231. [Google Scholar] [CrossRef]
- Borsboom, D. A network theory of mental disorders. World Psychiatry 2017, 16, 5–13. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease ID | Disease Name | Seeds Match | DIAMOnD Match | SCA Match | DIMSUM Match |
|---|---|---|---|---|---|
| D1 | Coronary artery disease | 8 | 1 | 0 | 10 |
| D2 | Diabetes mellitus, Type 2 | 14 | 0 | 1 | 2 |
| D3 | Macular degeneration | 17 | 0 | 0 | 3 |
| D4 | Osteoporosis | 5 | 1 | 1 | 0 |
| D5 | Alzheimer’s disease | 8 | 0 | 0 | 3 |
| D6 | Rheumatoid arthritis | 19 | 0 | 0 | 4 |
| D7 | Bipolar disorder | 12 | 0 | 0 | 8 |
| D8 | Schizophrenia | 29 | 0 | 0 | 14 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, H.; Srinivasan, S.; Korkin, D. Enriching Human Interactome with Functional Mutations to Detect High-Impact Network Modules Underlying Complex Diseases. Genes 2019, 10, 933. https://doi.org/10.3390/genes10110933
Cui H, Srinivasan S, Korkin D. Enriching Human Interactome with Functional Mutations to Detect High-Impact Network Modules Underlying Complex Diseases. Genes. 2019; 10(11):933. https://doi.org/10.3390/genes10110933
Chicago/Turabian StyleCui, Hongzhu, Suhas Srinivasan, and Dmitry Korkin. 2019. "Enriching Human Interactome with Functional Mutations to Detect High-Impact Network Modules Underlying Complex Diseases" Genes 10, no. 11: 933. https://doi.org/10.3390/genes10110933
APA StyleCui, H., Srinivasan, S., & Korkin, D. (2019). Enriching Human Interactome with Functional Mutations to Detect High-Impact Network Modules Underlying Complex Diseases. Genes, 10(11), 933. https://doi.org/10.3390/genes10110933