Abstract
This study aimed to investigate GJB2 (connexin 26) and GJB6 (connexin 30) mutations associated with familial non-syndromic childhood hearing impairment (HI) in Cameroon. We selected only families segregating HI, with at least two affected individuals and with strong evidence of non-environmental causes. DNA was extracted from peripheral blood, and the entire coding region of GJB2 was interrogated using Sanger sequencing. Multiplex PCR and Sanger sequencing were used to analyze the prevalence of the GJB6-D3S1830 deletion. A total of 93 patients, belonging to 41 families, were included in the analysis. Hearing impairment was sensorineural in 51 out of 54 (94.4%) patients. Pedigree analysis suggested autosomal recessive inheritance in 85.4% (35/41) of families. Hearing impairment was inherited in an autosomal dominant and mitochondrial mode in 12.2% (5/41) and 2.4% (1/41) of families, respectively. Most HI participants were non-syndromic (92.5%; 86/93). Four patients from two families presented with type 2 Waardenburg syndrome, and three cases of type 2 Usher syndrome were identified in one family. No GJB2 mutations were found in any of the 29 families with non-syndromic HI. Additionally, the GJB6-D3S1830 deletion was not identified in any of the HI patients. This study confirms that mutations in the GJB2 gene and the del(GJB6-D13S1830) mutation do not contribute to familial HI in Cameroon.
1. Introduction
Hearing impairment (HI) is a disabling congenital disease with the highest rate for age-standardized disability of life in the world [1]. Globally, congenital HI has a prevalence of 1.3 per 1000 population [2], and is accounted for in about 1 per 1000 live births in developed countries, with a much higher incidence of up to 6 per 1000 live births in sub-Saharan Africa [3]. Genetic factors contribute from 30 to 50% of hearing impairment cases in sub-Saharan Africa [4]. In 70% of neonates who fail newborn hearing screens (NBHS) and are presumed to have inherited HI, there are no other distinguishing physical findings and the HI is classified as non-syndromic [5]. Among non-syndromic (NS) HI, nearly 80% of cases are inherited in an autosomal recessive (AR) mode [6,7].
Non-syndromic hearing impairment (NSHI) is an extremely heterogeneous trait, with approximately 170 NSHI loci and 112 genes identified to date [8]. Nevertheless, studies in European and Asian populations have identified pathogenic variants in GJB2 (encoding connexin 26) and GJB6 (encoding connexin 30) as the major contributors to autosomal recessive NSHI (ARNSHI) [9], with GJB2-c.35delG being the most prevalent variant (20–50%) found in cases of autosomal recessive non-syndromic hearing impairment (ARNSHI) [7,10]. The GJB6-D13S1830 deletion was identified in up to 9.7% of cases, and thus is the second largest contributor to the genetic etiology of NSHI in European populations, either with homozygous presentation, or when present in addition to a GJB2 mutation on the opposite allele [11,12].
Cameroon is a sub-Saharan African country, covering an area of 475,442 km2, with a 2017 census reporting a population of 24,053,727 [13]. Cameroon is frequently referred to as “Africa in miniature”, because of its many geographical and cultural attributes, its population and linguistic diversity (there are more than 200 distinct local languages in the country) [14], and its vast genetic diversity that mimics that of Africa [15].
Previous studies have found no contribution of the GJB2 and GJB6 genes to HI in Cameroon [16,17,18]. However, the patients included in those studies were chosen indiscriminately, and consisted of both familial and isolated cases, with a high likelihood of environmental causes in many cases. In this study we revisit the contribution of the GJB2 and GJB6 genes to HI in Cameroon by only focusing investigations into cases that showed a clear pattern of inheritance within families.
2. Materials and Methods
2.1. Ethical Approval
The study was performed in accordance with the Declaration of Helsinki. Ethical approval was obtained from the Institutional Research Ethics Committee for Human Health of the Gyneco-Obstetric and Pediatric Hospital of Yaoundé, Cameroon (No. 723/CIERSH/DM/2018), and the University of Cape Town’s Faculty of Health Sciences’ Human Research Ethics Committee (HREC 104/2018). Written and signed informed consent was obtained from all participants who were 21 years of age or older, and from parents or guardians in cases of minors, with verbal assent from participants, including permission to publish photographs.
2.2. Participant Selection
Hearing impaired patients were recruited from eight of the ten administrative regions of Cameroon, from schools for the deaf, and in the community, following procedures previously reported in Cameroon [14]. Briefly, all participants’ detailed personal and family histories were obtained, medical records were reviewed by a general practitioner, a medical geneticist, and an ENT specialist when possible, and relevant data were extracted, including three-generation pedigrees and perinatal histories. A general systemic and otological examination and audiological evaluation were performed, including pure tone audiometry. We followed the recommendation number 02/1 of the Bureau International d’Audiophonologie (BIAP), Belgium, to classify hearing levels [14,19]. Only HI individuals belonging to families with segregating hearing impairment, with at least two affected individuals, and with strong evidence of non-environmental causes were recruited.
2.3. Molecular Analysis
Genomic DNA samples were extracted from peripheral blood, following the manufacturer’s instructions for the available commercial kit (Puregene Blood Kit® (Qiagen, Alameda, CA, USA)), at the Biochemical Laboratory of the Centre Pasteur du Cameroun, Yaoundé, Cameroon, or using the Chemagic extraction protocol, in the division of Human Genetics, University of Cape Town, South Africa.
Previously reported primers for the GJB2 gene were evaluated using BLAST® (NIH, USA) and other software, as recommended [17]. The entire coding region of the GJB2 gene (exon 2) was amplified, followed by sequencing using an ABI 3130XL Genetic Analyzer® (Applied Biosystems, Foster City, CA, USA), in the Division of Human Genetics, University of Cape Town, South Africa.
Detection of del(GJB6-D13S1830) was performed using the method and primers described by del Castillo et al. [11,12]. The entire coding region of GJB6 was amplified using the method described by Chen et al. [20]. The PCR results were validated by Sanger sequencing of 10% of the samples.
2.4. Data Analysis
Data analysis was performed through the use of descriptive statistics.
3. Results
3.1. Participant Demographics
We recruited a total of 93 patients belonging to 41 families. Their mean age was 18 ± 10.4 (1–50) years. The male/female ratio was 0.82 (42/51). Hearing impairment was congenital in 62 patients (66.7%), and the mean age at medical diagnosis was 3.2 ± 3.3 (1–22) years.
3.2. Audiometric Characteristics
Pure tone audiometry was performed in 54 of our 93 patients. Hearing impairment was sensorineural in 51 out of 54 (94.4%) patients and mixed in 3 patients; no patients exhibited a conductive hearing impairment. All of our patients had bilateral hearing impairment, and the majority had profound to total hearing impairment (n = 51; 94.4%) (Table 1).

Table 1.
Audiometric classification of hearing impairment, according to the Bureau International d’Audiophonologie (BIAP) recommendation.
3.3. Inheritance Pattern
Pedigree analysis suggestive of autosomal recessive inheritance was the most frequently observed pattern of inheritance and accounted for 85.4% (35/41) of families (Figure 1A). In 12.2% (5/41) and 2.4% (1/41) of families, HI were likely inherited in an autosomal dominant and mitochondrial mode, respectively (Figure 1B). Consanguinity was present in three families (3/41; 7.3%). The inbreeding coefficient was 0.0625 in one of these families (Figure 1A), and 0.0156 in the other two families. A total of six participants (6/93; 6.5%) were thus born from consanguineous union.
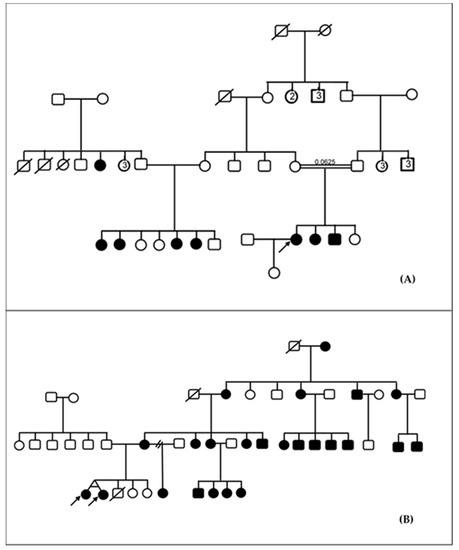
Figure 1.
Inheritance of familial hearing impairment in Cameroon. (A) Pedigree of a consanguineous family with autosomal recessive non-syndromic hearing impairment. (B) Pedigree of a family with non-syndromic hearing impairment suggestive of mitochondrial inheritance. Arrows here indicate the probands.
3.4. Non-Syndromic and Syndromic Hearing Impairment
The majority of our participants exhibited non-syndromic hearing impairment, which accounted for 92.5% (86/93) of cases, for a total of 38 families (Figure 2).
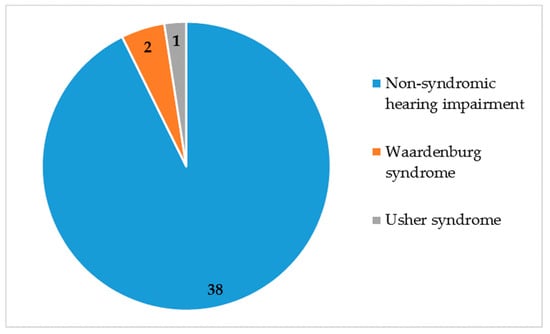
Figure 2.
Non-syndromic and syndromic hearing impairment. N = 41 families.
Four patients from two families in our cohort presented type 2 Waardenburg syndrome (without dystopia canthorum). The main clinical signs included: hearing impairment, patches of hypopigmented skin, sapphire-blue eyes, and premature white hair (Figure 3).
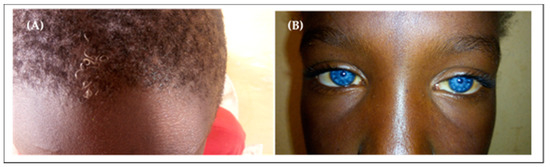
Figure 3.
Waardenburg syndrome in our cohort. (A) Premature white hair; (B) Sapphire-blue eyes.
Three cases of type 2 Usher syndrome (without vestibular areflexia), from one family, were identified; in addition to hearing impairment, clinical signs of retinitis pigmentosa were present, including night vision impairment and constricted visual field.
3.5. Molecular Analysis Results of GJB2 and GJB6
A total of 29 families with segregating recessive non-syndromic hearing impairment were tested for mutations in GJB2 and for the del(GJB6-D13S1830) mutation. None of the families exhibited the del(GJB6-D13S1830) mutation, or any of the reported disease-causing mutations in GJB2. However, a GJB2 variant of uncertain significance, NM_004004.6: c.499G>A (p.V167M), was present in one family in the heterozygous form (Supplementary Materials, Figure S1).
4. Discussion
This report is the most compressive study of the role of GJB2 and GJB6 in familial HI in Cameroon, and confirmed the non-implication of these genes in non-syndromic HI in that country; this is consistent with previous reports in selected isolated HI cases of putative genetic origin [16,17]. By carefully and stringently selecting only multiplex families in the current studies, our results consolidate previous findings. In addition, we recruited in nearly all the schools, as well as in the community, for the deaf in 8/10 provinces representing about 90% of the population; therefore, we are confident that the sample is representative of the population in Cameroon.
The low implication of the GJB2 gene in non-syndromic hearing impairment has also been demonstrated in other populations of African descent. The 35delG mutation, which constitutes almost 50% of all GJB2 mutations in Caucasians [21,22,23], was not reported in 406, 356, 182, and 126 probands from Kenya, Ghana, South Africa, and Uganda, respectively [24,25,26,27]. Moreover, no pathogenic mutations in GJB2 were found in a cohort of 44 probands from Nigeria with non-syndromic deafness [28], and only 7% of a cohort of 127 American probands of Hispanic or African descent with bilateral non-syndromic hearing impairment presented a disease causing mutation in GJB2 [29]. However, an African exception is the Ghanaian population, where the GJB2 founder mutation p.R143W (c.427C˃T) was shown to be highly prevalent in that population [30], and accounted for nearly 25% of familial cases and 8% of isolated cases of HI [31]. The GJB2 variant, c.499G>A (p.V167M), of uncertain significance according to the ClinVar database, was present in a family in our cohort, and has previously been described by our group in the Cameroonian population [17]. It has also been described in the USA [32] and in China [33]. The GJB2 variant c.499G>A (p.V167M) could thus be considered to require further investigation.
The most common mutation in GJB6 is a 342-kb deletion, GJB6-D13S1830, which causes non-syndromic HI when homozygous, or when present on the opposite allele to a GJB2 mutation [24,34]. The del(GJB6-D13S1830) mutation is the second most frequent genetic cause of non-syndromic prelingual hearing impairment in the Spanish population (after the 35delG mutation in GJB2) [11]. This deletion is also prevalent in France, Brazil, and Israel [12,35,36], but is rare or absent in Italy (i.e., Sicily), Romania, Iran, and India [7,37,38,39,40,41]. This deletion was also absent in Nigerians [28] and in Ghanaians [31]. In order to identify any other mutations different from del(GJB6-D13S1830), the coding region of GJB6 was sequenced in African probands from Cameroon, South Africa, and Uganda; however, this revealed no additional pathogenic mutations [18,26].
Our findings support previous reports that GJB2 and GJB6 do not play a significant role in non-syndromic hearing impairment in most populations of African descent. Interestingly, genetic testing through targeted genomic enrichment and massively parallel sequencing of 116 genes were used to screen 10 multiplex families with non-syndromic hearing impairment. In 7 of the 10 families (70%), 12 pathogenic variants were identified in 6 genes, and nearly half of these variants were novel [42]. Therefore, due to the highly heterogeneous genetic nature of NSHI, next generation sequencing would be the most effective way to identify variants associated with non-syndromic deafness in the African populations [4], and all the families investigated in the present study should be subjected to whole exome sequencing in order to potentially identify variants in known genes as well as novel genes. Indeed, based on the identification of specific inner ear transcripts, it is estimated that more than 1000 NSHI genes are still to be identified [43].
The study also confirms Waardenburg syndrome as the most common cause of syndromic hearing impairment in Cameroon, in line with previous reporting in other African populations [44]; these families, as well as the singular family displaying mitochondrial inheritance, should also warrant further molecular investigation.
5. Conclusions
The present study showed that hereditary hearing impairment in Cameroon is mostly non-syndromic, congenital, sensorineural, and inherited in an autosomal recessive mode. Additionally, this study identified Waardenburg and Usher syndromes as the most common syndromic hearing impairments in Cameroon. This study confirmed that mutations in the GJB2 gene and the del(GJB6-D13S1830) deletion are not implicated in familial non-syndromic hearing impairment in Cameroon. Future studies should employ whole genome sequencing approaches and functional genomics studies to identify other genes that may be implicated in the hearing impairment observed in these families.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4425/10/11/844/s1, Figure S1: GJB2 variant of uncertain significance, c.499G>A (p.V167M), present in a family in the heterozygous form, Figure S2: Regional distribution of families.
Author Contributions
A.W. conceived the project; E.T.W. performed the recruitment and molecular experiments. J.J.N. supervised the recruitment; E.T.W. and S.M.A. performed molecular analysis of the GJB2 and GJB6 genes. E.T.W. and J.V.F.F. interpreted audiometric data. A.W. and E.C. supervised the project. All the authors agreed to the final manuscript.
Funding
The study was funded by the Wellcome Trust, grant number 107755Z/15/Z to GAA and AW (co-applicants); NIH, USA, grant number U01-HG-009716 to AW, and the African Academy of Science/Wellcome Trust, grant number H3A/18/001 to AW. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Acknowledgments
We thank patients and their families for their participation in this project. We also thank schools and associations for the deaf for their help during the process of identification and recruitment of families.
Conflicts of Interest
We declare that no competing interests exist.
References
- Vos, T.; Allen, C.; Arora, M.; Barber, R.M.; Bhutta, Z.A.; Brown, A.; Carter, A.; Casey, D.C.; Charlson, F.J.; Chen, A.Z.; et al. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1545–1602. [Google Scholar] [CrossRef]
- James, M.; Praveen, K.P.; Ninan, P.J. A study on prevalence and risk factors of hearing impairment among newborns. Int. J. Contemp. Pediatrics 2018, 5, 304–309. [Google Scholar] [CrossRef][Green Version]
- Olusanya, B.O.; Neumann, K.J.; Saunders, J.E. The global burden of disabling hearing impairment: A call to action. Bull. World Health Organ. 2014, 92, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Lebeko, K.; Bosch, J.; Noubiap, J.J.N.; Dandara, C.; Wonkam, A. Genetics of hearing loss in Africans: Use of next generation sequencing is the best way forward. Pan Afr. Med. J. 2015, 20, 383. [Google Scholar] [CrossRef] [PubMed]
- Forty-six genes causing nonsyndromic hearing impairment: Which ones should be analyzed in DNA diagnostics? Mutat. Res. Rev. Mutat. Res. 2009, 681, 189–196. [CrossRef] [PubMed]
- Wu, X.; Wang, S.; Chen, S.; Wen, Y.; Liu, B.; Xie, W.; Li, D.; Liu, L.; Huang, X.; Sun, Y.; et al. Autosomal Recessive Congenital Sensorineural Hearing Loss due to a Novel Compound Heterozygous PTPRQ Mutation in a Chinese Family. Neural Plast. 2018, 2018, 9425725. [Google Scholar] [CrossRef]
- Adhikary, B.; Ghosh, S.; Paul, S.; Bankura, B.; Pattanayak, A.K.; Biswas, S.; Maity, B.; Das, M. Spectrum and frequency of GJB2, GJB6 and SLC26A4 gene mutations among nonsyndromic hearing loss patients in eastern part of India. Gene 2015, 573, 239–245. [Google Scholar] [CrossRef]
- Welcome to the Hereditary Hearing Loss Homepage|Hereditary Hearing Loss Homepage. Available online: https://hereditaryhearingloss.org/ (accessed on 19 November 2018).
- Chan, D.K.; Chang, K.W. GJB2-associated hearing loss: Systematic review of worldwide prevalence, genotype, and auditory phenotype. Laryngoscope 2014, 124, E34–E53. [Google Scholar] [CrossRef]
- Del Castillo, F.J.; Del Castillo, I. DFNB1 Non-syndromic Hearing Impairment: Diversity of Mutations and Associated Phenotypes. Front. Mol. Neurosci. 2017, 10, 428. [Google Scholar] [CrossRef]
- del Castillo, I.; Villamar, M.; Moreno-Pelayo, M.A.; del Castillo, F.J.; Alvarez, A.; Tellería, D.; Menéndez, I.; Moreno, F. A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N. Engl. J. Med. 2002, 346, 243–249. [Google Scholar] [CrossRef]
- del Castillo, I.; Moreno-Pelayo, M.A.; del Castillo, F.J.; Brownstein, Z.; Marlin, S.; Adina, Q.; Cockburn, D.J.; Pandya, A.; Siemering, K.R.; Chamberlin, G.P.; et al. Prevalence and Evolutionary Origins of the del(GJB6-D13S1830) Mutation in the DFNB1 Locus in Hearing-Impaired Subjects: A Multicenter Study. Am. J. Hum. Genet. 2003, 73, 1452–1458. [Google Scholar] [CrossRef] [PubMed]
- Population, Total|Data. Available online: https://data.worldbank.org/indicator/SP.POP.TOTL (accessed on 5 April 2019).
- Wonkam, A.; Noubiap, J.J.N.; Djomou, F.; Fieggen, K.; Njock, R.; Toure, G.B. Aetiology of childhood hearing loss in Cameroon (sub-Saharan Africa). Eur. J. Med Genet. 2013, 56, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Tishkoff, S.A.; Reed, F.A.; Friedlaender, F.R.; Ehret, C.; Ranciaro, A.; Froment, A.; Hirbo, J.B.; Awomoyi, A.A.; Bodo, J.-M.; Doumbo, O.; et al. The Genetic Structure and History of Africans and African Americans. Science 2009, 324, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Trotta, L.; Iacona, E.; Primignani, P.; Castorina, P.; Radaelli, C.; Bo, L.D.; Coviello, D.; Ambrosetti, U. GJB2 and MTRNR1 contributions in children with hearing impairment from Northern Cameroon. Int. J. Audiol. 2011, 50, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Bosch, J.; Noubiap, J.J.N.; Dandara, C.; Makubalo, N.; Wright, G.; Entfellner, J.-B.D.; Tiffin, N.; Wonkam, A. Sequencing of GJB2 in Cameroonians and Black South Africans and comparison to 1000 Genomes Project Data Support Need to Revise Strategy for Discovery of Nonsyndromic Deafness Genes in Africans. OMICS 2014, 18, 705–710. [Google Scholar] [CrossRef]
- Bosch, J.; Lebeko, K.; Nziale, J.J.N.; Dandara, C.; Makubalo, N.; Wonkam, A. In Search of Genetic Markers for Nonsyndromic Deafness in Africa: A Study in Cameroonians and Black South Africans with the GJB6 and GJA1 Candidate Genes. OMICS 2014, 18, 481–485. [Google Scholar] [CrossRef]
- BIAP—Bureau International d’Audiophonologie. Available online: https://www.biap.org/en/component/content/article/65-recommendations/ct-2-classification/5-biap-recommendation-021-bis (accessed on 18 November 2018).
- Chen, P.; Chen, H.; Fu, S.; Chen, G.; Dong, J. Prevalence of GJB6 mutations in Chinese patients with non-syndromic hearing loss. Int. J. Pediatric Otorhinolaryngol. 2012, 76, 265–267. [Google Scholar] [CrossRef]
- Pampanos, A.; Economides, J.; Iliadou, V.; Neou, P.; Leotsakos, P.; Voyiatzis, N.; Eleftheriades, N.; Tsakanikos, M.; Antoniadi, T.; Hatzaki, A.; et al. Prevalence of GJB2 mutations in prelingual deafness in the Greek population. Int. J. Pediatr. Otorhinolaryngol. 2002, 65, 101–108. [Google Scholar] [CrossRef]
- Dahl, H.H.; Saunders, K.; Kelly, T.M.; Osborn, A.H.; Wilcox, S.; Cone-Wesson, B.; Wunderlich, J.L.; Du Sart, D.; Kamarinos, M.; Gardner, R.J.; et al. Prevalence and nature of connexin 26 mutations in children with non-syndromic deafness. Med. J. Aust. 2001, 175, 191–194. [Google Scholar] [CrossRef]
- Neocleous, V.; Aspris, A.; Shahpenterian, V.; Nicolaou, V.; Panagi, C.; Ioannou, I.; Kyamides, Y.; Anastasiadou, V.; Phylactou, L.A. High frequency of 35delG GJB2 mutation and absence of del(GJB6-D13S1830) in Greek Cypriot patients with nonsyndromic hearing loss. Genet. Test. 2006, 10, 285–289. [Google Scholar] [CrossRef]
- Kabahuma, R.I.; Ouyang, X.; Du, L.L.; Yan, D.; Hutchin, T.; Ramsay, M.; Penn, C.; Liu, X.-Z. Absence of GJB2 gene mutations, the GJB6 deletion (GJB6-D13S1830) and four common mitochondrial mutations in nonsyndromic genetic hearing loss in a South African population. Int. J. Pediatric Otorhinolaryngol. 2011, 75, 611. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gasmelseed, N.M.A.; Schmidt, M.; Magzoub, M.M.A.; Macharia, M.; Elmustafa, O.M.; Ototo, B.; Winkler, E.; Ruge, G.; Horstmann, R.D.; Meyer, C.G. Low frequency of deafness-associated GJB2 variants in Kenya and Sudan and novel GJB2 variants. Hum. Mutat. 2004, 23, 206–207. [Google Scholar] [CrossRef] [PubMed]
- Javidnia, H.; Carson, N.; Awubwa, M.; Byaruhanga, R.; Mack, D.; Vaccani, J.-P. Connexin gene mutations among ugandan patients with nonsyndromic sensorineural hearing loss. Laryngoscope 2014, 124, E373–E376. [Google Scholar] [CrossRef] [PubMed]
- Hamelmann, C.; Amedofu, G.K.; Albrecht, K.; Muntau, B.; Gelhaus, A.; Brobby, G.W.; Horstmann, R.D. Pattern of connexin 26 (GJB2) mutations causing sensorineural hearing impairment in Ghana. Hum. Mutat. 2001, 18, 84–85. [Google Scholar] [CrossRef] [PubMed]
- Lasisi, A.O.; Bademci, G.; Foster, J.; Blanton, S.; Tekin, M. Common genes for non-syndromic deafness are uncommon in sub-Saharan Africa: A report from Nigeria. Int. J. Pediatric Otorhinolaryngol. 2014, 78, 1870–1873. [Google Scholar] [CrossRef][Green Version]
- Shan, J.; Chobot-Rodd, J.; Castellanos, R.; Babcock, M.; Shanske, A.; Parikh, S.R.; Morrow, B.E.; Samanich, J. GJB2 mutation spectrum in 209 hearing impaired individuals of predominantly Caribbean Hispanic and African descent. Int. J. Pediatr. Otorhinolaryngol. 2010, 74, 611–618. [Google Scholar] [CrossRef]
- Brobby, G.W.; Müller-Myhsok, B.; Horstmann, R.D. Connexin 26 R143W Mutation Associated with Recessive Nonsyndromic Sensorineural Deafness in Africa. N. Engl. J. Med. 1998, 338, 548–550. [Google Scholar] [CrossRef]
- Adadey, S.M.; Manyisa, N.; Mnika, K.; De Kock, C.; Nembaware, V.; Quaye, O.Q.; Amedofu, G.K.K.; Awandare, G.; Wonkam, A. GJB2 and GJB6 mutations in non-syndromic childhood hearing impairment in Ghana. Front. Genet. 2019, 10, 841. [Google Scholar] [CrossRef]
- Ross, S.A.; Novak, Z.; Kumbla, R.A.; Zhang, K.; Fowler, K.B.; Boppana, S. GJB2 and GJB6 mutations in children with congenital cytomegalovirus infection. Pediatr. Res. 2007, 61, 687–691. [Google Scholar] [CrossRef][Green Version]
- Shi, L.; Chen, J.; Li, J.; Wei, X.; Gao, X. Prevalence of GJB2 gene mutation in 330 cochlear implant patients in the Jiangsu province. J. Laryngol. Otol. 2016, 130, 902–906. [Google Scholar] [CrossRef]
- Rudman, J.R.; Kabahuma, R.I.; Bressler, S.E.; Feng, Y.; Blanton, S.H.; Yan, D.; Liu, X.-Z. The genetic basis of deafness in populations of African descent. J. Genet. Genom. 2017, 44, 285–294. [Google Scholar] [CrossRef]
- Feldmann, D.; Denoyelle, F.; Chauvin, P.; Garabédian, E.-N.; Couderc, R.; Odent, S.; Joannard, A.; Schmerber, S.; Delobel, B.; Leman, J.; et al. Large deletion of the GJB6 gene in deaf patients heterozygous for the GJB2 gene mutation: Genotypic and phenotypic analysis. Am. J. Med Genet. Part A 2004, 127A, 263–267. [Google Scholar] [CrossRef]
- Batissoco, A.C.; Abreu-Silva, R.S.; Braga, M.C.C.; Lezirovitz, K.; Della-Rosa, V.; Alfredo, T.; Otto, P.A.; Mingroni-Netto, R.C. Prevalence of GJB2 (connexin-26) and GJB6 (connexin-30) mutations in a cohort of 300 Brazilian hearing-impaired individuals: Implications for diagnosis and genetic counseling. Ear Hear. 2009, 30, 1–7. [Google Scholar] [CrossRef]
- Dragomir, C.; Stan, A.; Stefanescu, D.T.; Savu, L.; Severin, E. Prenatal screening for the 35delG GJB2, del (GJB6-D13S1830), and del (GJB6-D13S1854) mutations in the Romanian population. Genet. Test. Mol. Biomark. 2011, 15, 749–753. [Google Scholar] [CrossRef]
- Bhalla, S.; Sharma, R.; Khandelwal, G.; Panda, N.K.; Khullar, M. Absence of GJB6 mutations in Indian patients with non-syndromic hearing loss. Int. J. Pediatr. Otorhinolaryngol. 2011, 75, 356–359. [Google Scholar] [CrossRef]
- Naddafnia, H.; Noormohammadi, Z.; Irani, S.; Salahshoorifar, I. Frequency of GJB2 mutations, GJB6-D13S1830 and GJB6-D13S1854 deletions among patients with non-syndromic hearing loss from the central region of Iran. Mol. Genet. Genom. Med. 2019, 7, e00780. [Google Scholar]
- Amorini, M.; Romeo, P.; Bruno, R.; Galletti, F.; Di Bella, C.; Longo, P.; Briuglia, S.; Salpietro, C.; Rigoli, L. Prevalence of Deafness-Associated Connexin-26 (GJB2) and Connexin-30 (GJB6) Pathogenic Alleles in a Large Patient Cohort from Eastern Sicily. Ann. Hum. Genet. 2015, 79, 341–349. [Google Scholar] [CrossRef]
- Primignani, P.; Trotta, L.; Castorina, P.; Lalatta, F.; Sironi, F.; Radaelli, C.; Degiorgio, D.; Curcio, C.; Travi, M.; Ambrosetti, U.; et al. Analysis of the GJB2 and GJB6 genes in Italian patients with nonsyndromic hearing loss: Frequencies, novel mutations, genotypes, and degree of hearing loss. Genet. Test. Mol. Biomark. 2009, 13, 209–217. [Google Scholar] [CrossRef]
- Lebeko, K.; Sloan-Heggen, C.M.; Noubiap, J.J.N.; Dandara, C.; Kolbe, D.L.; Ephraim, S.S.; Booth, K.T.; Azaiez, H.; Santos-Cortez, R.L.P.; Leal, S.M.; et al. Targeted genomic enrichment and massively parallel sequencing identifies novel nonsyndromic hearing impairment pathogenic variants in Cameroonian families. Clin. Genet. 2016, 90, 288–290. [Google Scholar] [CrossRef]
- Hertzano, R.; Elkon, R. High throughput gene expression analysis of the inner ear. Hear. Res. 2012, 288, 77–88. [Google Scholar] [CrossRef]
- Noubiap, J.-J.; Djomou, F.; Njock, R.; Toure, G.B.; Wonkam, A. Waardenburg syndrome in childhood deafness in Cameroon. S. Afr. J. Child Health 2014, 8, 3–5. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).