Type II DNA Topoisomerases Cause Spontaneous Double-Strand Breaks in Genomic DNA


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
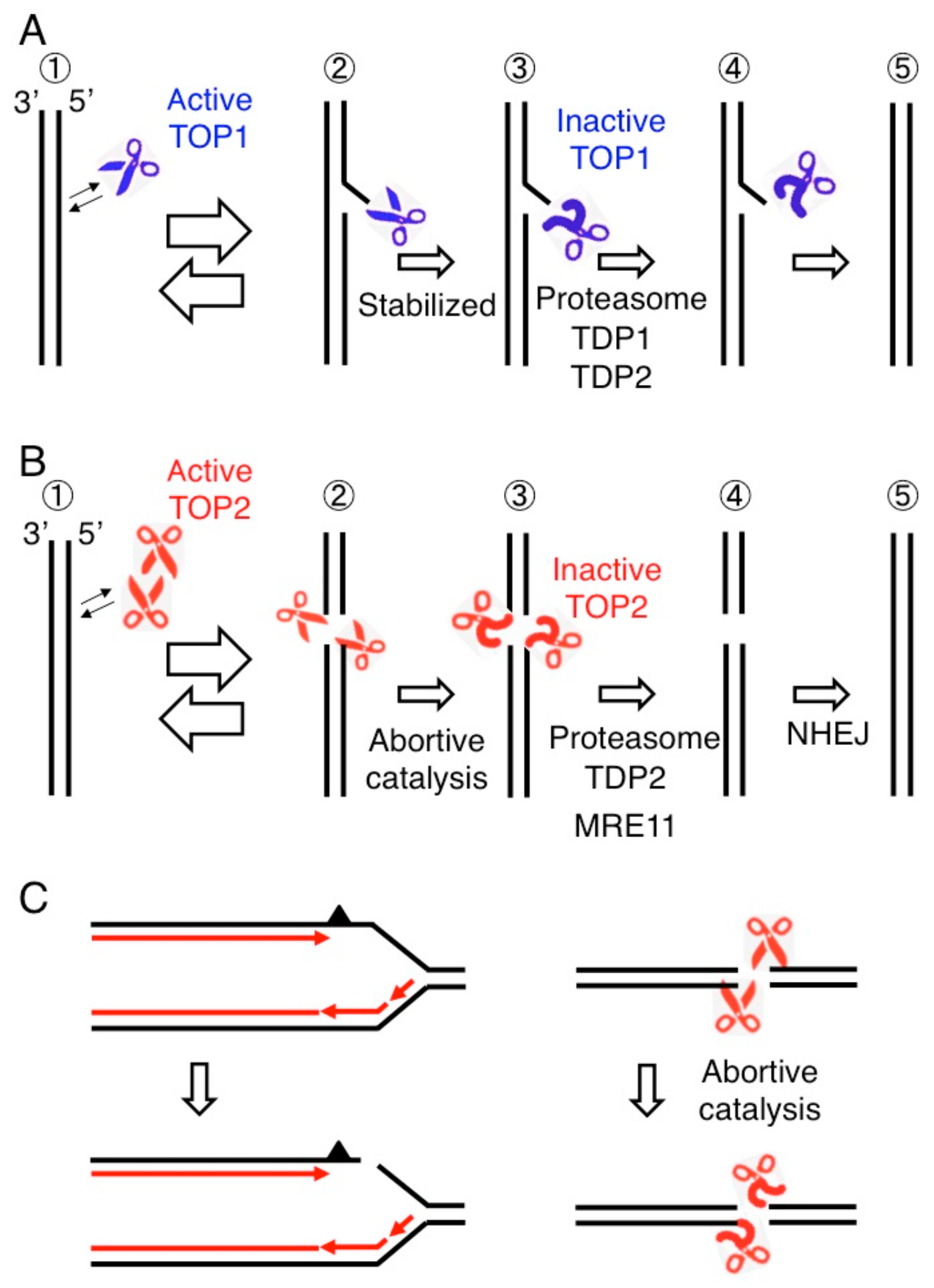
2. The Consequences of Failure of Catalysis by DNA Topoisomerase I and II
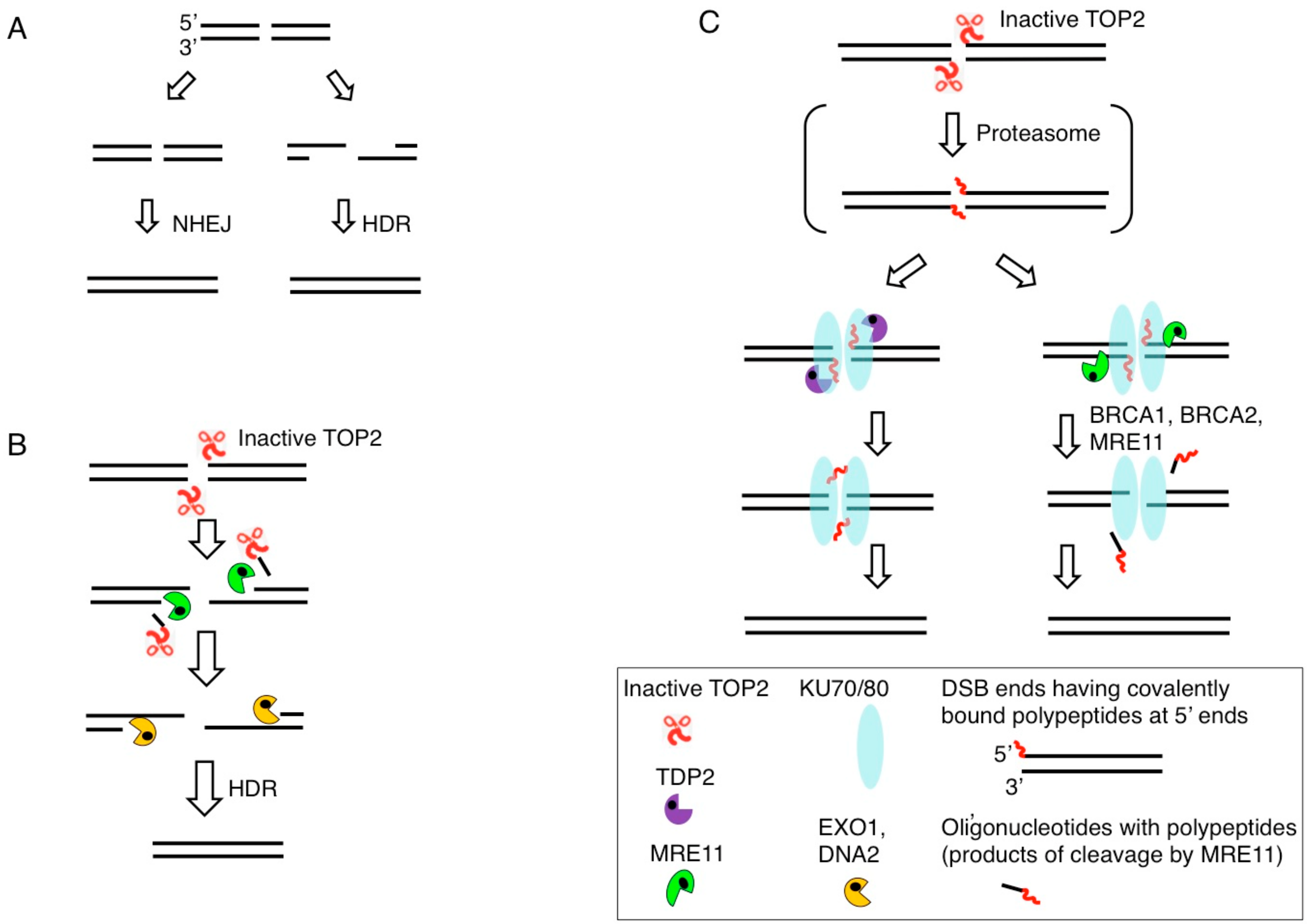
3. Removal of 5′ TOP2 Adducts to Restore Ligatable DNA Ends is Necessary for DSB Repair by NHEJ
4. MRE11 Contributes to DSB Repair through Two Distinct Mechanisms: The Removal of Stalled TOP2ccs and DSB Resection in HDR
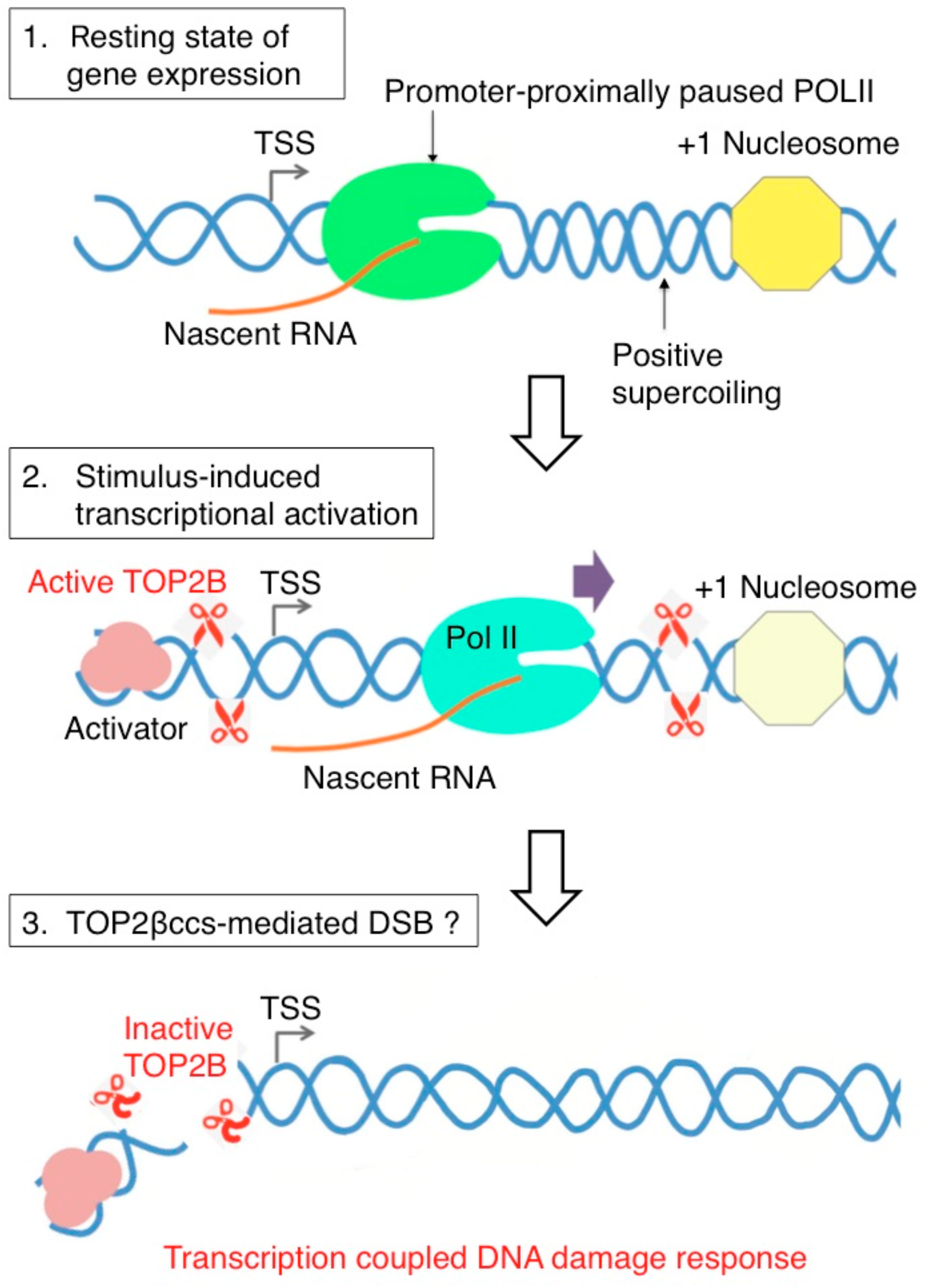
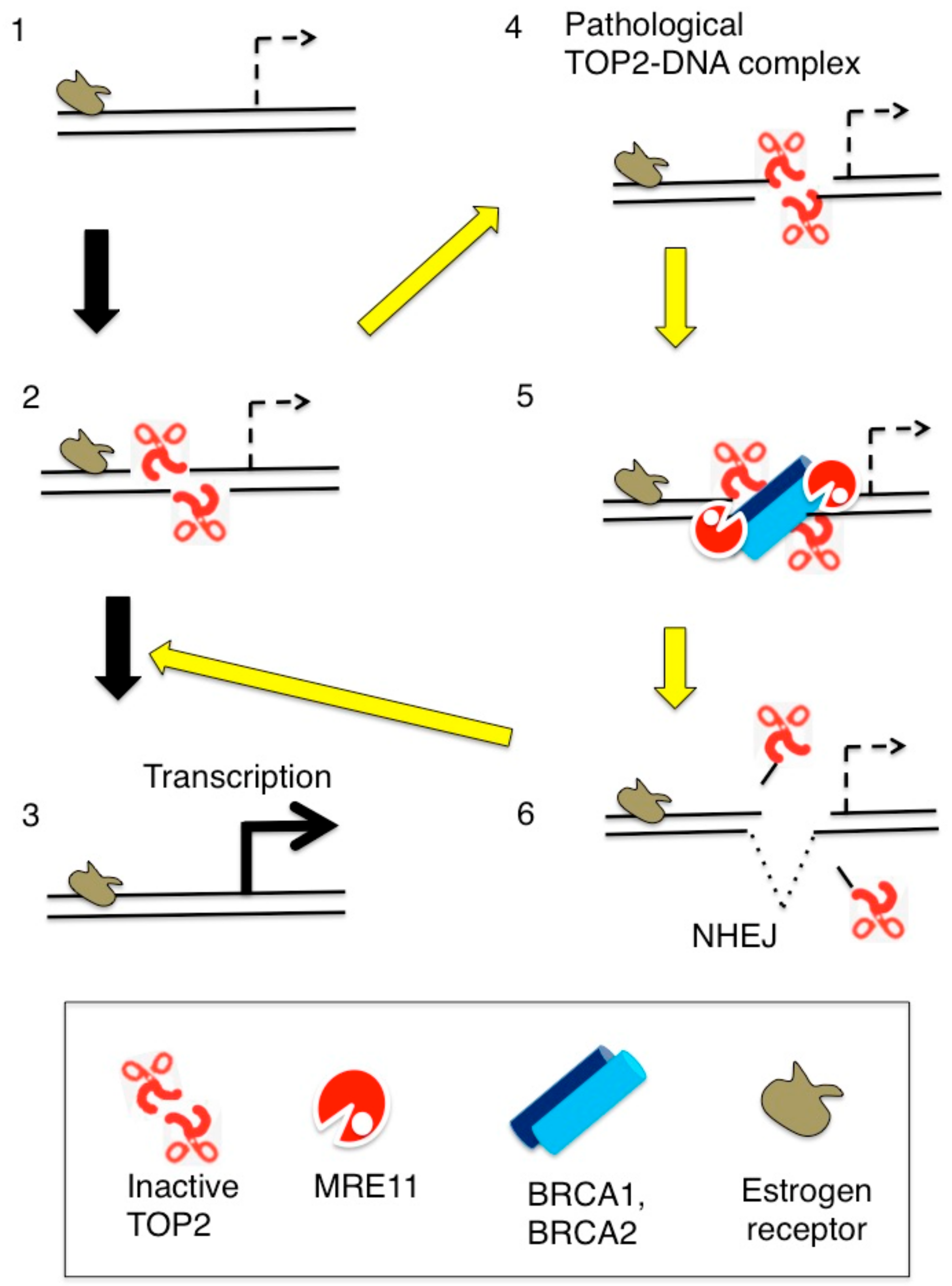
5. Abortive TOP2B Catalysis Occurs during Early Transcriptional Responses to Estrogens
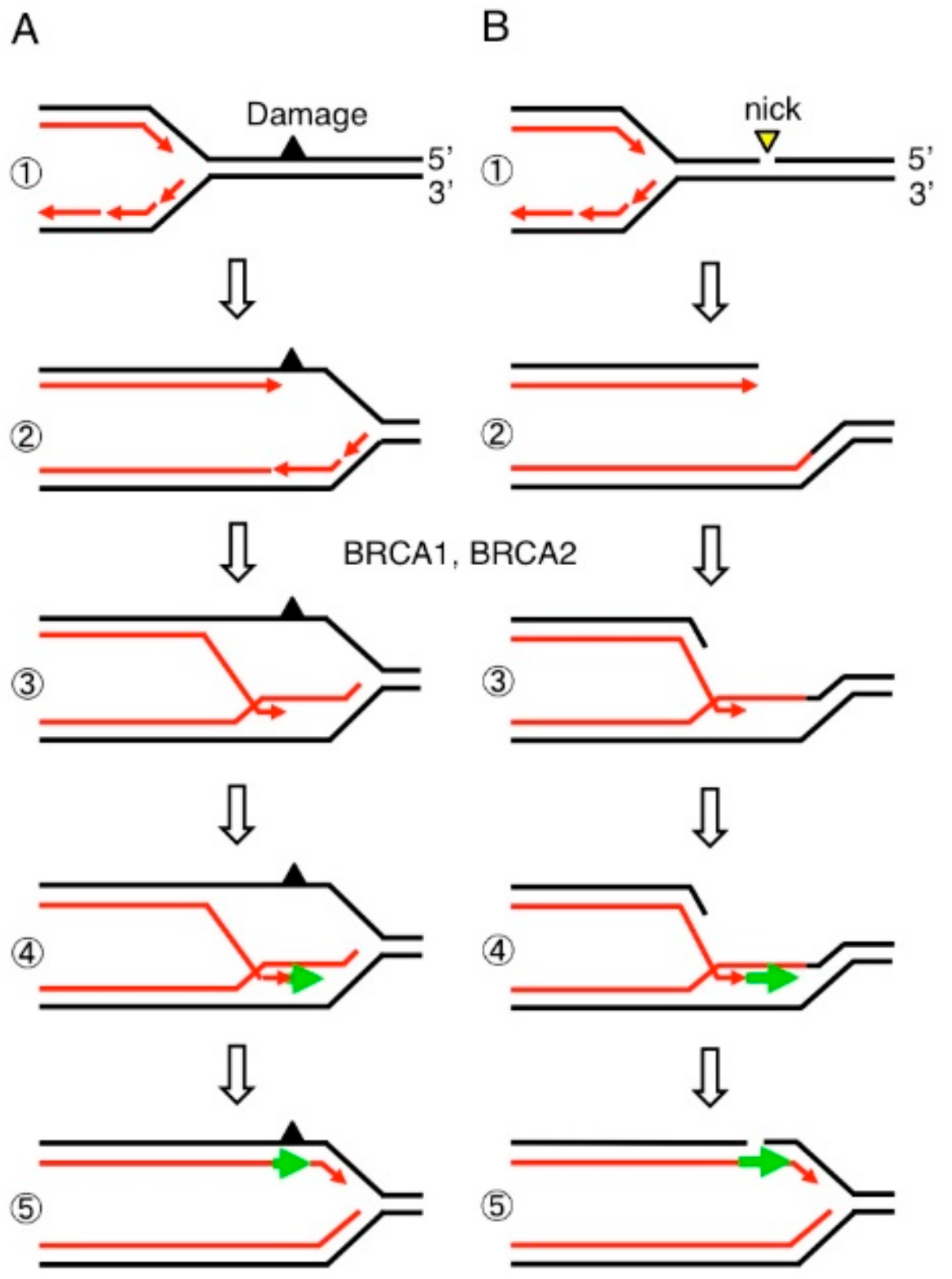
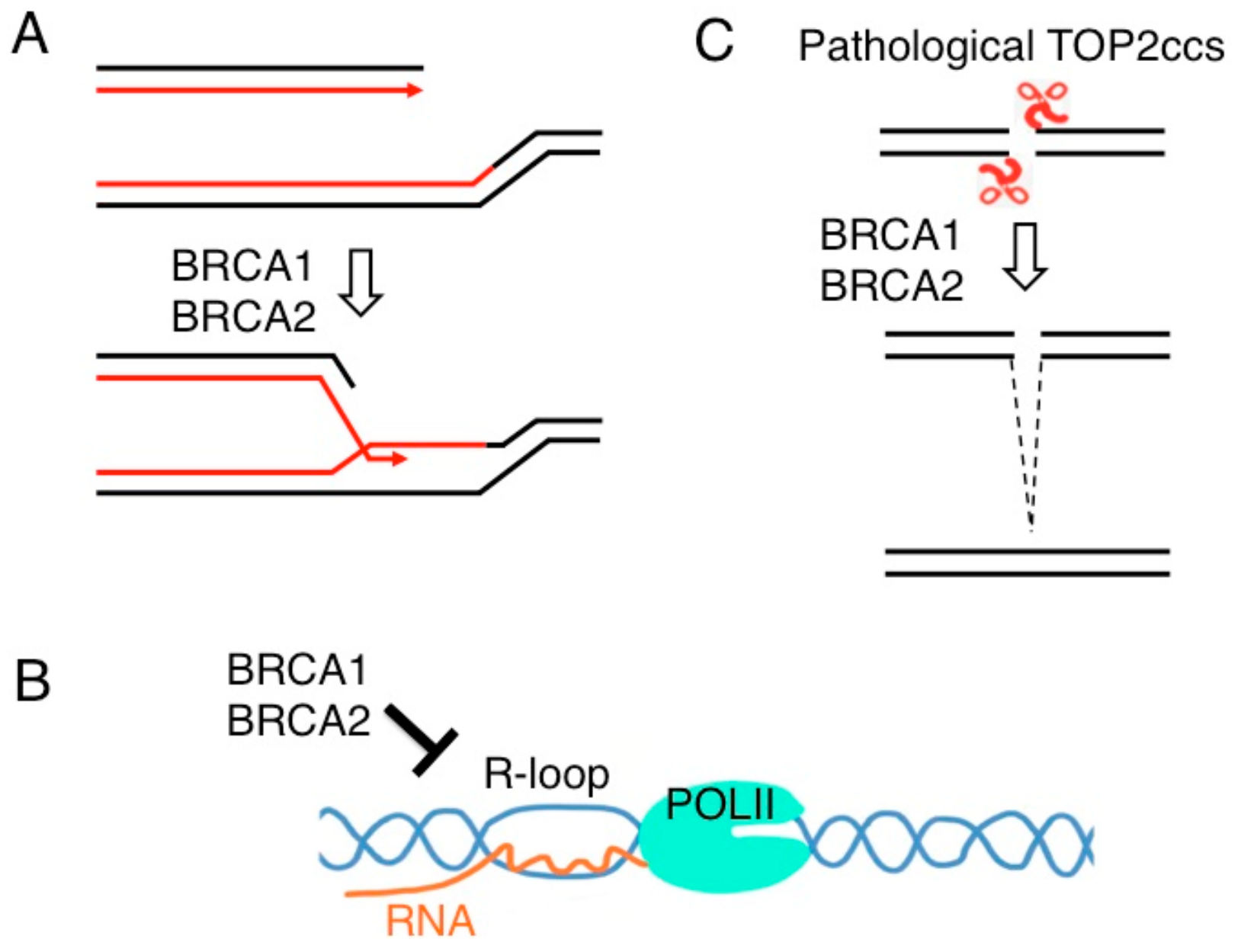
6. BRCA1 and BRCA2 Promote MRE11-Mediated Removal of 5′ TOP2 Adducts from Pathological TOP2ccs
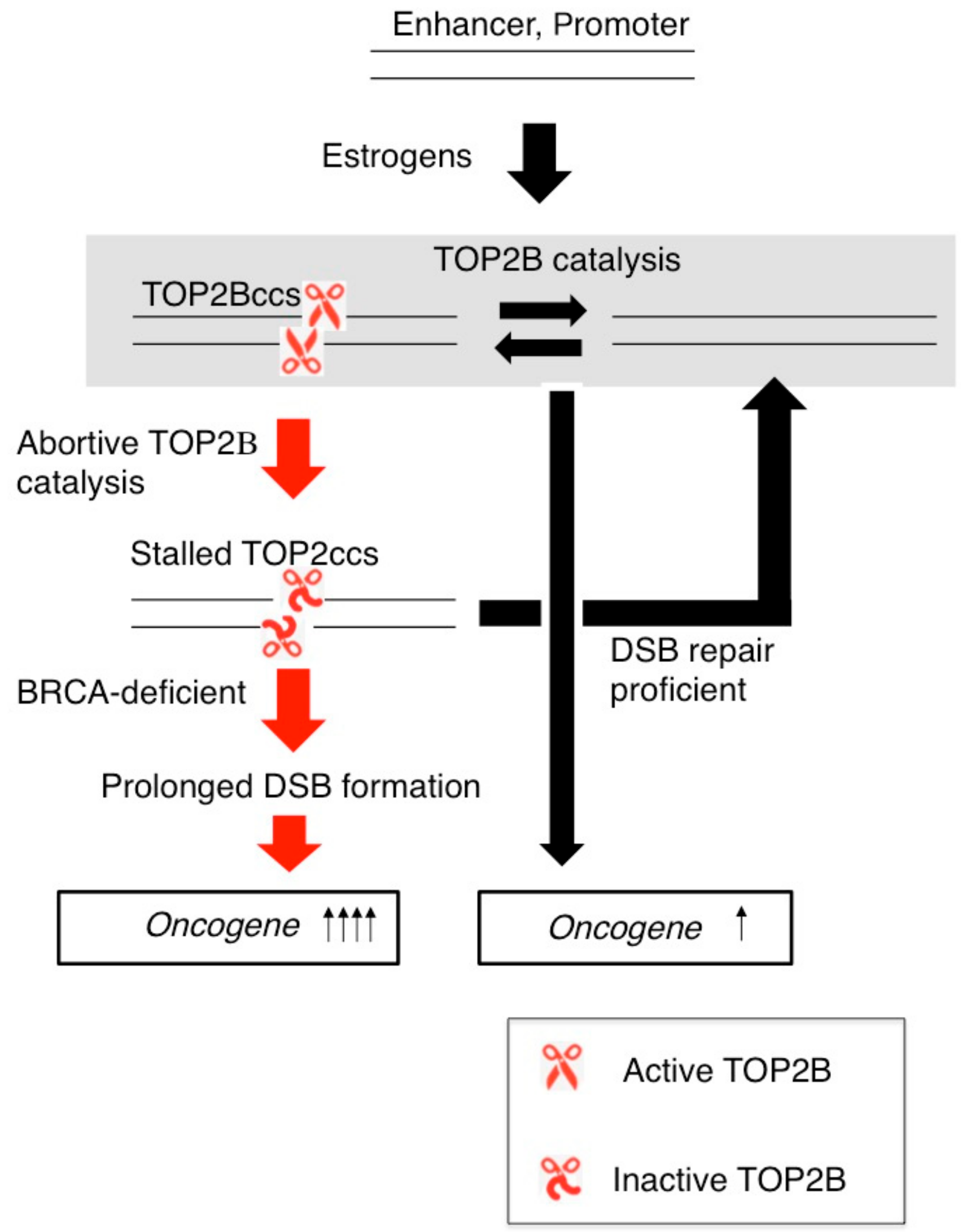
7. Estrogens Cause Accumulation of Stalled TOP2ccs in BRCA1-Deficient Mammary Epithelial Cells
8. Conclusions and Unsolved Questions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bunting, S.F.; Nussenzweig, A. End-joining, translocations and cancer. Nat. Rev. Cancer 2013, 13, 443–454. [Google Scholar] [CrossRef]
- Mehta, A.; Haber, J.E. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb. Perspect. Biol. 2014, 6, a016428. [Google Scholar] [CrossRef]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef]
- Oh, J.; Symington, L.S. Role of the Mre11 Complex in Preserving Genome Integrity. Genes 2018, 9, 589. [Google Scholar] [CrossRef]
- Chen, C.-C.; Feng, W.; Lim, P.X.; Kass, E.M.; Jasin, M. Homology-Directed Repair and the Role of BRCA1, BRCA2, and Related Proteins in Genome Integrity and Cancer. Annu. Rev. Cancer Biol. 2018, 2, 313–336. [Google Scholar] [CrossRef] [PubMed]
- Venkitaraman, A.R. How do mutations affecting the breast cancer genes BRCA1 and BRCA2 cause cancer susceptibility? DNA Repair 2019, 81, 102668. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Sun, Y.; Huang, S.N.; Nitiss, J.L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol. 2016, 17, 703–721. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-Y.N.; Williams, J.S.; Arana, M.E.; Kunkel, T.A.; Pommier, Y. Topoisomerase I-mediated cleavage at unrepaired ribonucleotides generates DNA double-strand breaks. EMBO J. 2017, 36, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.S.; Clausen, A.R.; Lujan, S.A.; Marjavaara, L.; Clark, A.B.; Burgers, P.M.; Chabes, A.; Kunkel, T.A. Evidence that processing of ribonucleotides in DNA by topoisomerase 1 is leading-strand specific. Nat. Struct. Mol. Biol. 2015, 22, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Huang, S.N.; Williams, J.S.; Li, Y.C.; Clark, A.B.; Cho, J.-E.; Kunkel, T.A.; Pommier, Y.; Jinks-Robertson, S. Mutagenic processing of ribonucleotides in DNA by yeast topoisomerase I. Science 2011, 332, 1561–1564. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.W.; Burgin, A.B.; Huizenga, B.N.; Robertson, C.A.; Yao, K.C.; Nash, H.A. A eukaryotic enzyme that can disjoin dead-end covalent complexes between DNA and type I topoisomerases. Proc. Natl. Acad. Sci. USA 1996, 93, 11534–11539. [Google Scholar] [CrossRef] [PubMed]
- Takashima, H.; Boerkoel, C.F.; John, J.; Saifi, G.M.; Salih, M.A.M.; Armstrong, D.; Mao, Y.; Quiocho, F.A.; Roa, B.B.; Nakagawa, M.; et al. Mutation of TDP1, encoding a topoisomerase I–dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nat. Genet. 2002, 32, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Sharma, A.; Ju, L.; Murai, J.; Umans, L.; Vermeire, L.; Pommier, Y.; Takeda, S.; Huylebroeck, D.; Caldecott, K.W.; et al. TDP2 promotes repair of topoisomerase I-mediated DNA damage in the absence of TDP1. Nucleic Acids Res. 2012, 40, 8371–8380. [Google Scholar] [CrossRef] [PubMed]
- El-Khamisy, S.F.; Saifi, G.M.; Weinfeld, M.; Johansson, F.; Helleday, T.; Lupski, J.R.; Caldecott, K.W. Defective DNA single-strand break repair in spinocerebellar ataxia with axonal neuropathy-1. Nature 2005, 434, 108–113. [Google Scholar] [CrossRef]
- Interthal, H.; Chen, H.J.; Kehl-Fie, T.E.; Zotzmann, J.; Leppard, J.B.; Champoux, J.J. SCAN1 mutant Tdp1 accumulates the enzyme—DNA intermediate and causes camptothecin hypersensitivity. EMBO J. 2005, 24, 2224–2233. [Google Scholar] [CrossRef]
- Das, B.B.; Antony, S.; Gupta, S.; Dexheimer, T.S.; Redon, C.E.; Garfield, S.; Shiloh, Y.; Pommier, Y. Optimal function of the DNA repair enzyme TDP1 requires its phosphorylation by ATM and/or DNA-PK. EMBO J. 2009, 28, 3667–3680. [Google Scholar] [CrossRef]
- Alagoz, M.; Chiang, S.-C.; Sharma, A.; El-Khamisy, S.F. ATM deficiency results in accumulation of DNA-topoisomerase I covalent intermediates in neural cells. PLoS ONE 2013, 8, e58239. [Google Scholar] [CrossRef]
- Katyal, S.; Lee, Y.; Nitiss, K.C.; Downing, S.M.; Li, Y.; Shimada, M.; Zhao, J.; Russell, H.R.; Petrini, J.H.J.; Nitiss, J.L.; et al. Aberrant topoisomerase-1 DNA lesions are pathogenic in neurodegenerative genome instability syndromes. Nat. Neurosci. 2014, 17, 813–821. [Google Scholar] [CrossRef]
- Austin, C.A.; Lee, K.C.; Swan, R.L.; Khazeem, M.M.; Manville, C.M.; Cridland, P.; Treumann, A.; Porter, A.; Morris, N.J.; Cowell, I.G. TOP2B: The First Thirty Years. Int. J. Mol. Sci. 2018, 19, 2765. [Google Scholar] [CrossRef]
- Madabhushi, R. The Roles of DNA Topoisomerase IIβ in Transcription. Int. J. Mol. Sci. 2018, 19, 1917. [Google Scholar] [CrossRef]
- Gale, K.C.; Osheroff, N. Intrinsic intermolecular DNA ligation activity of eukaryotic topoisomerase II. Potential roles in recombination. J. Biol. Chem. 1992, 267, 12090–12097. [Google Scholar] [PubMed]
- Pommier, Y.; Marchand, C. Interfacial inhibitors: Targeting macromolecular complexes. Nat. Rev. Drug Discov. 2011, 11, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Cowell, I.G.; Sondka, Z.; Smith, K.; Lee, K.C.; Manville, C.M.; Sidorczuk-Lesthuruge, M.; Rance, H.A.; Padget, K.; Jackson, G.H.; Adachi, N.; et al. Model for MLL translocations in therapy-related leukemia involving topoisomerase IIβ-mediated DNA strand breaks and gene proximity. Proc. Natl. Acad. Sci. USA 2012, 109, 8989–8994. [Google Scholar] [CrossRef] [PubMed]
- Karras, G.I.; Yi, S.; Sahni, N.; Fischer, M.; Xie, J.; Vidal, M.; D’Andrea, A.D.; Whitesell, L.; Lindquist, S. HSP90 Shapes the Consequences of Human Genetic Variation. Cell 2017, 168, 856–866.e12. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Herreros, F.; Schuurs-Hoeijmakers, J.H.M.; McCormack, M.; Greally, M.T.; Rulten, S.; Romero-Granados, R.; Counihan, T.J.; Chaila, E.; Conroy, J.; Ennis, S.; et al. TDP2 protects transcription from abortive topoisomerase activity and is required for normal neural function. Nat. Genet. 2014, 46, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Herreros, F.; Romero-Granados, R.; Zeng, Z.; Alvarez-Quilón, A.; Quintero, C.; Ju, L.; Umans, L.; Vermeire, L.; Huylebroeck, D.; Caldecott, K.W.; et al. TDP2-dependent non-homologous end-joining protects against topoisomerase II-induced DNA breaks and genome instability in cells and in vivo. PLoS Genet. 2013, 9, e1003226. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Herreros, F.; Zagnoli-Vieira, G.; Ntai, I.; Martínez-Macías, M.I.; Anderson, R.M.; Herrero-Ruíz, A.; Caldecott, K.W. TDP2 suppresses chromosomal translocations induced by DNA topoisomerase II during gene transcription. Nat. Commun. 2017, 8, 233. [Google Scholar] [CrossRef]
- Hoa, N.N.; Shimizu, T.; Zhou, Z.W.; Wang, Z.-Q.; Deshpande, R.A.; Paull, T.T.; Akter, S.; Tsuda, M.; Furuta, R.; Tsutsui, K.; et al. Mre11 Is Essential for the Removal of Lethal Topoisomerase 2 Covalent Cleavage Complexes. Mol. Cell 2016, 64, 1010. [Google Scholar] [CrossRef]
- Beucher, A.; Birraux, J.; Tchouandong, L.; Barton, O.; Shibata, A.; Conrad, S.; Goodarzi, A.A.; Krempler, A.; Jeggo, P.A.; Löbrich, M. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J. 2009, 28, 3413–3427. [Google Scholar] [CrossRef]
- Shibata, A.; Moiani, D.; Arvai, A.S.; Perry, J.; Harding, S.M.; Genois, M.-M.; Maity, R.; van Rossum-Fikkert, S.; Kertokalio, A.; Romoli, F.; et al. DNA Double-Strand Break Repair Pathway Choice Is Directed by Distinct MRE11 Nuclease Activities. Mol. Cell 2014, 53, 7–18. [Google Scholar] [CrossRef]
- Chappell, C.; Hanakahi, L.A.; Karimi-Busheri, F.; Weinfeld, M.; West, S.C. Involvement of human polynucleotide kinase in double-strand break repair by non-homologous end joining. EMBO J. 2002, 21, 2827–2832. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Robins, P.; Lindahl, T. DNA ligase IV from HeLa cell nuclei. J. Biol. Chem. 1996, 271, 24257–24261. [Google Scholar] [CrossRef] [PubMed]
- Pryor, J.M.; Conlin, M.P.; Carvajal-Garcia, J.; Luedeman, M.E.; Luthman, A.J.; Small, G.W.; Ramsden, D.A. Ribonucleotide incorporation enables repair of chromosome breaks by nonhomologous end joining. Science 2018, 361, 1126–1129. [Google Scholar] [CrossRef]
- Schellenberg, M.J.; Lieberman, J.A.; Herrero-Ruiz, A.; Butler, L.R.; Williams, J.G.; Muñoz-Cabello, A.M.; Mueller, G.A.; London, R.E.; Cortés-Ledesma, F.; Williams, R.S. ZATT (ZNF451)-mediated resolution of topoisomerase 2 DNA-protein cross-links. Science 2017, 357, 1412–1416. [Google Scholar] [CrossRef]
- Lee, K.C.; Swan, R.L.; Sondka, Z.; Padget, K.; Cowell, I.G.; Austin, C.A. Effect of TDP2 on the Level of TOP2-DNA Complexes and SUMOylated TOP2-DNA Complexes. Int. J. Mol. Sci. 2018, 19, 2056. [Google Scholar] [CrossRef]
- Yamaguchi-Iwai, Y.; Sonoda, E.; Sasaki, M.S.; Morrison, C.; Haraguchi, T.; Hiraoka, Y.; Yamashita, Y.M.; Yagi, T.; Takata, M.; Price, C.; et al. Mre11 is essential for the maintenance of chromosomal DNA in vertebrate cells. EMBO J. 1999, 18, 6619–6629. [Google Scholar] [CrossRef]
- Buis, J.; Wu, Y.; Deng, Y.; Leddon, J.; Westfield, G.; Eckersdorff, M.; Sekiguchi, J.M.; Chang, S.; Ferguson, D.O. Mre11 Nuclease Activity Has Essential Roles in DNA Repair and Genomic Stability Distinct from ATM Activation. Cell 2008, 135, 85–96. [Google Scholar] [CrossRef]
- Hoa, N.N.; Akagawa, R.; Yamasaki, T.; Hirota, K.; Sasa, K.; Natsume, T.; Kobayashi, J.; Sakuma, T.; Yamamoto, T.; Komatsu, K.; et al. Relative contribution of four nucleases, CtIP, Dna2, Exo1 and Mre11, to the initial step of DNA double-strand break repair by homologous recombination in both the chicken DT40 and human TK6 cell lines. Genes Cells 2015, 20, 1059–1076. [Google Scholar] [CrossRef]
- Llorente, B.; Symington, L.S. The Mre11 nuclease is not required for 5′ to 3′ resection at multiple HO-induced double-strand breaks. Mol. Cell. Biol. 2004, 24, 9682–9694. [Google Scholar] [CrossRef]
- Westmoreland, J.W.; Resnick, M.A. Coincident resection at both ends of random, γ-induced double-strand breaks requires MRX (MRN), Sae2 (Ctp1), and Mre11-nuclease. PLoS Genet. 2013, 9, e1003420. [Google Scholar] [CrossRef] [PubMed]
- Hartsuiker, E.; Neale, M.J.; Carr, A.M. Distinct requirements for the Rad32(Mre11) nuclease and Ctp1(CtIP) in the removal of covalently bound topoisomerase I and II from DNA. Mol. Cell 2009, 33, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.C.; Padget, K.; Curtis, H.; Cowell, I.G.; Moiani, D.; Sondka, Z.; Morris, N.J.; Jackson, G.H.; Cockell, S.J.; Tainer, J.A.; et al. MRE11 facilitates the removal of human topoisomerase II complexes from genomic DNA. Biol. Open 2012, 1, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.; Jasrotia, A.; Bundschuh, D.; Howard, S.M.; Ranjha, L.; Stucki, M.; Cejka, P. NBS1 promotes the endonuclease activity of the MRE11-RAD50 complex by sensing CtIP phosphorylation. EMBO J. 2019, 38. [Google Scholar] [CrossRef]
- Deshpande, R.A.; Lee, J.-H.; Arora, S.; Paull, T.T. Nbs1 Converts the Human Mre11/Rad50 Nuclease Complex into an Endo/Exonuclease Machine Specific for Protein-DNA Adducts. Mol. Cell 2016, 64, 593–606. [Google Scholar] [CrossRef]
- Paull, T.T. 20 Years of Mre11 Biology: No End in Sight. Mol. Cell 2018, 71, 419–427. [Google Scholar] [CrossRef]
- Adelman, C.A.; De, S.; Petrini, J.H.J. Rad50 is dispensable for the maintenance and viability of postmitotic tissues. Mol. Cell. Biol. 2009, 29, 483–492. [Google Scholar] [CrossRef]
- Frappart, P.-O.; Tong, W.-M.; Demuth, I.; Radovanovic, I.; Herceg, Z.; Aguzzi, A.; Digweed, M.; Wang, Z.-Q. An essential function for NBS1 in the prevention of ataxia and cerebellar defects. Nat. Med. 2005, 11, 538–544. [Google Scholar] [CrossRef]
- Yang, X.; Li, W.; Prescott, E.D.; Burden, S.J.; Wang, J.C. DNA Topoisomerase II and Neural Development. Science 2000, 287, 131–134. [Google Scholar] [CrossRef]
- Lyu, Y.L.; Wang, J.C. Aberrant lamination in the cerebral cortex of mouse embryos lacking DNA topoisomerase IIbeta. Proc. Natl. Acad. Sci. USA 2003, 100, 7123–7128. [Google Scholar] [CrossRef]
- King, I.F.; Yandava, C.N.; Mabb, A.M.; Hsiao, J.S.; Huang, H.-S.; Pearson, B.L.; Calabrese, J.M.; Starmer, J.; Parker, J.S.; Magnuson, T.; et al. Topoisomerases facilitate transcription of long genes linked to autism. Nature 2013, 501, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Uusküla-Reimand, L.; Wilson, M.D. Break Check: Transcription-Driven Topoisomerase II Collisions near Chromatin Loop Anchors Are Hotspots for DNA Damage and Translocations. Mol. Cell 2019, 75, 203–205. [Google Scholar] [CrossRef] [PubMed]
- Uusküla-Reimand, L.; Hou, H.; Samavarchi-Tehrani, P.; Rudan, M.V.; Liang, M.; Medina-Rivera, A.; Mohammed, H.; Schmidt, D.; Schwalie, P.; Young, E.J.; et al. Topoisomerase II beta interacts with cohesin and CTCF at topological domain borders. Genome Biol. 2016, 17, 182. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Kawauchi, D.; Körkel-Qu, H.; Deng, H.; Serger, E.; Sieber, L.; Lieberman, J.A.; Jimeno-González, S.; Lambo, S.; Hanna, B.S.; et al. Chd7 is indispensable for mammalian brain development through activation of a neuronal differentiation programme. Nat. Commun. 2017, 8, 14758. [Google Scholar] [CrossRef] [PubMed]
- Manville, C.M.; Smith, K.; Sondka, Z.; Rance, H.; Cockell, S.; Cowell, I.G.; Lee, K.C.; Morris, N.J.; Padget, K.; Jackson, G.H.; et al. Genome-wide ChIP-seq analysis of human TOP2B occupancy in MCF7 breast cancer epithelial cells. Biol. Open 2015, 4, 1436–1447. [Google Scholar] [CrossRef] [PubMed]
- Kouzine, F.; Gupta, A.; Baranello, L.; Wojtowicz, D.; Ben-Aissa, K.; Liu, J.; Przytycka, T.M.; Levens, D. Transcription-dependent dynamic supercoiling is a short-range genomic force. Nat. Struct. Mol. Biol. 2013, 20, 396–403. [Google Scholar] [CrossRef]
- Naughton, C.; Avlonitis, N.; Corless, S.; Prendergast, J.G.; Mati, I.K.; Eijk, P.P.; Cockroft, S.L.; Bradley, M.; Ylstra, B.; Gilbert, N. Transcription forms and remodels supercoiling domains unfolding large-scale chromatin structures. Nat. Struct. Mol. Biol. 2013, 20, 387–395. [Google Scholar] [CrossRef]
- Baranello, L.; Kouzine, F.; Wojtowicz, D.; Cui, K.; Przytycka, T.M.; Zhao, K.; Levens, D. DNA break mapping reveals topoisomerase II activity genome-wide. Int. J. Mol. Sci. 2014, 15, 13111–13122. [Google Scholar] [CrossRef]
- Madabhushi, R.; Gao, F.; Pfenning, A.R.; Pan, L.; Yamakawa, S.; Seo, J.; Rueda, R.; Phan, T.X.; Yamakawa, H.; Pao, P.-C.; et al. Activity-Induced DNA Breaks Govern the Expression of Neuronal Early-Response Genes. Cell 2015, 161, 1592–1605. [Google Scholar] [CrossRef]
- Vian, L.; Pękowska, A.; Rao, S.S.P.; Kieffer-Kwon, K.-R.; Jung, S.; Baranello, L.; Huang, S.-C.; El Khattabi, L.; Dose, M.; Pruett, N.; et al. The Energetics and Physiological Impact of Cohesin Extrusion. Cell 2018, 175, 292–294. [Google Scholar] [CrossRef]
- Dellino, G.I.; Palluzzi, F.; Chiariello, A.M.; Piccioni, R.; Bianco, S.; Furia, L.; De Conti, G.; Bouwman, B.A.M.; Melloni, G.; Guido, D.; et al. Release of paused RNA polymerase II at specific loci favors DNA double-strand-break formation and promotes cancer translocations. Nat. Genet. 2019, 51, 1011–1023. [Google Scholar] [CrossRef] [PubMed]
- McNamara, S.; Wang, H.; Hanna, N.; Miller, W.H. Topoisomerase IIbeta negatively modulates retinoic acid receptor alpha function: A novel mechanism of retinoic acid resistance. Mol. Cell. Biol. 2008, 28, 2066–2077. [Google Scholar] [CrossRef]
- Haffner, M.C.; Aryee, M.J.; Toubaji, A.; Esopi, D.M.; Albadine, R.; Gurel, B.; Isaacs, W.B.; Bova, G.S.; Liu, W.; Xu, J.; et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat. Genet. 2010, 42, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.H.F.; Chang, I.; Hudak, C.S.S.; Hyun, S.; Kwan, H.-Y.; Sul, H.S. A role of DNA-PK for the metabolic gene regulation in response to insulin. Cell 2009, 136, 1056–1072. [Google Scholar] [CrossRef] [PubMed]
- Trotter, K.W.; King, H.A.; Archer, T.K. Glucocorticoid Receptor Transcriptional Activation via the BRG1-Dependent Recruitment of TOP2β and Ku70/86. Mol. Cell. Biol. 2015, 35, 2799–2817. [Google Scholar] [CrossRef]
- Bunch, H.; Lawney, B.P.; Lin, Y.-F.; Asaithamby, A.; Murshid, A.; Wang, Y.E.; Chen, B.P.C.; Calderwood, S.K. Transcriptional elongation requires DNA break-induced signalling. Nat. Commun. 2015, 6, 10191. [Google Scholar] [CrossRef]
- Liu, X.; Kraus, W.L.; Bai, X. Ready, pause, go: Regulation of RNA polymerase II pausing and release by cellular signaling pathways. Trends Biochem. Sci. 2015, 40, 516–525. [Google Scholar] [CrossRef]
- Jonkers, I.; Lis, J.T. Getting up to speed with transcription elongation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 2015, 16, 167–177. [Google Scholar] [CrossRef]
- Bunch, H. Gene regulation of mammalian long non-coding RNA. Mol. Genet. Genom. 2018, 293, 1–15. [Google Scholar] [CrossRef]
- Ju, B.-G.; Lunyak, V.V.; Perissi, V.; Garcia-Bassets, I.; Rose, D.W.; Glass, C.K.; Rosenfeld, M.G. A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science 2006, 312, 1798–1802. [Google Scholar] [CrossRef]
- Perillo, B.; Ombra, M.N.; Bertoni, A.; Cuozzo, C.; Sacchetti, S.; Sasso, A.; Chiariotti, L.; Malorni, A.; Abbondanza, C.; Avvedimento, E. V DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science 2008, 319, 202–206. [Google Scholar] [CrossRef] [PubMed]
- McNamara, S.; Nichol, J.N.; Wang, H.; Miller, W.H. Targeting PKC delta-mediated topoisomerase II beta overexpression subverts the differentiation block in a retinoic acid-resistant APL cell line. Leukemia 2010, 24, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Puc, J.; Aggarwal, A.K.; Rosenfeld, M.G. Physiological functions of programmed DNA breaks in signal-induced transcription. Nat. Rev. Mol. Cell Biol. 2017, 18, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Bunch, H. Role of genome guardian proteins in transcriptional elongation. FEBS Lett. 2016, 590, 1064–1075. [Google Scholar] [CrossRef]
- Core, L.; Adelman, K. Promoter-proximal pausing of RNA polymerase II: A nexus of gene regulation. Genes Dev. 2019, 33, 960–982. [Google Scholar] [CrossRef]
- Ma, J.; Bai, L.; Wang, M.D. Transcription Under Torsion. Science 2013, 340, 1580–1583. [Google Scholar] [CrossRef]
- Ma, J.; Tan, C.; Gao, X.; Fulbright, R.M.; Roberts, J.W.; Wang, M.D. Transcription factor regulation of RNA polymerase’s torque generation capacity. Proc. Natl. Acad. Sci. USA 2019, 116, 2583–2588. [Google Scholar] [CrossRef]
- Teves, S.S.; Henikoff, S. DNA torsion as a feedback mediator of transcription and chromatin dynamics. Nucleus 2014, 5, 211–218. [Google Scholar] [CrossRef]
- Zlotorynski, E. Pol II and topoisomerase 1 hand-in-hand. Nat. Rev. Mol. Cell Biol. 2016, 17, 265. [Google Scholar] [CrossRef]
- Crisona, N.J.; Strick, T.R.; Bensimon, D.; Croquette, V.; Cozzarelli, N.R. Preferential relaxation of positively supercoiled DNA by E. coli topoisomerase IV in single-molecule and ensemble measurements. Genes Dev. 2000, 14, 2881–2892. [Google Scholar] [CrossRef]
- Liu, L.F.; Wang, J.C. Supercoiling of the DNA template during transcription. Proc. Natl. Acad. Sci. USA 1987, 84, 7024–7027. [Google Scholar] [CrossRef] [PubMed]
- Farnung, L.; Vos, S.M.; Cramer, P. Structure of transcribing RNA polymerase II-nucleosome complex. Nat. Commun. 2018, 9, 5432. [Google Scholar] [CrossRef] [PubMed]
- Jimeno-González, S.; Ceballos-Chávez, M.; Reyes, J.C. A positioned +1 nucleosome enhances promoter-proximal pausing. Nucleic Acids Res. 2015, 43, 3068–3078. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, P.J. Topoisomerases and the regulation of neural function. Nat. Rev. Neurosci. 2016, 17, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Macneil, L.T.; Walhout, A.J.M. Gene regulatory networks and the role of robustness and stochasticity in the control of gene expression. Genome Res. 2011, 21, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Teng, G.; Schatz, D.G. Regulation and Evolution of the RAG Recombinase. Adv. Immunol. 2015, 128, 1–39. [Google Scholar]
- Lange, J.; Yamada, S.; Tischfield, S.E.; Pan, J.; Kim, S.; Zhu, X.; Socci, N.D.; Jasin, M.; Keeney, S. The Landscape of Mouse Meiotic Double-Strand Break Formation, Processing, and Repair. Cell 2016, 167, 695–708.e16. [Google Scholar] [CrossRef]
- Keeney, S.; Giroux, C.N.; Kleckner, N. Meiosis-Specific DNA Double-Strand Breaks Are Catalyzed by Spo11, a Member of a Widely Conserved Protein Family. Cell 1997, 88, 375–384. [Google Scholar] [CrossRef]
- Gennery, A. Recent advances in understanding RAG deficiencies. F1000Res. 2019, 8, 148. [Google Scholar] [CrossRef]
- Ngo, J.; Matsuyama, M.; Kim, C.; Poventud-Fuentes, I.; Bates, A.; Siedlak, S.L.; Lee, H.; Doughman, Y.Q.; Watanabe, M.; Liner, A.; et al. Bax deficiency extends the survival of Ku70 knockout mice that develop lung and heart diseases. Cell Death Dis. 2015, 6, e1706. [Google Scholar] [CrossRef]
- Li, H.; Vogel, H.; Holcomb, V.B.; Gu, Y.; Hasty, P. Deletion of Ku70, Ku80, or Both Causes Early Aging without Substantially Increased Cancer. Mol. Cell. Biol. 2007, 27, 8205–8214. [Google Scholar] [CrossRef] [PubMed]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.-A.; Mooij, T.M.; Roos-Blom, M.-J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402. [Google Scholar] [CrossRef] [PubMed]
- Moynahan, M.E.; Chiu, J.W.; Koller, B.H.; Jasin, M. Brca1 controls homology-directed DNA repair. Mol. Cell 1999, 4, 511–518. [Google Scholar] [CrossRef]
- Qing, Y.; Yamazoe, M.; Hirota, K.; Dejsuphong, D.; Sakai, W.; Yamamoto, K.N.; Bishop, D.K.; Wu, X.; Takeda, S. The Epistatic Relationship between BRCA2 and the Other RAD51 Mediators in Homologous Recombination. PLoS Genet. 2011, 7, e1002148. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.-E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016, 535, 382–387. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef]
- Rheinbay, E.; Parasuraman, P.; Grimsby, J.; Tiao, G.; Engreitz, J.M.; Kim, J.; Lawrence, M.S.; Taylor-Weiner, A.; Rodriguez-Cuevas, S.; Rosenberg, M.; et al. Recurrent and functional regulatory mutations in breast cancer. Nature 2017, 547, 55–60. [Google Scholar] [CrossRef]
- García-Muse, T.; Aguilera, A. R Loops: From Physiological to Pathological Roles. Cell 2019, 179, 604–618. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, J.-Y.; Zhang, X.; Gu, Y.; Xiao, R.; Shao, C.; Tang, P.; Qian, H.; Luo, D.; Li, H.; et al. R-ChIP Using Inactive RNase H Reveals Dynamic Coupling of R-loops with Transcriptional Pausing at Gene Promoters. Mol. Cell 2017, 68, 745–757.e5. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A.; García-Muse, T. R loops: From transcription byproducts to threats to genome stability. Mol. Cell 2012, 46, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Hatchi, E.; Skourti-Stathaki, K.; Ventz, S.; Pinello, L.; Yen, A.; Kamieniarz-Gdula, K.; Dimitrov, S.; Pathania, S.; McKinney, K.M.; Eaton, M.L.; et al. BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol. Cell 2015, 57, 636–647. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.J.; Rolland, T.; Adelmant, G.; Xia, X.; Owen, M.S.; Dricot, A.; Zack, T.I.; Sahni, N.; Jacob, Y.; Hao, T.; et al. Systematic screening reveals a role for BRCA1 in the response to transcription-associated DNA damage. Genes Dev. 2014, 28, 1957–1975. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, V.; Barroso, S.I.; García-Rubio, M.L.; Tumini, E.; Herrera-Moyano, E.; Aguilera, A. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 2014, 511, 362–365. [Google Scholar] [CrossRef]
- Tan, S.L.W.; Chadha, S.; Liu, Y.; Gabasova, E.; Perera, D.; Ahmed, K.; Constantinou, S.; Renaudin, X.; Lee, M.; Aebersold, R.; et al. A Class of Environmental and Endogenous Toxins Induces BRCA2 Haploinsufficiency and Genome Instability. Cell 2017, 169, 1105–1118.e15. [Google Scholar] [CrossRef]
- Shivji, M.K.K.; Renaudin, X.; Williams, Ç.H.; Venkitaraman, A.R. BRCA2 Regulates Transcription Elongation by RNA Polymerase II to Prevent R-Loop Accumulation. Cell Rep. 2018, 22, 1031–1039. [Google Scholar] [CrossRef]
- Scully, R.; Anderson, S.F.; Chao, D.M.; Wei, W.; Ye, L.; Young, R.A.; Livingston, D.M.; Parvin, J.D. BRCA1 is a component of the RNA polymerase II holoenzyme. Proc. Natl. Acad. Sci. USA 1997, 94, 5605–5610. [Google Scholar] [CrossRef]
- Zhang, X.; Chiang, H.-C.; Wang, Y.; Zhang, C.; Smith, S.; Zhao, X.; Nair, S.J.; Michalek, J.; Jatoi, I.; Lautner, M.; et al. Attenuation of RNA polymerase II pausing mitigates BRCA1-associated R-loop accumulation and tumorigenesis. Nat. Commun. 2017, 8, 15908. [Google Scholar] [CrossRef]
- Bunting, S.F.; Callén, E.; Kozak, M.L.; Kim, J.M.; Wong, N.; López-Contreras, A.J.; Ludwig, T.; Baer, R.; Faryabi, R.B.; Malhowski, A.; et al. BRCA1 functions independently of homologous recombination in DNA interstrand crosslink repair. Mol. Cell 2012, 46, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Sasanuma, H.; Tsuda, M.; Morimoto, S.; Saha, L.K.; Rahman, M.M.; Kiyooka, Y.; Fujiike, H.; Cherniack, A.D.; Itou, J.; Callen Moreu, E.; et al. BRCA1 ensures genome integrity by eliminating estrogen-induced pathological topoisomerase II–DNA complexes. Proc. Natl. Acad. Sci. USA 2018, 115, E10642–E10651. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Goodrich, D.W. The retinoblastoma tumor suppressor protein is required for efficient processing and repair of trapped topoisomerase II-DNA-cleavable complexes. Oncogene 2005, 24, 8105–8113. [Google Scholar] [CrossRef] [PubMed]
- Polanowska, J.; Martin, J.S.; Garcia-Muse, T.; Petalcorin, M.I.R.; Boulton, S.J. A conserved pathway to activate BRCA1-dependent ubiquitylation at DNA damage sites. EMBO J. 2006, 25, 2178–2188. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, R.A.; Sobhian, B.; Pathania, S.; Cantor, S.B.; Nakatani, Y.; Livingston, D.M. Multifactorial contributions to an acute DNA damage response by BRCA1/BARD1-containing complexes. Genes Dev. 2006, 20, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.Y.; Sonoda, E.; Barber, L.J.; Oka, H.; Murakawa, Y.; Yamada, K.; Ikura, T.; Wang, X.; Kobayashi, M.; Yamamoto, K.; et al. A critical role for the ubiquitin-conjugating enzyme Ubc13 in initiating homologous recombination. Mol. Cell 2007, 25, 663–675. [Google Scholar] [CrossRef]
- Sano, K.; Miyaji-Yamaguchi, M.; Tsutsui, K.M.; Tsutsui, K. Topoisomerase IIbeta activates a subset of neuronal genes that are repressed in AT-rich genomic environment. PLoS ONE 2008, 3, e4103. [Google Scholar] [CrossRef]
- Williamson, L.M.; Lees-Miller, S.P. Estrogen receptor α-mediated transcription induces cell cycle-dependent DNA double-strand breaks. Carcinogenesis 2011, 32, 279–285. [Google Scholar] [CrossRef]
- Stork, C.T.; Bocek, M.; Crossley, M.P.; Sollier, J.; Sanz, L.A.; Chédin, F.; Swigut, T.; Cimprich, K.A. Co-transcriptional R-loops are the main cause of estrogen-induced DNA damage. Elife 2016, 5, e17548. [Google Scholar] [CrossRef]
- Harding, S.M.; Boiarsky, J.A.; Greenberg, R.A. ATM Dependent Silencing Links Nucleolar Chromatin Reorganization to DNA Damage Recognition. Cell Rep. 2015, 13, 251–259. [Google Scholar] [CrossRef]
- Larsen, D.H.; Stucki, M. Nucleolar responses to DNA double-strand breaks. Nucleic Acids Res. 2016, 44, 538–544. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morimoto, S.; Tsuda, M.; Bunch, H.; Sasanuma, H.; Austin, C.; Takeda, S. Type II DNA Topoisomerases Cause Spontaneous Double-Strand Breaks in Genomic DNA. Genes 2019, 10, 868. https://doi.org/10.3390/genes10110868
Morimoto S, Tsuda M, Bunch H, Sasanuma H, Austin C, Takeda S. Type II DNA Topoisomerases Cause Spontaneous Double-Strand Breaks in Genomic DNA. Genes. 2019; 10(11):868. https://doi.org/10.3390/genes10110868
Chicago/Turabian StyleMorimoto, Suguru, Masataka Tsuda, Heeyoun Bunch, Hiroyuki Sasanuma, Caroline Austin, and Shunichi Takeda. 2019. "Type II DNA Topoisomerases Cause Spontaneous Double-Strand Breaks in Genomic DNA" Genes 10, no. 11: 868. https://doi.org/10.3390/genes10110868
APA StyleMorimoto, S., Tsuda, M., Bunch, H., Sasanuma, H., Austin, C., & Takeda, S. (2019). Type II DNA Topoisomerases Cause Spontaneous Double-Strand Breaks in Genomic DNA. Genes, 10(11), 868. https://doi.org/10.3390/genes10110868