Gut Microbiota Influences Experimental Outcomes in Mouse Models of Colorectal Cancer


Abstract
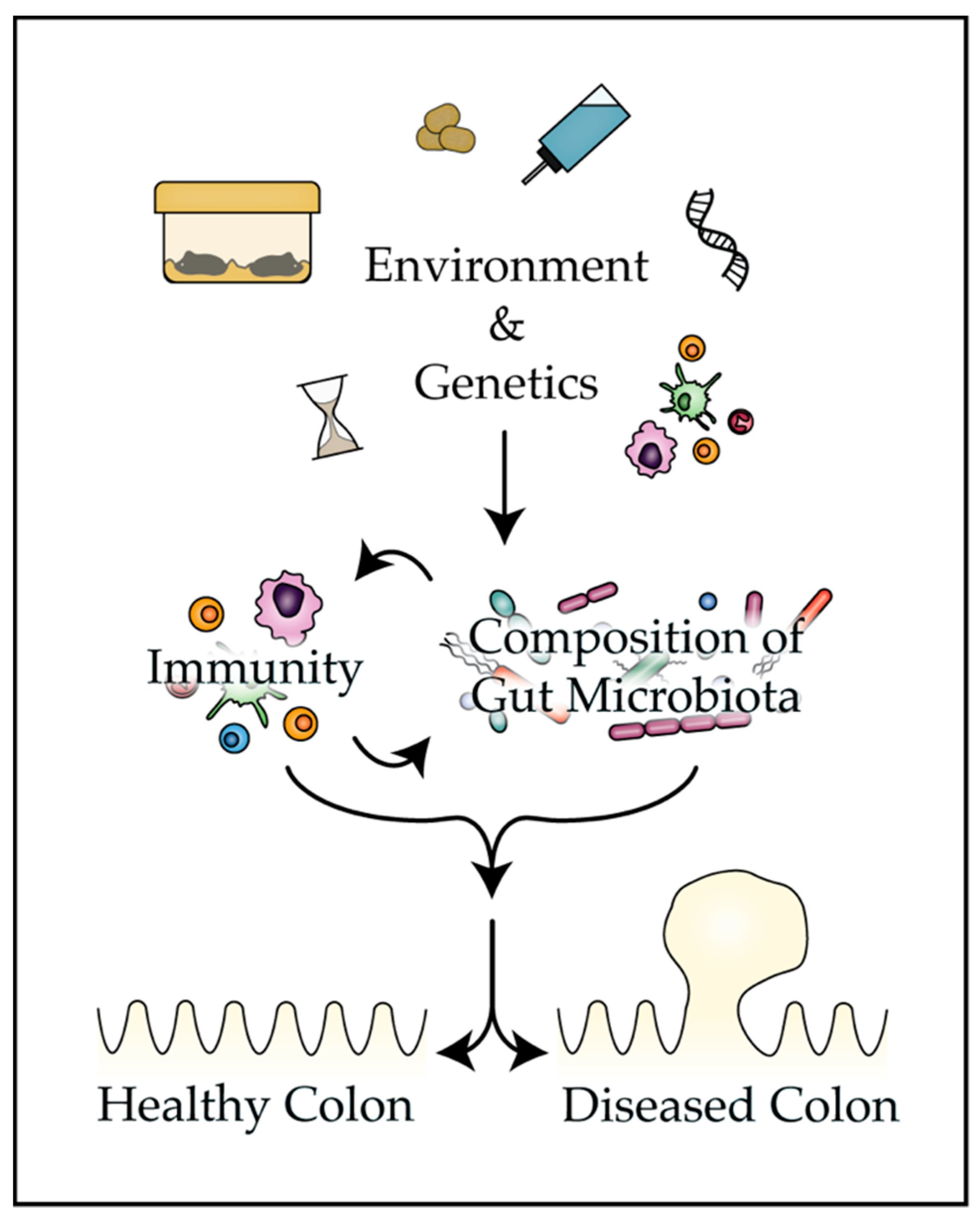
1. Introduction
2. Gut Microbiota Modulate Colon Tumorigenesis
2.1. Evidence that Microbiota Can Restrain Colon Tumorigenesis
2.2. Evidence that Microbiota Can Promote Colon Tumorigenesis
2.3. Contribution of Specific Bacteria to Colon Tumorigenesis
2.3.1. Fusobacterium Nucleatum
2.3.2. Bacteroides Fragilis
2.3.3. Escherichia Coli
2.4. Activities of the Microbiota that Impact the Homeostasis of the Colonic Mucosa
2.4.1. Biofilm Formation
2.4.2. Metabolites Produced by Microbiota
2.4.3. Interactions with Tissue-Resident Immune Cells
3. Gut Microbiota Modulate the Response of Colon Tumors to Chemotherapy
3.1. Activation of Autophagy in Cancer Cells
3.2. Metabolism of Chemotherapeutic Agents
3.3. Promotion of Antitumor Immunity
4. Factors that Influence the Composition of the Gut Microbiota
4.1. Genetics
4.2. Birth Mother
4.3. Age
4.4. Housing
4.5. Diet
4.6. Institution
4.7. Immune System
5. Implications for Model Selection and Experimental Design
5.1. Characterizing Microbiota to Improve Mouse Models of CRC
5.2. Modifying Microbiota to Improve Mouse Models of CRC
5.3. Experimental Design—Correcting for Factors that Influence Gut Microbiota in Experiments
5.4. Reporting Experimental Details
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Ciardiello, D.; Vitiello, P.P.; Cardone, C.; Martini, G.; Troiani, T.; Martinelli, E.; Ciardiello, F. Immunotherapy of colorectal cancer: Challenges for therapeutic efficacy. Cancer Treat. Rev. 2019, 76, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Tintelnot, J.; Stein, A. Immunotherapy in colorectal cancer: Available clinical evidence, challenges and novel approaches. WJG 2019, 25, 3920–3928. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.-C.L.; Jackson, C.; Riel, S.; Cooper, H.S.; Devarajan, K.; Hensley, H.H.; Zhou, Y.; Vanderveer, L.A.; Nguyen, M.T.; Clapper, M.L. Differential preventive activity of sulindac and atorvastatin in Apc +/Min-FCCCmice with or without colorectal adenomas. Gut 2018, 67, 1290–1298. [Google Scholar] [CrossRef] [PubMed]
- Kaur, K.; Saxena, A.; Debnath, I.; O’Brien, J.L.; Ajami, N.J.; Auchtung, T.A.; Petrosino, J.F.; Sougiannis, A.-J.; Depaep, S.; Chumanevich, A.; et al. Antibiotic-mediated bacteriome depletion in Apc Min/+ mice is associated with reduction in mucus-producing goblet cells and increased colorectal cancer progression. Cancer Med. 2018, 7, 2003–2012. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Rhee, K.-J.; Albesiano, E.; Rabizadeh, S.; Wu, X.; Yen, H.-R.; Huso, D.L.; Brancati, F.L.; Wick, E.; Mcallister, F.; et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 2009, 15, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- Destefano Shields, C.E.; Van Meerbeke, S.W.; Housseau, F.; Wang, H.; Huso, D.L.; Casero, R.A.; O’Hagan, H.M.; Sears, C.L. Reduction of Murine Colon Tumorigenesis Driven by Enterotoxigenic Bacteroides fragilis Using Cefoxitin Treatment. J. Infect. Dis. 2016, 214, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Chung, L.; Thiele Orberg, E.; Geis, A.L.; Chan, J.L.; Fu, K.; Destefano Shields, C.E.; Dejea, C.M.; Fathi, P.; Chen, J.; Finard, B.B.; et al. Bacteroides fragilis Toxin Coordinates a Pro-carcinogenic Inflammatory Cascade via Targeting of Colonic Epithelial Cells. Cell Host Microbe 2018, 23, 203–214.e5. [Google Scholar] [CrossRef]
- Dejea, C.M.; Fathi, P.; Craig, J.M.; Boleij, A.; Taddese, R.; Geis, A.L.; Wu, X.; Destefano Shields, C.E.; Hechenbleikner, E.M.; Huso, D.L.; et al. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science 2018, 359, 592–597. [Google Scholar] [CrossRef]
- Arthur, J.C.; Perez-Chanona, E.; Muhlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.J.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal Inflammation Targets Cancer-Inducing Activity of the Microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef]
- Yang, I.; Eibach, D.; Kops, F.; Brenneke, B.; Woltemate, S.; Schulze, J.; Bleich, A.; Gruber, A.D.; Muthupalani, S.; Fox, J.G.; et al. Intestinal Microbiota Composition of Interleukin-10 Deficient C57BL/6J Mice and Susceptibility to Helicobacter hepaticus-Induced Colitis. PLoS ONE 2013, 8, e70783-13. [Google Scholar] [CrossRef] [PubMed]
- Kullberg, M.C.; Ward, J.M.; Gorelick, P.L.; Caspar, P.; Hieny, S.; Cheever, A.; Jankovic, D.; Sher, A. Helicobacter hepaticus triggers colitis in specific-pathogen-free interleukin-10 (IL-10)-deficient mice through an IL-12- and gamma interferon-dependent mechanism. Infect. Immun. 1998, 66, 5157–5166. [Google Scholar] [PubMed]
- Dieleman, L.A.; Arends, A.; Tonkonogy, S.L.; Goerres, M.S.; Craft, D.W.; Grenther, W.; Sellon, R.K.; Balish, E.; Sartor, R.B. Helicobacter hepaticus does not induce or potentiate colitis in interleukin-10-deficient mice. Infect. Immun. 2000, 68, 5107–5113. [Google Scholar] [CrossRef] [PubMed]
- Belcheva, A.; Irrazabal, T.; Robertson, S.J.; Streutker, C.; Maughan, H.; Rubino, S.; Moriyama, E.H.; Copeland, J.K.; Surendra, A.; Kumar, S.; et al. Gut Microbial Metabolism Drives Transformation of Msh2-Deficient Colon Epithelial Cells. Cell 2014, 158, 288–299. [Google Scholar] [CrossRef]
- Zackular, J.P.; Baxter, N.T.; Iverson, K.D.; Sadler, W.D.; Petrosino, J.F.; Chen, G.Y.; Schloss, P.D. The Gut Microbiome Modulates Colon Tumorigenesis. mBio 2013, 4, e00692-13. [Google Scholar] [CrossRef]
- Zhan, Y.; Chen, P.-J.; Sadler, W.D.; Wang, F.; Poe, S.; Nunez, G.; Eaton, K.A.; Chen, G.Y. Gut microbiota protects against gastrointestinal tumorigenesis caused by epithelial injury. Cancer Res. 2013, 73, 7199–7210. [Google Scholar] [CrossRef]
- Rosshart, S.P.; Vassallo, B.G.; Angeletti, D.; Hutchinson, D.S.; Morgan, A.P.; Takeda, K.; Hickman, H.D.; Mcculloch, J.A.; Badger, J.H.; Ajami, N.J.; et al. Wild Mouse Gut Microbiota Promotes Host Fitness and Improves Disease Resistance. Cell 2017, 171, 1015–1028.e13. [Google Scholar] [CrossRef]
- Lehouritis, P.; Cummins, J.; Stanton, M.; Murphy, C.T.; McCarthy, F.O.; Reid, G.; Urbaniak, C.; Byrne, W.L.; Tangney, M. Local bacteria affect the efficacy of chemotherapeutic drugs. Sci. Rep. 2015, 5, 14554. [Google Scholar] [CrossRef]
- Iida, N.; Dzutsev, A.; Stewart, C.A.; Smith, L.; Bouladoux, N.; Weingarten, R.A.; Molina, D.A.; Salcedo, R.; Back, T.; Cramer, S.; et al. Commensal Bacteria Control Cancer Response to Therapy by Modulating the Tumor Microenvironment. Science 2013, 342, 967–970. [Google Scholar] [CrossRef]
- Vetizou, M.; Pitt, J.M.; Daillere, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.M.; et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef]
- Hooper, L.V.; Littman, D.R.; Macpherson, A.J. Interactions Between the Microbiota and the Immune System. Science 2012, 336, 1268–1273. [Google Scholar] [CrossRef]
- Carroll, I.M.; Threadgill, D.W.; Threadgill, D.S. The gastrointestinal microbiome: A malleable, third genome of mammals. Mamm. Genome 2009, 20, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Son, J.S.; Khair, S.; Pettet, D.W.; Ouyang, N.; Tian, X.; Zhang, Y.; Zhu, W.; Mackenzie, G.G.; Robertson, C.E.; Ir, D.; et al. Altered Interactions between the Gut Microbiome and Colonic Mucosa Precede Polyposis in APCMin/+ Mice. PLoS ONE 2015, 10, e0127985. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Haines, C.; Watson, A.J.M.; Hart, A.R.; Platt, M.J.; Pardoll, D.M.; Cosgrove, S.E.; Gebo, K.A.; Sears, C.L. Oral antibiotic use and risk of colorectal cancer in the United Kingdom, 1989–2012: A matched case-control study. Gut 2019, 68, 1971–1978. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-S.-E.; Li, W.-B.; Wang, H.-Y.; Ma, Y.-M.; Zhao, X.-H.; Yang, H.; Qian, J.-M.; Li, J.-N. VSL#3 can prevent ulcerative colitis-associated carcinogenesis in mice. WJG 2018, 24, 4254–4262. [Google Scholar]
- Do, E.-J.; Hwang, S.W.; Kim, S.-Y.; Ryu, Y.-M.; Cho, E.A.; Chung, E.-J.; Park, S.; Lee, H.J.; Byeon, J.-S.; Ye, B.D.; et al. Suppression of colitis-associated carcinogenesis through modulation of IL-6/STAT3 pathway by balsalazide and VSL#3. J. Gastroenterol. Hepatol. 2016, 31, 1453–1461. [Google Scholar]
- Grivennikov, S.I.; Wang, K.; Mucida, D.; Stewart, C.A.; Schnabl, B.; Jauch, D.; Taniguchi, K.; Yu, G.-Y.; Österreicher, C.H.; Hung, K.E.; et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 2012, 491, 254–258. [Google Scholar] [CrossRef]
- Wong, S.H.; Zhao, L.; Zhang, X.; Nakatsu, G.; Han, J.; Xu, W.; Xiao, X.; Kwong, T.N.Y.; Tsoi, H.; Wu, W.K.K.; et al. Gavage of Fecal Samples From Patients With Colorectal Cancer Promotes Intestinal Carcinogenesis in Germ-Free and Conventional Mice. Gastroenterology 2017, 153, 1621–1633. [Google Scholar] [CrossRef]
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef]
- Arthur, J.C.; Gharaibeh, R.Z.; Mühlbauer, M.; Perez-Chanona, E.; Uronis, J.M.; Mccafferty, J.; Fodor, A.A.; Jobin, C. Microbial genomic analysis reveals the essential role of inflammation in bacteria-induced colorectal cancer. Nat. Commun. 2014, 5, 4724. [Google Scholar] [CrossRef]
- Cougnoux, A.; Dalmasso, G.; Martinez, R.; Buc, E.; Delmas, J.; Gibold, L.; Sauvanet, P.; Darcha, C.; Déchelotte, P.; Bonnet, M.; et al. Bacterial genotoxin colibactin promotes colon tumour growth by inducing a senescence-associated secretory phenotype. Gut 2014, 63, 1932–1942. [Google Scholar] [CrossRef] [PubMed]
- Tytgat, H.L.P.; Nobrega, F.L.; van der Oost, J.; de Vos, W.M. Bowel Biofilms: Tipping Points between a Healthy and Compromised Gut? Trends Microbiol. 2019, 27, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Dejea, C.M.; Wick, E.C.; Hechenbleikner, E.M.; White, J.R.; Mark Welch, J.L.; Rossetti, B.J.; Peterson, S.N.; Snesrud, E.C.; Borisy, G.G.; Lazarev, M.; et al. Microbiota organization is a distinct feature of proximal colorectal cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 18321–18326. [Google Scholar] [CrossRef] [PubMed]
- Zarco, M.F.; Vess, T.J.; Ginsburg, G.S. The oral microbiome in health and disease and the potential impact on personalized dental medicine. Oral Dis. 2012, 18, 109–120. [Google Scholar] [CrossRef]
- Han, S.; Gao, J.; Zhou, Q.; Liu, S.; Wen, C.; Yang, X. Role of intestinal flora in colorectal cancer from the metabolite perspective: A systematic review. CMAR 2018, 10, 199–206. [Google Scholar] [CrossRef]
- Seidel, D.V.; Azcarate-Peril, M.A.; Chapkin, R.S.; Turner, N.D. Shaping functional gut microbiota using dietary bioactives to reduce colon cancer risk. Semin. Cancer Biol. 2017, 46, 191–204. [Google Scholar] [CrossRef]
- Wilson, M.R.; Jiang, Y.; Villalta, P.W.; Stornetta, A.; Boudreau, P.D.; Carrá, A.; Brennan, C.A.; Chun, E.; Ngo, L.; Samson, L.D.; et al. The human gut bacterial genotoxin colibactin alkylates DNA. Science 2019, 363, eaar7785. [Google Scholar] [CrossRef]
- Donohoe, D.R.; Holley, D.; Collins, L.B.; Montgomery, S.A.; Whitmore, A.C.; Hillhouse, A.; Curry, K.P.; Renner, S.W.; Greenwalt, A.; Ryan, E.P.; et al. A Gnotobiotic Mouse Model Demonstrates That Dietary Fiber Protects against Colorectal Tumorigenesis in a Microbiota- and Butyrate-Dependent Manner. Cancer Discov. 2014, 4, 1387–1397. [Google Scholar] [CrossRef]
- Kim, M.; Friesen, L.; Park, J.; Kim, H.M.; Kim, C.H. Microbial metabolites, short-chain fatty acids, restrain tissue bacterial load, chronic inflammation, and associated cancer in the colon of mice. Eur. J. Immunol. 2018, 48, 1235–1247. [Google Scholar] [CrossRef]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly-Y, M.; Glickman, J.N.; Garrett, W.S. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef]
- Chapkin, R.S.; Davidson, L.A.; Ly, L.; Weeks, B.R.; Lupton, J.R.; McMurray, D.N. Immunomodulatory effects of (n-3) fatty acids: Putative link to inflammation and colon cancer. J. Nutr. 2007, 137, 200S–204S. [Google Scholar] [CrossRef] [PubMed]
- Bauer, H.; Horowitz, R.E.; Levenson, S.M.; Popper, H. The response of the lymphatic tissue to the microbial flora. Studies on germfree mice. Am. J. Pathol. 1963, 42, 471–483. [Google Scholar] [PubMed]
- Gensollen, T.; Iyer, S.S.; Kasper, D.L.; Blumberg, R.S. How colonization by microbiota in early life shapes the immune system. Science 2016, 352, 539–544. [Google Scholar] [CrossRef]
- Knoop, K.A.; Gustafsson, J.K.; McDonald, K.G.; Kulkarni, D.H.; Coughlin, P.E.; McCrate, S.; Kim, D.; Hsieh, C.-S.; Hogan, S.P.; Elson, C.O.; et al. Microbial antigen encounter during a preweaning interval is critical for tolerance to gut bacteria. Sci. Immunol. 2017, 2, eaao1314. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, U.; Wei, D.; Goldfarb, K.C.; Santee, C.A.; Lynch, S.V.; et al. Induction of Intestinal Th17 Cells by Segmented Filamentous Bacteria. Cell 2009, 139, 485–498. [Google Scholar] [CrossRef]
- Duan, J.; Chung, H.; Troy, E.; Kasper, D.L. Microbial colonization drives expansion of IL-1 receptor 1-expressing and IL-17-producing gamma/delta T cells. Cell Host Microbe 2010, 7, 140–150. [Google Scholar] [CrossRef]
- Bhattacharya, N.; Yuan, R.; Prestwood, T.R.; Penny, H.L.; DiMaio, M.A.; Reticker-Flynn, N.E.; Krois, C.R.; Kenkel, J.A.; Pham, T.D.; Carmi, Y.; et al. Normalizing Microbiota-Induced Retinoic Acid Deficiency Stimulates Protective CD8 + T Cell-Mediated Immunity in Colorectal Cancer. Immunity 2016, 45, 641–655. [Google Scholar] [CrossRef]
- Britton, G.J.; Contijoch, E.J.; Mogno, I.; Vennaro, O.H.; Llewellyn, S.R.; Ng, R.; Li, Z.; Mortha, A.; Merad, M.; Das, A.; et al. Microbiotas from Humans with Inflammatory Bowel Disease Alter the Balance of Gut Th17 and RORγt+ Regulatory T Cells and Exacerbate Colitis in Mice. Immunity 2019, 50, 212–224.e4. [Google Scholar] [CrossRef]
- Housseau, F.; Wu, S.; Wick, E.C.; Fan, H.; Wu, X.; Llosa, N.J.; Smith, K.N.; Tam, A.; Ganguly, S.; Wanyiri, J.W.; et al. Redundant Innate and Adaptive Sources of IL17 Production Drive Colon Tumorigenesis. Cancer Res. 2016, 76, 2115–2124. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 2011, 331, 337–341. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Oshima, K.; Suda, W.; Nagano, Y.; Nishikawa, H.; Fukuda, S.; Saito, T.; Narushima, S.; Hase, K.; et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 2013, 500, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Pastille, E.; Bardini, K.; Fleissner, D.; Adamczyk, A.; Frede, A.; Wadwa, M.; Von Smolinski, D.; Kasper, S.; Sparwasser, T.; Gruber, A.D.; et al. Transient Ablation of Regulatory T cells Improves Antitumor Immunity in Colitis-Associated Colon Cancer. Cancer Res. 2014, 74, 4258–4269. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Guo, F.; Yu, Y.; Sun, T.; Ma, D.; Han, J.; Qian, Y.; Kryczek, I.; Sun, D.; Nagarsheth, N.; et al. Fusobacterium nucleatum Promotes Chemoresistance to Colorectal Cancer by Modulating Autophagy. Cell 2017, 170, 548–563.e16. [Google Scholar] [CrossRef] [PubMed]
- Kunzmann, A.T.; Proença, M.A.; Jordao, H.W.; Jiraskova, K.; Schneiderova, M.; Levy, M.; Liska, V.; Buchler, T.; Vodickova, L.; Vymetalkova, V.; et al. Fusobacterium nucleatum tumor DNA levels are associated with survival in colorectal cancer patients. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 1891–1899. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, K.; Izumi, D.; Kandimalla, R.; Sonohara, F.; Baba, Y.; Yoshida, N.; Kodera, Y.; Baba, H.; Goel, A. Intratumoral Fusobacterium Nucleatum Levels Predict Therapeutic Response to Neoadjuvant Chemotherapy in Esophageal Squamous Cell Carcinoma. Clin. Cancer Res. 2019, 25, 6170–6179. [Google Scholar] [CrossRef] [PubMed]
- Panebianco, C.; Andriulli, A.; Pazienza, V. Pharmacomicrobiomics: Exploiting the drug-microbiota interactions in anticancer therapies. Microbiome 2018, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Cong, J.; Zhang, X. Roles of intestinal microbiota in response to cancer immunotherapy. Eur. J. Clin. Microbiol. Infect. Dis. 2018, 37, 2235–2240. [Google Scholar] [CrossRef]
- Hagan, T.; Cortese, M.; Rouphael, N.; Boudreau, C.; Linde, C.; Maddur, M.S.; Das, J.; Wang, H.; Guthmiller, J.; Zheng, N.-Y.; et al. Antibiotics-Driven Gut Microbiome Perturbation Alters Immunity to Vaccines in Humans. Cell 2019, 178, 1313–1328.e13. [Google Scholar] [CrossRef]
- Kalyan, A.; Kircher, S.; Shah, H.; Mulcahy, M.; Benson, A. Updates on immunotherapy for colorectal cancer. J. Gastrointest. Oncol. 2018, 9, 160–169. [Google Scholar] [CrossRef]
- Rioux, C.R.; Clapper, M.L.; Cooper, H.S.; Michaud, J.; St Amant, N.; Koohsari, H.; Workman, L.; Kaunga, E.; Hensley, H.; Pilorget, A.; et al. Self-antigen MASH2 combined with the AS15 immunostimulant induces tumor protection in colorectal cancer mouse models. PLoS ONE 2019, 14, e0210261. [Google Scholar] [CrossRef]
- Kwon, S.; Kim, Y.-E.; Park, J.-A.; Kim, D.-S.; Kwon, H.-J.; Lee, Y. Therapeutic effect of a TM4SF5-specific peptide vaccine against colon cancer in a mouse model. BMB Rep. 2014, 47, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Zheng, X.-J.; Liu, C.-C.; Zhou, Y.; Ye, X.-S. A cancer vaccine based on fluorine-modified sialyl-Tn induces robust immune responses in a murine model. Oncotarget 2017, 8, 47330–47343. [Google Scholar] [CrossRef] [PubMed]
- Kurilshikov, A.; Wijmenga, C.; Fu, J.; Zhernakova, A. Host Genetics and Gut Microbiome: Challenges and Perspectives. Trends Immunol. 2017, 38, 633–647. [Google Scholar] [CrossRef] [PubMed]
- Benson, A.K.; Kelly, S.A.; Legge, R.; Ma, F.; Low, S.J.; Kim, J.; Zhang, M.; Oh, P.L.; Nehrenberg, D.; Hua, K.; et al. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc. Natl. Acad. Sci. USA 2010, 107, 18933–18938. [Google Scholar] [CrossRef]
- Deloris Alexander, A.; Orcutt, R.P.; Henry, J.C.; Baker, J.; Bissahoyo, A.C.; Threadgill, D.W. Quantitative PCR assays for mouse enteric flora reveal strain-dependent differences in composition that are influenced by the microenvironment. Mamm. Genome 2006, 17, 1093–1104. [Google Scholar] [CrossRef]
- Friswell, M.K.; Gika, H.; Stratford, I.J.; Theodoridis, G.; Telfer, B.; Wilson, I.D.; Mcbain, A.J. Site and strain-specific variation in gut microbiota profiles and metabolism in experimental mice. PLoS ONE 2010, 5, e8584. [Google Scholar] [CrossRef]
- Pantoja-Feliciano, I.G.; Clemente, J.C.; Costello, E.K.; Perez, M.E.; Blaser, M.J.; Knight, R.; Dominguez-Bello, M.G. Biphasic assembly of the murine intestinal microbiota during early development. ISME J. 2013, 7, 1112–1115. [Google Scholar] [CrossRef]
- Hufeldt, M.R.; Nielsen, D.S.; Vogensen, F.K.; Midtvedt, T.; Hansen, A.K. Family relationship of female breeders reduce the systematic inter-individual variation in the gut microbiota of inbred laboratory mice. Lab. Anim. 2010, 44, 283–289. [Google Scholar] [CrossRef]
- Lundberg, R.; Bahl, M.I.; Licht, T.R.; Toft, M.F.; Hansen, A.K. Microbiota composition of simultaneously colonized mice housed under either a gnotobiotic isolator or individually ventilated cage regime. Sci. Rep. 2017, 7, 42245. [Google Scholar] [CrossRef]
- Mccafferty, J.; Mühlbauer, M.; Gharaibeh, R.Z.; Arthur, J.C.; Perez-Chanona, E.; Sha, W.; Jobin, C.; Fodor, A.A. Stochastic changes over time and not founder effects drive cage effects in microbial community assembly in a mouse model. ISME J. 2013, 7, 2116–2125. [Google Scholar] [CrossRef]
- Scott, K.A.; Ida, M.; Peterson, V.L.; Prenderville, J.A.; Moloney, G.M.; Izumo, T.; Murphy, K.; Murphy, A.; Ross, R.P.; Stanton, C.; et al. Revisiting Metchnikoff: Age-related alterations in microbiota-gut-brain axis in the mouse. Brain Behav. Immun. 2017, 65, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Fransen, F.; van Beek, A.A.; Borghuis, T.; Aidy, S.E.; Hugenholtz, F.; van der Gaast de Jongh, C.; Savelkoul, H.F.J.; De Jonge, M.I.; Boekschoten, M.V.; Smidt, H.; et al. Aged Gut Microbiota Contributes to Systemical Inflammaging after Transfer to Germ-Free Mice. Front. Immunol. 2017, 8, 1385. [Google Scholar] [CrossRef] [PubMed]
- Choo, J.M.; Trim, P.J.; Leong, L.E.X.; Abell, G.C.J.; Brune, C.; Jeffries, N.; Wesselingh, S.; Dear, T.N.; Snel, M.F.; Rogers, G.B. Inbred Mouse Populations Exhibit Intergenerational Changes in Intestinal Microbiota Composition and Function Following Introduction to a Facility. Front. Microbiol. 2017, 8, 608. [Google Scholar] [CrossRef] [PubMed]
- Llewellyn, S.R.; Britton, G.J.; Contijoch, E.J.; Vennaro, O.H.; Mortha, A.; Colombel, J.-F.; Grinspan, A.; Clemente, J.C.; Merad, M.; Faith, J.J. Interactions Between Diet and the Intestinal Microbiota Alter Intestinal Permeability and Colitis Severity in Mice. Gastroenterology 2018, 154, 1037–1046.e2. [Google Scholar] [CrossRef] [PubMed]
- Rausch, P.; Basic, M.; Batra, A.; Bischoff, S.C.; Blaut, M.; Clavel, T.; Gläsner, J.; Gopalakrishnan, S.; Grassl, G.A.; Günther, C.; et al. Analysis of factors contributing to variation in the C57BL/6J fecal microbiota across German animal facilities. Int. J. Med. Microbiol. 2016, 306, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Bidot, W.A.; Ericsson, A.C.; Franklin, C.L. Effects of water decontamination methods and bedding material on the gut microbiota. PLoS ONE 2018, 13, e0198305. [Google Scholar] [CrossRef] [PubMed]
- Sofi, M.H.; Gudi, R.; Karumuthil-Melethil, S.; Perez, N.; Johnson, B.M.; Vasu, C. pH of drinking water influences the composition of gut microbiome and type 1 diabetes incidence. Diabetes 2014, 63, 632–644. [Google Scholar] [CrossRef]
- Franklin, C.L.; Ericsson, A.C. Microbiota and reproducibility of rodent models. Lab. Anim. 2017, 46, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Kühn, R.; Löhler, J.; Rennick, D.; Rajewsky, K.; Muller, W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell 1993, 75, 263–274. [Google Scholar] [CrossRef]
- Zhang, H.; Sparks, J.B.; Karyala, S.V.; Settlage, R.; Luo, X.M. Host adaptive immunity alters gut microbiota. ISME J. 2015, 9, 770–781. [Google Scholar] [CrossRef]
- Larsson, E.; Tremaroli, V.; Lee, Y.S.; Koren, O.; Nookaew, I.; Fricker, A.; Nielsen, J.; Ley, R.E.; Bäckhed, F. Analysis of gut microbial regulation of host gene expression along the length of the gut and regulation of gut microbial ecology through MyD88. Gut 2012, 61, 1124–1131. [Google Scholar] [CrossRef]
- Vijay-Kumar, M.; Aitken, J.D.; Carvalho, F.A.; Cullender, T.C.; Mwangi, S.; Srinivasan, S.; Sitaraman, S.V.; Knight, R.; Ley, R.E.; Gewirtz, A.T. Metabolic Syndrome and Altered Gut Microbiota in Mice Lacking Toll-Like Receptor 5. Science 2010, 328, 228–231. [Google Scholar] [CrossRef]
- Zhao, X.; Li, L.; Starr, T.K.; Subramanian, S. Tumor location impacts immune response in mouse models of colon cancer. Oncotarget 2017, 8, 54775–54787. [Google Scholar] [CrossRef]
- Rosshart, S.P.; Herz, J.; Vassallo, B.G.; Hunter, A.; Wall, M.K.; Badger, J.H.; Mcculloch, J.A.; Anastasakis, D.G.; Sarshad, A.A.; Leonardi, I.; et al. Laboratory mice born to wild mice have natural microbiota and model human immune responses. Science 2019, 365, eaaw4361. [Google Scholar] [CrossRef]
- Arnold, J.W.; Roach, J.; Azcarate-Peril, M.A. Emerging Technologies for Gut Microbiome Research. Trends Microbiol. 2016, 24, 887–901. [Google Scholar] [CrossRef] [PubMed]
- Panek, M.; Čipčić Paljetak, H.; Barešić, A.; Perić, M.; Matijašić, M.; Lojkić, I.; Vranešić Bender, D.; Krznarić, Ž.; Verbanac, D. Methodology challenges in studying human gut microbiota—Effects of collection, storage, DNA extraction and next generation sequencing technologies. Sci. Rep. 2018, 8, 5143. [Google Scholar] [CrossRef]
- Rintala, A.; Pietilä, S.; Munukka, E.; Eerola, E.; Pursiheimo, J.-P.; Laiho, A.; Pekkala, S.; Huovinen, P. Gut Microbiota Analysis Results Are Highly Dependent on the 16S rRNA Gene Target Region, Whereas the Impact of DNA Extraction Is Minor. J. Biomol. Tech. 2017, 28, 19–30. [Google Scholar] [CrossRef]
- Ranjan, R.; Rani, A.; Metwally, A.; McGee, H.S.; Perkins, D.L. Analysis of the microbiome: Advantages of whole genome shotgun versus 16S amplicon sequencing. Biochem. Biophys. Res. Commun. 2016, 469, 967–977. [Google Scholar] [CrossRef] [PubMed]
- Laukens, D.; Brinkman, B.M.; Raes, J.; De Vos, M.; Vandenabeele, P. Heterogeneity of the gut microbiome in mice: Guidelines for optimizing experimental design. FEMS Microbiol. Rev. 2016, 40, 117–132. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-G.; Wu, G.-Z.; Wang, L.; Zhang, Y.-Y.; Li, Z.; Li, D.-C. Tumor cell lysate-pulsed dendritic cells induce a T cell response against colon cancer in vitro and in vivo. Med. Oncol. 2010, 27, 736–742. [Google Scholar] [CrossRef] [PubMed]
- Vo, M.-C.; Nguyen-Pham, T.-N.; Lee, H.-J.; Jaya Lakshmi, T.; Yang, S.; Jung, S.-H.; Kim, H.-J.; Lee, J.-J. Combination therapy with dendritic cells and lenalidomide is an effective approach to enhance antitumor immunity in a mouse colon cancer model. Oncotarget 2017, 8, 27252–27262. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Chow, K.-H.; Fleming, E.; Oh, J. Selective colonization ability of human fecal microbes in different mouse gut environments. ISME J. 2019, 13, 805–823. [Google Scholar] [CrossRef]
- Staley, C.; Kaiser, T.; Beura, L.K.; Hamilton, M.J.; Weingarden, A.R.; Bobr, A.; Kang, J.; Masopust, D.; Sadowsky, M.J.; Khoruts, A. Stable engraftment of human microbiota into mice with a single oral gavage following antibiotic conditioning. Microbiome 2017, 5, 87. [Google Scholar] [CrossRef] [PubMed]
- Bailoo, J.D.; Reichlin, T.S.; Wurbel, H. Refinement of Experimental Design and Conduct in Laboratory Animal Research. ILAR J. 2014, 55, 383–391. [Google Scholar] [CrossRef]
- Bodden, C.; von Kortzfleisch, V.T.; Karwinkel, F.; Kaiser, S.; Sachser, N.; Richter, S.H. Heterogenising study samples across testing time improves reproducibility of behavioural data. Sci. Rep. 2019, 9, 8247. [Google Scholar] [CrossRef] [PubMed]
- Voelkl, B.; Vogt, L.; Sena, E.S.; Würbel, H. Reproducibility of preclinical animal research improves with heterogeneity of study samples. PLoS Biol. 2018, 16, e2003693. [Google Scholar] [CrossRef] [PubMed]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biol. 2010, 8, e1000412. [Google Scholar] [CrossRef]


{kind=link}
| Tumor Induction | Mouse Model | Impact of Microbiota on Colon Phenotype | References |
|---|---|---|---|
| Sporadic Familial Adenomatous Polyposis | ApcMin/+ApcMinΔ716/+Cdx2-Cre Apcflox/+ | Mice administered continuous broad-spectrum antibiotics develop fewer colon tumors, whereas mice administered intermittent antibiotics develop more tumors. Infection of ApcMin/+ mice by Fusobacterium nucleatum, or ApcMin716/+ mice by enterotoxigenic Bacteroides fragilis and/or pks+ Escherichia coli increases tumor multiplicity. | [5,6,7,8,9] |
| Inflammation | Il10−/− | Germ-free mice do not develop intestinal inflammation. Differences in the composition of microbiota influence severity of sporadic colitis in mice housed at different institutions. Infection with E. coli increases tumorigenesis following azoxymethane (AOM) treatment. | [10,11,12,13] |
| DNA mismatch repair deficiency | Msh2−/− | Germ-free and antibiotic (broad-spectrum)-treated ApcMin/+ Msh2−/− mice develop fewer colon tumors than ‘conventional’ untreated mice (bearing natural microbiota). | [14] |
| Chemical induction | AOM/DSS | Treatment with either AOM or dextran sodium sulfate (DSS) changes the composition of the gut microbiota. Germ-free mice exhibit delayed tissue repair and develop more tumors than conventional mice. Conventional C57BL/6 mice develop more tumors than the genetically identical mice colonized with microbiota from wild-caught mice. | [15,16,17] |
| Transplantation | CT26 MC38 | E. coli modifies the response of tumors to chemotherapy. Depletion of microbiota by broad-spectrum antibiotics attenuates the response of tumors to immunotherapeutics. | [18,19,20] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leystra, A.A.; Clapper, M.L. Gut Microbiota Influences Experimental Outcomes in Mouse Models of Colorectal Cancer. Genes 2019, 10, 900. https://doi.org/10.3390/genes10110900
Leystra AA, Clapper ML. Gut Microbiota Influences Experimental Outcomes in Mouse Models of Colorectal Cancer. Genes. 2019; 10(11):900. https://doi.org/10.3390/genes10110900
Chicago/Turabian StyleLeystra, Alyssa A., and Margie L. Clapper. 2019. "Gut Microbiota Influences Experimental Outcomes in Mouse Models of Colorectal Cancer" Genes 10, no. 11: 900. https://doi.org/10.3390/genes10110900
APA StyleLeystra, A. A., & Clapper, M. L. (2019). Gut Microbiota Influences Experimental Outcomes in Mouse Models of Colorectal Cancer. Genes, 10(11), 900. https://doi.org/10.3390/genes10110900