Fine-Mapping Array Design for Multi-Ethnic Studies of Multiple Sclerosis


{kind=link}
Abstract
1. Introduction
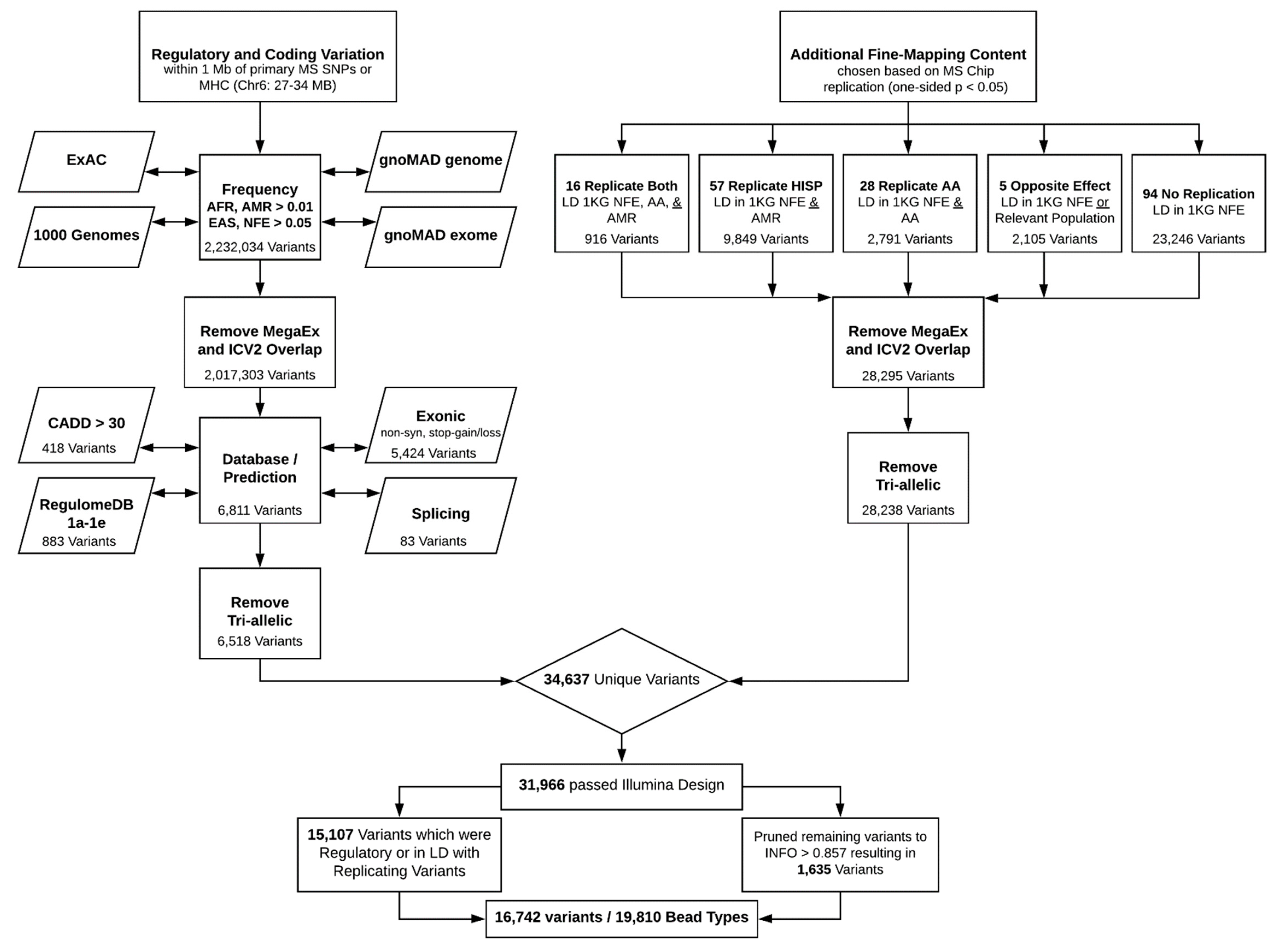
2. Coding and Regulatory Variation
3. Fine-Mapping Content
4. Final Pruning
5. Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- International Multiple Sclerosis Genetics Consortium (IMSGC). Multiple Sclerosis Genomic Map implicates peripheral immune cells and microglia in susceptibility. Science 2019, 365. [Google Scholar] [CrossRef]
- Hafler, D.A.; Compston, A.; Sawcer, S.; Lander, E.S.; Daly, M.J.; De Jager, P.L.; de Bakker, P.I.; Gabriel, S.B.; Mirel, D.B.; Ivinson, A.J.; et al. International Multiple Sclerosis Genetics Consortium. Risk alleles for multiple sclerosis identified by a genomewide study. N. Engl. J. Med. 2007, 357, 851–862. [Google Scholar] [PubMed]
- Sawcer, S.; Hellenthal, G.; Pirinen, M.; Spencer, C.C.; Patsopoulos, N.A.; Moutsianas, L.; Dilthey, A.; Su, Z.; Freeman, C.; Hunt, S.E.; et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011, 476, 214–219. [Google Scholar] [PubMed]
- Patsopoulos, N.A.; Bayer Pharma, M.S.; Esposito, F.; Reischl, J.; Lehr, S.; Bauer, D.; Heubach, J.; Sandbrink, R.; Pohl, C.; Edan, G.; et al. Genome-wide meta-analysis identifies novel multiple sclerosis susceptibility loci. Ann. Neurol. 2011, 70, 897–912. [Google Scholar] [CrossRef] [PubMed]
- Schaid, D.J.; Chen, W.; Larson, N.B. From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat. Rev. Genet. 2018, 19, 491–504. [Google Scholar] [CrossRef]
- Beecham, A.H.; Patsopoulos, N.A.; Xifara, D.K.; Davis, M.F.; Kemppinen, A.; Cotsapas, C.; Shah, T.S.; Spencer, C.; Booth, D.; Goris, A.; et al. International Multiple Sclerosis Genetics Consortium (IMSGC); Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat. Genet. 2013, 45, 1353–1360. [Google Scholar]
- Cortes, A.; Brown, M.A. Promise and pitfalls of the Immunochip. Arthritis Res. Ther. 2011, 13, 101. [Google Scholar] [CrossRef]
- Beecham, A.H.; Amezcua, L.; Chinea, A.; Manrique, C.P.; Rubi, C.; Isobe, N.; Lund, B.T.; Santaniello, A.; Beecham, G.W.; Burchard, E.G.; et al. The genetic diversity of multiple sclerosis risk among Hispanic and African American populations living in the United States. Mult. Scler. 2019. [Google Scholar] [CrossRef]
- Isobe, N.; Madireddy, L.; Khankhanian, P.; Matsushita, T.; Caillier, S.J.; More, J.M.; Gourraud, P.A.; McCauley, J.L.; Beecham, A.H.; International Multiple Sclerosis Genetics Consortium; et al. An ImmunoChip study of multiple sclerosis risk in African Americans. Brain 2015, 138, 1518–1530. [Google Scholar] [CrossRef]
- Karczewski, K.; Francioli, L.; Tiao, G.; Cummings, B. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv 2019. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Exome Aggregation Consortium Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Abecasis, G.R.; Auton, A.; Brooks, L.D.; DePristo, M.A.; Durbin, R.M.; Handsaker, R.E.; Kang, H.M.; Marth, G.T.; McVean, G.A. 1000 Genomes Project Consortium; An integrated map of genetic variation from 1,092 human genomes. Nature 2012, 491, 56–65. [Google Scholar] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Boyle, A.P.; Hong, E.L.; Hariharan, M.; Cheng, Y.; Schaub, M.A.; Kasowski, M.; Karczewski, K.J.; Park, J.; Hitz, B.C.; Weng, S.; et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012, 22, 1790–1797. [Google Scholar] [CrossRef] [PubMed]
- GTEx Consortium. Genetic effects on gene expression across human tissues. Nature 2017, 550, 204–213. [Google Scholar] [CrossRef]
- Zheng, X.; Shen, J.; Cox, C.; Wakefield, J.C.; Ehm, M.G.; Nelson, M.R.; Weir, B.S. HIBAG—HLA genotype imputation with attribute bagging. Pharmacogenom. J. 2014, 14, 192–200. [Google Scholar] [CrossRef]
- Illumina Infinium® iSelect® CustomGenotypingAssays. Available online: https://www.illumina.com/Documents/products/technotes/technote_iselect_design.pdf (accessed on 29 September 2019).
- Bustamante, C.D.; Burchard, E.G.; De la Vega, F.M. Genomics for the world. Nature 2011, 475, 163–165. [Google Scholar] [CrossRef]
- Popejoy, A.B.; Fullerton, S.M. Genomics is failing on diversity. Nature 2016, 538, 161–164. [Google Scholar] [CrossRef]
- Need, A.C.; Goldstein, D.B. Next generation disparities in human genomics: Concerns and remedies. Trends Genet. 2009, 25, 489–494. [Google Scholar] [CrossRef]
- Bentley, A.R.; Callier, S.; Rotimi, C.N. Diversity and inclusion in genomic research: Why the uneven progress? J. Community Genet. 2017, 8, 255–266. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beecham, A.H.; McCauley, J.L. Fine-Mapping Array Design for Multi-Ethnic Studies of Multiple Sclerosis. Genes 2019, 10, 903. https://doi.org/10.3390/genes10110903
Beecham AH, McCauley JL. Fine-Mapping Array Design for Multi-Ethnic Studies of Multiple Sclerosis. Genes. 2019; 10(11):903. https://doi.org/10.3390/genes10110903
Chicago/Turabian StyleBeecham, Ashley H., and Jacob L. McCauley. 2019. "Fine-Mapping Array Design for Multi-Ethnic Studies of Multiple Sclerosis" Genes 10, no. 11: 903. https://doi.org/10.3390/genes10110903
APA StyleBeecham, A. H., & McCauley, J. L. (2019). Fine-Mapping Array Design for Multi-Ethnic Studies of Multiple Sclerosis. Genes, 10(11), 903. https://doi.org/10.3390/genes10110903