Development of Tissue-Specific Age Predictors Using DNA Methylation Data


Abstract
1. Introduction
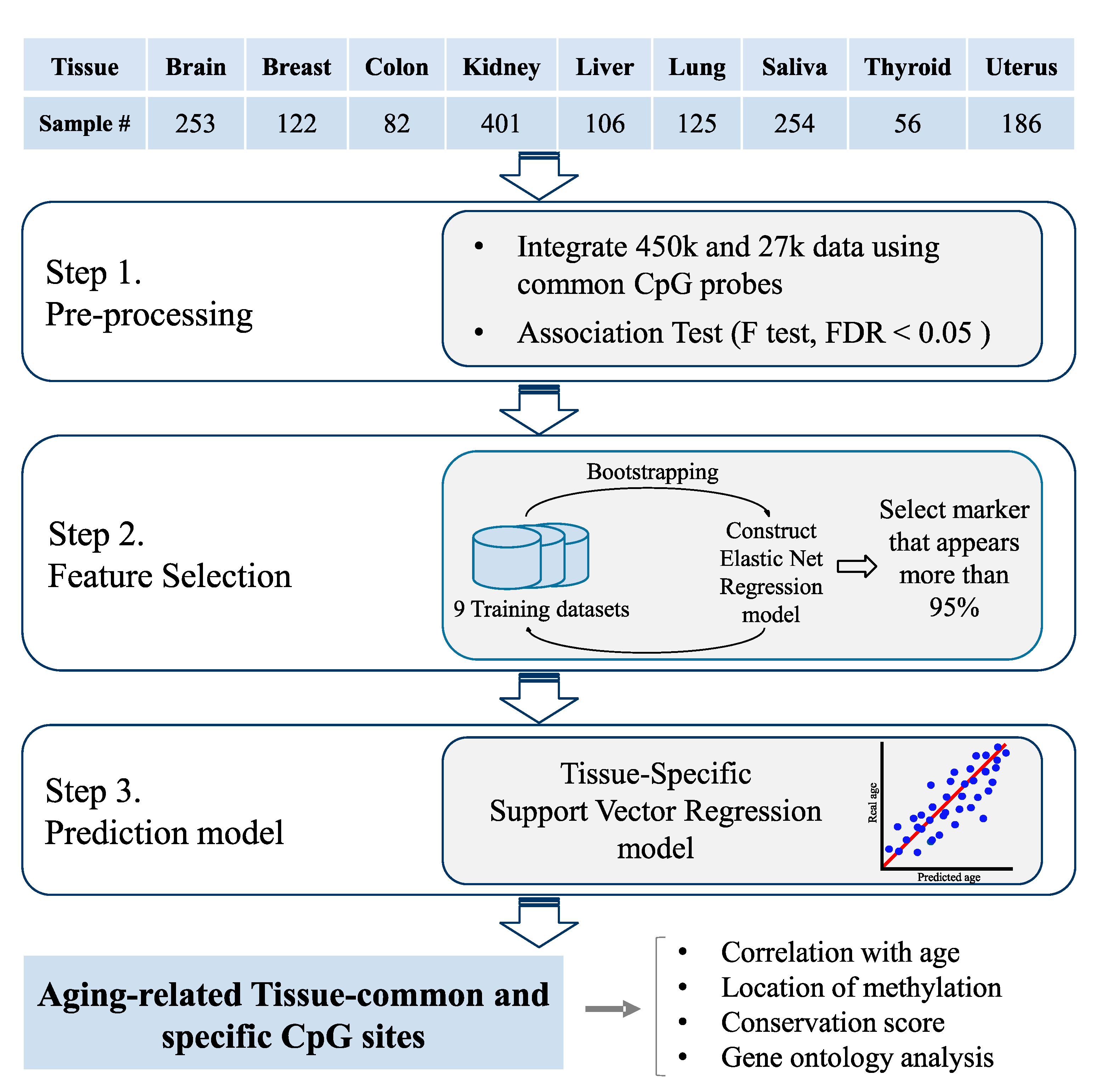
2. Materials and Methods
2.1. Tissue-Specific Methylation Datasets
2.2. Association Test
2.3. Aging Model Construction
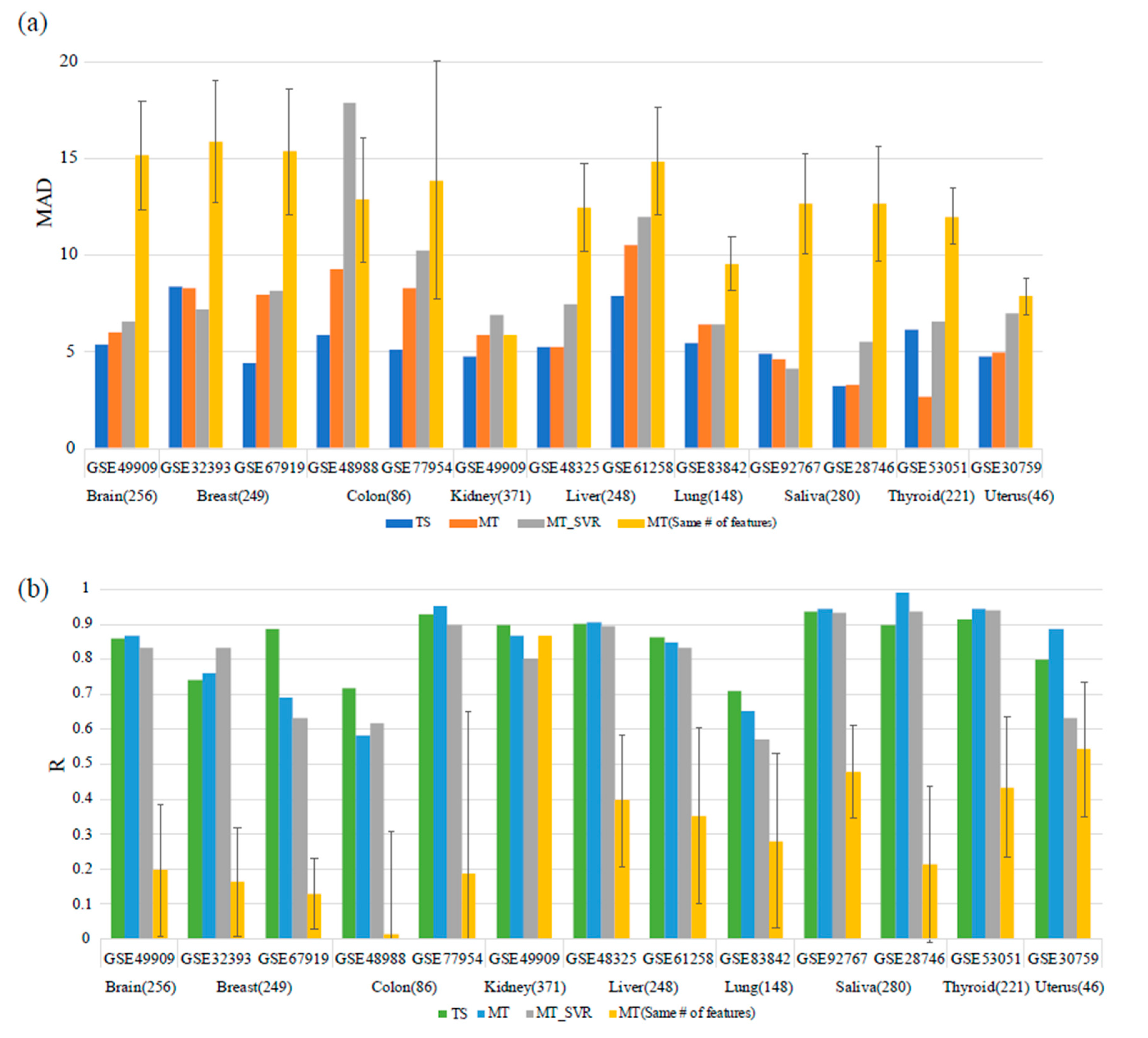
2.4. Performance Comparison with Multi-Tissue Predictors
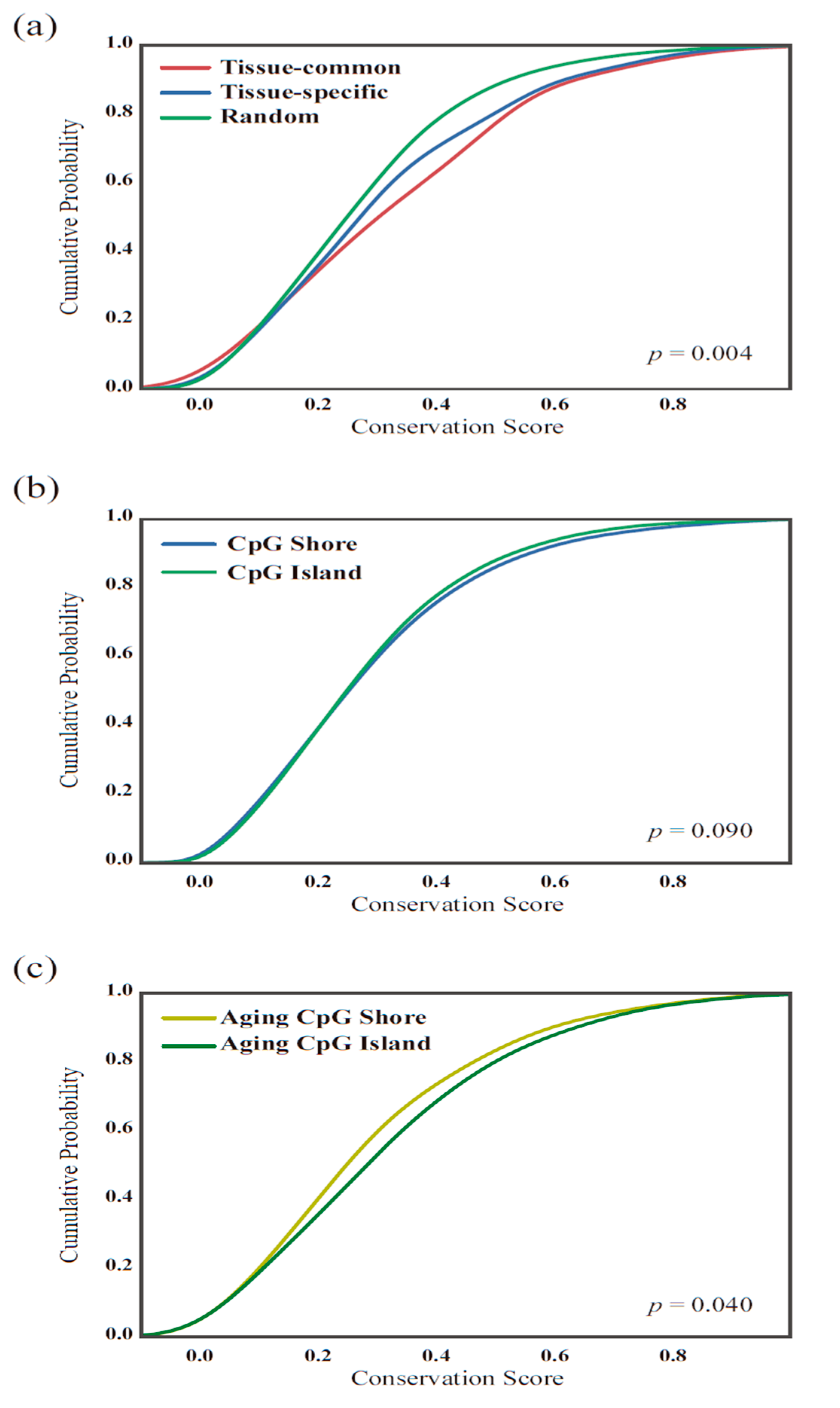
2.5. Conservation Score Calculation
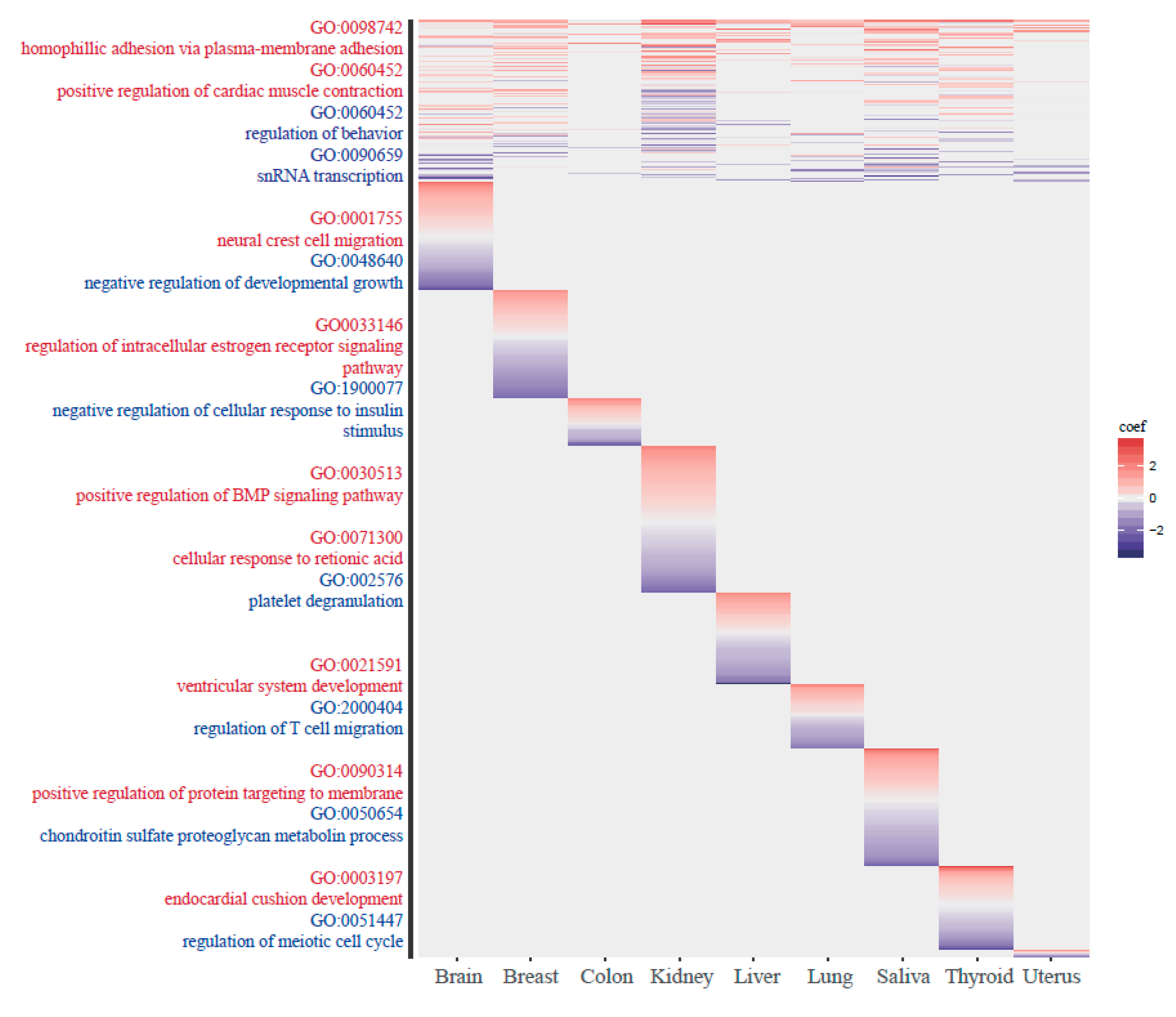
2.6. GO Analysis
3. Results
3.1. Tissue-Specific Age Prediction Model
3.2. Comparison with the Multi-Tissue Age Predictor
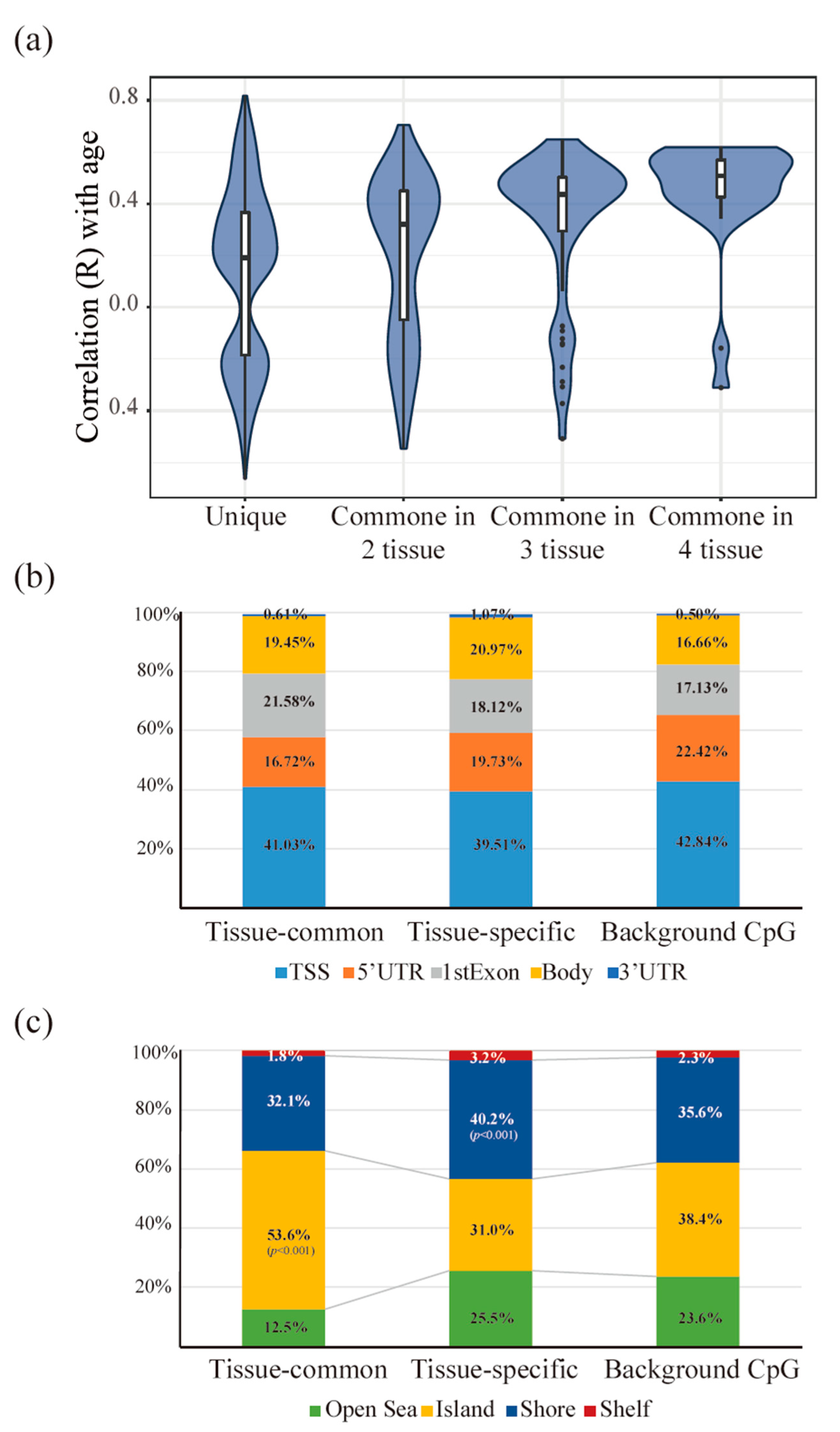
3.3. Tissue-Common and Tissue-Specific Features of Age-Dependent Methylation in Tissues
3.4. The Functionality of Tissue-Common and Specific Methylation Regions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- West, M.D.; Sternberg, H.; Labat, I.; Janus, J.; Chapman, K.B.; Malik, N.N.; de Grey, A.D.; Larocca, D. Toward a unified theory of aging and regeneration. Regen. Med. 2019, 14, 867–886. [Google Scholar] [CrossRef]
- Baker, G.T., 3rd; Sprott, R.L. Biomarkers of aging. Exp. Gerontol. 1988, 23, 223–239. [Google Scholar] [CrossRef]
- Hudgins, A.D.; Tazearslan, C.; Tare, A.; Zhu, Y.; Huffman, D.; Suh, Y. Age-and tissue-specific expression of senescence biomarkers in mice. Front. Genet. 2018, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Peters, M.J.; Joehanes, R.; Pilling, L.C.; Schurmann, C.; Conneely, K.N.; Powell, J.; Reinmaa, E.; Sutphin, G.L.; Zhernakova, A.; Schramm, K.; et al. The transcriptional landscape of age in human peripheral blood. Nat. Commun. 2015, 6, 8570. [Google Scholar] [CrossRef] [PubMed]
- De Magalhaes, J.P.; Curado, J.; Church, G.M. Meta-analysis of age-related gene expression profiles identifies common signatures of aging. Bioinformatics 2009, 25, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345, 458–460. [Google Scholar] [CrossRef]
- Heidinger, B.J.; Blount, J.D.; Boner, W.; Griffiths, K.; Metcalfe, N.B.; Monaghan, P. Telomere length in early life predicts lifespan. Proc. Natl. Acad. Sci. USA 2012, 109, 1743–1748. [Google Scholar] [CrossRef]
- Blackburn, E.H.; Gall, J.G. A tandemly repeated sequence at the termini of the extrachromosomal ribosomal RNA genes in Tetrahymena. J. Mol. Biol. 1978, 120, 33–53. [Google Scholar] [CrossRef]
- Jung, M.; Pfeifer, G.P. Aging and DNA methylation. BMC Biol. 2015, 13, 7. [Google Scholar] [CrossRef]
- Teschendorff, A.E.; Menon, U.; Gentry-Maharaj, A.; Ramus, S.J.; Weisenberger, D.J.; Shen, H.; Campan, M.; Noushmehr, H.; Bell, C.G.; Maxwell, A.P.; et al. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010, 20, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Zhang, Y.; Langfelder, P.; Kahn, R.S.; Boks, M.P.; van Eijk, K.; van den Berg, L.H.; Ophoff, R.A. Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biol. 2012, 13, R97. [Google Scholar] [CrossRef] [PubMed]
- Bocklandt, S.; Lin, W.; Sehl, M.E.; Sanchez, F.J.; Sinsheimer, J.S.; Horvath, S.; Vilain, E. Epigenetic predictor of age. PLoS ONE 2011, 6, e14821. [Google Scholar] [CrossRef] [PubMed]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.B.; Gao, Y.; et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Weidner, C.I.; Lin, Q.; Koch, C.M.; Eisele, L.; Beier, F.; Ziegler, P.; Bauerschlag, D.O.; Jockel, K.H.; Erbel, R.; Muhleisen, T.W.; et al. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 2014, 15, R24. [Google Scholar] [CrossRef]
- Marioni, R.E.; Shah, S.; McRae, A.F.; Chen, B.H.; Colicino, E.; Harris, S.E.; Gibson, J.; Henders, A.K.; Redmond, P.; Cox, S.R.; et al. DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol. 2015, 16, 25. [Google Scholar] [CrossRef]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef]
- Horvath, S.; Pirazzini, C.; Bacalini, M.G.; Gentilini, D.; Di Blasio, A.M.; Delledonne, M.; Mari, D.; Arosio, B.; Monti, D.; Passarino, G.; et al. Decreased epigenetic age of PBMCs from Italian semi-supercentenarians and their offspring. Aging 2015, 7, 1159–1170. [Google Scholar] [CrossRef]
- Chen, B.H.; Marioni, R.E.; Colicino, E.; Peters, M.J.; Ward-Caviness, C.K.; Tsai, P.C.; Roetker, N.S.; Just, A.C.; Demerath, E.W.; Guan, W.; et al. DNA methylation-based measures of biological age: Meta-analysis predicting time to death. Aging 2016, 8, 1844–1865. [Google Scholar] [CrossRef]
- Li, X.; Li, W.; Xu, Y. Human age prediction based on DNA methylation using a gradient boosting regressor. Genes (Basel) 2018, 9, 424. [Google Scholar] [CrossRef]
- Jung, S.E.; Shin, K.J.; Lee, H.Y. DNA methylation-based age prediction from various tissues and body fluids. BMB Rep. 2017, 50, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Maegawa, S.; Hinkal, G.; Kim, H.S.; Shen, L.; Zhang, L.; Zhang, J.; Zhang, N.; Liang, S.; Donehower, L.A.; Issa, J.P. Widespread and tissue specific age-related DNA methylation changes in mice. Genome Res. 2010, 20, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Day, K.; Waite, L.L.; Thalacker-Mercer, A.; West, A.; Bamman, M.M.; Brooks, J.D.; Myers, R.M.; Absher, D. Differential DNA methylation with age displays both common and dynamic features across human tissues that are influenced by CpG landscape. Genome Biol. 2013, 14, R102. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, J.R.; van der Brug, M.P.; Hernandez, D.G.; Traynor, B.J.; Nalls, M.A.; Lai, S.L.; Arepalli, S.; Dillman, A.; Rafferty, I.P.; Troncoso, J.; et al. Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genet. 2010, 6, e1000952. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Wang, S.; Zhang, Y.J.; Kappil, M.; Wu, H.C.; Kibriya, M.G.; Wang, Q.; Jasmine, F.; Ahsan, H.; Lee, P.H.; et al. Genome-wide DNA methylation profiles in hepatocellular carcinoma. Hepatology 2012, 55, 1799–1808. [Google Scholar] [CrossRef]
- Gopalan, S.; Carja, O.; Fagny, M.; Patin, E.; Myrick, J.W.; McEwen, L.M.; Mah, S.M.; Kobor, M.S.; Froment, A.; Feldman, M.W.; et al. Trends in DNA methylation with age replicate across diverse human populations. Genetics 2017, 206, 1659–1674. [Google Scholar] [CrossRef]
- Liu, J.; Morgan, M.; Hutchison, K.; Calhoun, V.D. A study of the influence of sex on genome wide methylation. PLoS ONE 2010, 5, e10028. [Google Scholar] [CrossRef]
- Zhuang, J.; Jones, A.; Lee, S.H.; Ng, E.; Fiegl, H.; Zikan, M.; Cibula, D.; Sargent, A.; Salvesen, H.B.; Jacobs, I.J.; et al. The dynamics and prognostic potential of DNA methylation changes at stem cell gene loci in women’s cancer. PLoS Genet. 2012, 8, e1002517. [Google Scholar] [CrossRef]
- Hair, B.Y.; Xu, Z.; Kirk, E.L.; Harlid, S.; Sandhu, R.; Robinson, W.R.; Wu, M.C.; Olshan, A.F.; Conway, K.; Taylor, J.A.; et al. Body mass index associated with genome-wide methylation in breast tissue. Breast Cancer Res. Treat. 2015, 151, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Sandmann, T.; Frierson, H.; Fu, L.; Fuentes, E.; Walter, K.; Okrah, K.; Rumpel, C.; Moskaluk, C.; Lu, S.; et al. Integrated genomic analysis of colorectal cancer progression reveals activation of EGFR through demethylation of the EREG promoter. Oncogene 2016, 35, 6403–6415. [Google Scholar] [CrossRef] [PubMed]
- Noreen, F.; Roosli, M.; Gaj, P.; Pietrzak, J.; Weis, S.; Urfer, P.; Regula, J.; Schar, P.; Truninger, K. Modulation of age- and cancer-associated DNA methylation change in the healthy colon by aspirin and lifestyle. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Erhart, W.; Brosch, M.; Ammerpohl, O.; von Schonfels, W.; Ahrens, M.; Heits, N.; Bell, J.T.; Tsai, P.C.; Spector, T.D.; et al. Obesity accelerates epigenetic aging of human liver. Proc. Natl. Acad. Sci. USA 2014, 111, 15538–15543. [Google Scholar] [CrossRef] [PubMed]
- Ahrens, M.; Ammerpohl, O.; von Schonfels, W.; Kolarova, J.; Bens, S.; Itzel, T.; Teufel, A.; Herrmann, A.; Brosch, M.; Hinrichsen, H.; et al. DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery. Cell Metab. 2013, 18, 296–302. [Google Scholar] [CrossRef]
- Kajiura, K.; Masuda, K.; Naruto, T.; Kohmoto, T.; Watabnabe, M.; Tsuboi, M.; Takizawa, H.; Kondo, K.; Tangoku, A.; Imoto, I. Frequent silencing of the candidate tumor suppressor TRIM58 by promoter methylation in early-stage lung adenocarcinoma. Oncotarget 2017, 8, 2890–2905. [Google Scholar] [CrossRef]
- Hong, S.R.; Jung, S.E.; Lee, E.H.; Shin, K.J.; Yang, W.I.; Lee, H.Y. DNA methylation-based age prediction from saliva: High age predictability by combination of 7 CpG markers. Forensic Sci. Int. Genet. 2017, 29, 118–125. [Google Scholar] [CrossRef]
- Timp, W.; Bravo, H.C.; McDonald, O.G.; Goggins, M.; Umbricht, C.; Zeiger, M.; Feinberg, A.P.; Irizarry, R.A. Large hypomethylated blocks as a universal defining epigenetic alteration in human solid tumors. Genome Med. 2014, 6, 61. [Google Scholar] [CrossRef]
- Weisenberger, D.; Van Den Berg, D.; Pan, F.; Berman, B.; Laird, P.J.I. Comprehensive DNA Methylation Analysis on the Illumina Infinium Assay Platform; Illumina: San Diego, CA, USA, 2008. [Google Scholar]
- Illumina. Infinium HumanMethylation450 BeadChip. Available online: https://support.illumina.com/array/array_kits/infinium_humanmethylation450_beadchip_kit/documentation.html (accessed on 4 August 2018).
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Benjamini, Y.; Hochberg, Y.J. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Lomax, R.G.; Hahs-Vaughn, D.L. Statistical Concepts: A Second Course; Routledge: New York, NY, USA, 2013. [Google Scholar]
- Zou, H.; Hastie, T. Regularization and variable selection via the elastic net. J. R. Stat. Soc. Ser. B (Stat. Methodol.) 2005, 67, 768. [Google Scholar] [CrossRef]
- Cortes, C.; Vapnik, V.J. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Geisser, S. Predictive Inference; Routledge: London, UK, 2017. [Google Scholar]
- Kohavi, R. A study of cross-validation and bootstrap for accuracy estimation and model selection. Proceedings of of the 14th International Joint Conference on Artificial Intelligence, Montreal, QC, Canada, 20–25 August 1995; pp. 1137–1145. [Google Scholar]
- Devijver, P.A.; Kittler, J. Pattern Recognition: A statistical Approach; Prentice Hall: Upper Saddle River, NJ, USA, 1982. [Google Scholar]
- Tofallis, C. A better measure of relative prediction accuracy for model selection and model estimation. J. Operat. Res. Soc. 2015, 66, 1352–1362. [Google Scholar] [CrossRef]
- Theil, H. Economic Forecasts and Policy; Cambridge University Press: Cambridge, UK, 1958. [Google Scholar]
- Margulies, E.H.; Blanchette, M.; Haussler, D.; Green, E.D.; Progra, N.C.S. Identification and characterization of multi-species conserved sequences. Genome Res. 2003, 13, 2507–2518. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pages, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef]
- Xu, C.; Qu, H.; Wang, G.; Xie, B.; Shi, Y.; Yang, Y.; Zhao, Z.; Hu, L.; Fang, X.; Yan, J.; et al. A novel strategy for forensic age prediction by DNA methylation and support vector regression model. Sci. Rep. 2015, 5, 17788. [Google Scholar] [CrossRef]
- Jones, M.J.; Goodman, S.J.; Kobor, M.S. DNA methylation and healthy human aging. Aging Cell 2015, 14, 924–932. [Google Scholar] [CrossRef]
- Ishimoto, Y.; Tomiyama-Miyaji, C.; Watanabe, H.; Yokoyama, H.; Ebe, K.; Tsubata, S.; Aoyagi, Y.; Abo, T. Age-dependent variation in the proportion and number of intestinal lymphocyte subsets, especially natural killer T cells, double-positive CD4+ CD8+ cells and B220+ T cells, in mice. Immunology 2004, 113, 371–377. [Google Scholar] [CrossRef]
- Horvath, S.; Mah, V.; Lu, A.T.; Woo, J.S.; Choi, O.W.; Jasinska, A.J.; Riancho, J.A.; Tung, S.; Coles, N.S.; Braun, J.; et al. The cerebellum ages slowly according to the epigenetic clock. Aging-Us 2015, 7, 294–306. [Google Scholar] [CrossRef]
- Sehl, M.E.; Henry, J.E.; Storniolo, A.M.; Ganz, P.A.; Horvath, S. DNA methylation age is elevated in breast tissue of healthy women. Breast Cancer Res. Treat. 2017, 164, 209–219. [Google Scholar] [CrossRef]
- Lee, J.-H.; Park, S.-J.; Nakai, K.J. Differential landscape of non-CpG methylation in embryonic stem cells and neurons caused by DNMT3s. Sci. Rep. 2017, 7, 11295. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Wyckoff, J.; Morris, A.T.; Succop, A.; Avery, A.; Duncan, G.E.; Jazwinski, S.M. DNA methylation associated with healthy aging of elderly twins. Geroscience 2018, 40, 469–484. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.H.; Ritchie, S.J.; Bastin, M.E.; Valdes Hernandez, M.C.; Munoz Maniega, S.; Royle, N.; Corley, J.; Pattie, A.; Harris, S.E.; Zhang, Q.; et al. Brain age predicts mortality. Mol. Psychiatry 2018, 23, 1385–1392. [Google Scholar] [CrossRef]
- Steiner, L.; Hopp, L.; Wirth, H.; Galle, J.; Binder, H.; Prohaska, S.J.; Rohlf, T. A global genome segmentation method for exploration of epigenetic patterns. PLoS ONE 2012, 7, e46811. [Google Scholar] [CrossRef]
- Mamoshina, P.; Kochetov, K.; Putin, E.; Cortese, F.; Aliper, A.; Lee, W.S.; Ahn, S.M.; Uhn, L.; Skjodt, N.; Kovalchuk, O.; et al. Population Specific Biomarkers of Human Aging: A Big Data Study Using South Korean, Canadian, and Eastern European Patient Populations. J. Gerontol. A Biol. Sci. Med. Sci. 2018, 73, 1482–1490. [Google Scholar] [CrossRef] [PubMed]
- Pyrkov, T.V.; Slipensky, K.; Barg, M.; Kondrashin, A.; Zhurov, B.; Zenin, A.; Pyatnitskiy, M.; Menshikov, L.; Markov, S.; Fedichev, P.O. Extracting biological age from biomedical data via deep learning: Too much of a good thing? Sci. Rep. 2018, 8, 5210. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Set | Tissue Type | The Number of Patients | Age Range | Platform | |
|---|---|---|---|---|---|
| GSE15745 [26] | Brain | 253 | 16–95 | HumanMethylation 27 K | |
| TCGA [24] | Breast | 95 | 122 | 28–90 | HumanMethylation 450 K |
| 27 | 35–88 | HumanMethylation 27 K | |||
| TCGA [24] | Colon | 45 | 82 | 40–90 | HumanMethylation 450 K |
| 37 | 43–90 | HumanMethylation 27 K | |||
| TCGA [24] | Kidney | 205 | 401 | 31–90 | HumanMethylation 450 K |
| 196 | 33–86 | HumanMethylation 27 K | |||
| TCGA [24] | Liver | 49 | 106 | 20–81 | HumanMethylation 450 K |
| GSE37988 [27] | 57 | 20–79 | HumanMethylation 27 K | ||
| TCGA [24] | Lung | 74 | 125 | 40–86 | HumanMethylation 450 K |
| 51 | 51–83 | HumanMethylation 27 K | |||
| GSE99029 [28] | Saliva | 57 | 254 | 21–91 | HumanMethylation 27 K |
| GSE34035 [29] | 197 | 21–55 | HumanMethylation 27 K | ||
| TCGA [24] | Thyroid | 56 | 56 | 15–81 | HumanMethylation 450 K |
| TCGA [24] | Uterus | 34 | 186 | 36–90 | HumanMethylation 450 K |
| GSE30758 [30] | 152 | 19–55 | HumanMethylation 27 K | ||
| Brain | Breast | Colon | Kidney | Liver | Lung | Saliva | Thyroid | Uterus | Total | |
|---|---|---|---|---|---|---|---|---|---|---|
| Total | 256 | 249 | 86 | 371 | 248 | 148 | 280 | 221 | 46 | 1460 |
| Common | 93 | 92 | 17 | 153 | 53 | 49 | 103 | 99 | 35 | 247 |
| Specific | 163 | 157 | 69 | 218 | 195 | 99 | 177 | 122 | 11 | 1213 |
| Positive | 170 | 202 | 54 | 281 | 186 | 67 | 232 | 151 | 35 | 989 |
| Negative | 86 | 47 | 32 | 90 | 62 | 81 | 48 | 70 | 11 | 471 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, H.; Joe, S.; Nam, H. Development of Tissue-Specific Age Predictors Using DNA Methylation Data. Genes 2019, 10, 888. https://doi.org/10.3390/genes10110888
Choi H, Joe S, Nam H. Development of Tissue-Specific Age Predictors Using DNA Methylation Data. Genes. 2019; 10(11):888. https://doi.org/10.3390/genes10110888
Chicago/Turabian StyleChoi, Heeyeon, Soobok Joe, and Hojung Nam. 2019. "Development of Tissue-Specific Age Predictors Using DNA Methylation Data" Genes 10, no. 11: 888. https://doi.org/10.3390/genes10110888
APA StyleChoi, H., Joe, S., & Nam, H. (2019). Development of Tissue-Specific Age Predictors Using DNA Methylation Data. Genes, 10(11), 888. https://doi.org/10.3390/genes10110888