Diversity Analysis of Sweet Potato Genetic Resources Using Morphological and Qualitative Traits and Molecular Markers





Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Molecular Analysis
2.2.1. Genomic DNA Isolation
2.2.2. SSR Genotyping
2.2.3. Marker Data Analysis
2.3. Morphological and Chemical Analyses
2.3.1. Extraction of Phenols for Analysis
2.3.2. Determination of TP Content by the FC Assay
2.3.3. Determination of Total Antioxidant Activity by FRAP
2.3.4. Quantitative Determination of Ions by IC and Organic Nitrogen
2.3.5. Brix Content
2.3.6. Starch
2.3.7. Quantitative Determination of Sugars by HPLC
2.4. Statistical Analysis
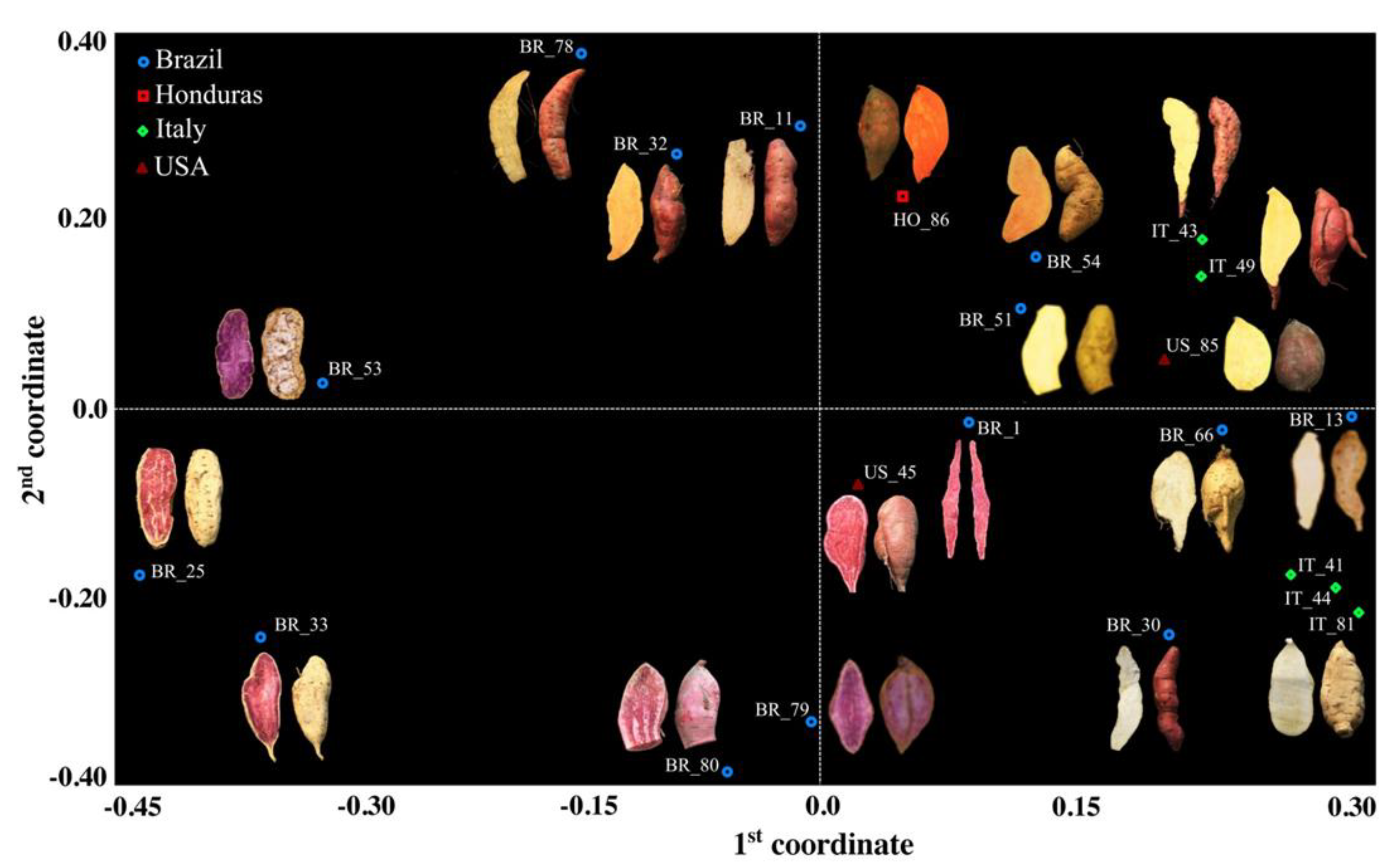
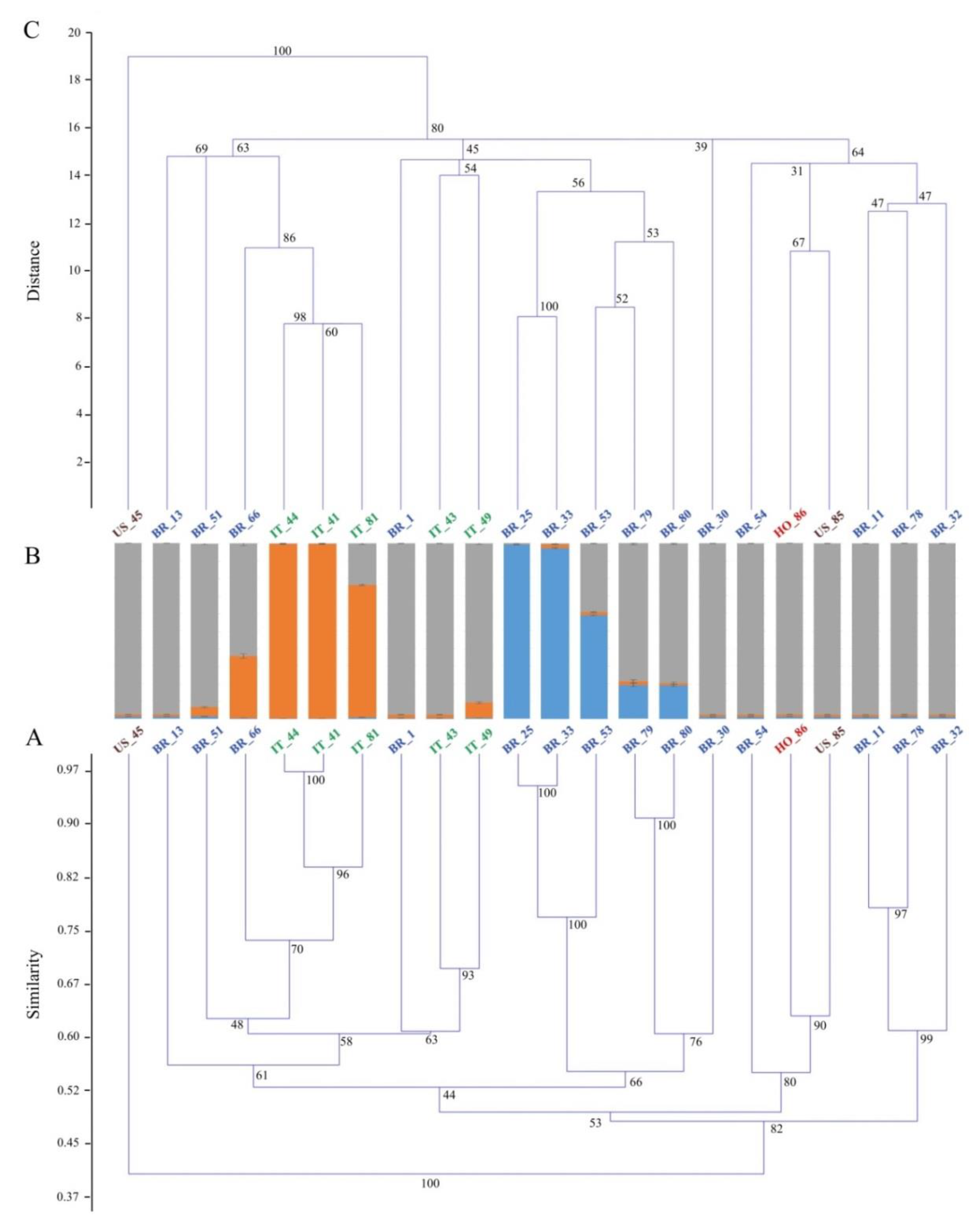
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Woolfe, J.A. Sweet Potato: An Untapped Food Resource; Cambridge University Press: Cambridge, UK, 1992; Volume 366–372. [Google Scholar]
- CBI Market Intelligence CBI Product Factsheet: Fresh Sweet Potatoes in Europe; CBI Market Intelligence: The Hague, The Netherlands, 2015.
- Nicoletto, C.; Vianello, F.; Sambo, P. Effect of different home-cooking methods on textural and nutritional properties of sweet potato genotypes grown in temperate climate conditions. J. Sci. Food Agric. 2018, 98, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Veasey, E.A.; Borges, A.; Rosa, M.S.; Queiroz-Silva, J.R.; Bressan, E.D.A.; Peroni, N. Genetic diversity in Brazilian sweet potato (Ipomoea batatas (L.) Lam., Solanales, Convolvulaceae) landraces assessed with microsatellite markers. Genet. Mol. Biol. 2008, 31, 725–733. [Google Scholar] [CrossRef]
- Roullier, C.; Rossel, G.; Tay, D.; McKey, D.; Lebot, V. Combining chloroplast and nuclear microsatellites to investigate origin and dispersal of New World sweet potato landraces. Mol. Ecol. 2011, 20, 3963–3977. [Google Scholar] [CrossRef] [PubMed]
- Roullier, C.; Duputié, A.; Wennekes, P.; Benoit, L.; Fernández Bringas, V.M.; Rossel, G.; Tay, D.; McKey, D.; Lebot, V. Disentangling the Origins of Cultivated Sweet Potato (Ipomoea batatas (L.) Lam.). PLoS ONE 2013, 8, e62707. [Google Scholar] [CrossRef]
- Yang, X.S.; Su, W.J.; Wang, L.J.; Lei, J.; Chai, S.S.; Liu, Q. chang Molecular diversity and genetic structure of 380 sweetpotato accessions as revealed by SSR markers. J. Integr. Agric. 2015, 14, 633–641. [Google Scholar] [CrossRef]
- Wadl, P.A.; Olukolu, B.A.; Branham, S.E.; Jarret, R.L.; Yencho, G.C.; Jackson, D.M. Genetic Diversity and Population Structure of the USDA Sweetpotato (Ipomoea batatas) Germplasm Collections Using GBSpoly. Front. Plant Sci. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Austin, D. The taxonomy, evolution and genetic diversity of sweet potatoes and related wild species. In Exploration, Maintenance and Utilization of Sweet Potato Genetic Resources: Report of the First Sweet Potato Planning Conference; International Potato Center (CIP): Lima, Perù, 1987. [Google Scholar]
- Wang, Z.; Li, J.; Luo, Z.; Huang, L.; Chen, X.; Fang, B.; Li, Y.; Chen, J.; Zhang, X. Characterization and development of EST-derived SSR markers in cultivated sweetpotato (Ipomoea batatas). BMC Plant Biol. 2011, 11. [Google Scholar] [CrossRef]
- Gao, M.; Ashu, G.M.; Stewart, L.; Akwe, W.A.; Njiti, V.; Barnes, S. Wx intron variations support an allohexaploid origin of the sweetpotato [Ipomoea batatas (L.) Lam]. Euphytica 2011, 177, 111–133. [Google Scholar] [CrossRef]
- Magoon, M.; Krishnan, R.; Vijaya Bai, K. Cytological evidence on the origin of sweet potato. Theor. Appl. Genet. 1970, 40, 360–366. [Google Scholar] [CrossRef]
- Soltis, D.E.; Soltis, P.S. Polyploidy: Recurrent formation and genome evolution. Trends Ecol. Evol. 1999, 14, 348–352. [Google Scholar] [CrossRef]
- Silva Ritschel, P.; Huamán, Z. Variabilidade morfologica da coleçao de germoplasma de batata doce da Embrapa Centro Nacional de Pesquisas de Hortaliças. Pesqui. Agropecu. Bras. 2002, 37, 485–492. [Google Scholar] [CrossRef]
- Palumbo, F.; Galla, G.; Martinez-Bello, L.; Barcaccia, G. Venetian Local Corn (Zea mays L.) Germplasm: Disclosing the Genetic Anatomy of Old Landraces Suited for Typical Cornmeal Mush Production. Diversity 2017, 9, 32. [Google Scholar] [CrossRef]
- Cruz, C.D.; Carneiro, P.C.S.; Regazzi, A.J. Modelos Biométricos Aplicados ao Melhoramento Genético—Volume 1, 3rd ed.; Editora UFV: Viçosa, Brazil, 2012; ISBN 9788572694339. [Google Scholar]
- Palumbo, F.; Galla, G.; Barcaccia, G. Developing a molecular identification assay of old landraces for the genetic authentication of typical agro-food products: The case study of the barley “agordino”. Food Technol. Biotechnol. 2017, 55, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Crinò, P.; Pagnotta, M.A. Phenotyping, genotyping, and selections within italian local landraces of romanesco globe artichoke. Diversity 2017, 9, 14. [Google Scholar] [CrossRef]
- Mercati, F.; Longo, C.; Poma, D.; Araniti, F.; Lupini, A.; Mammano, M.M.; Fiore, M.C.; Abenavoli, M.R.; Sunseri, F. Genetic variation of an Italian long shelf-life tomato (Solanum lycopersicon L.) collection by using SSR and morphological fruit traits. Genet. Resour. Crop Evol. 2014, 62, 721–732. [Google Scholar] [CrossRef]
- Andreakis, N.; Kooistra, W.H.C.F.; Procaccini, G. High genetic diversity and connectivity in the polyploid invasive seaweed Asparagopsis taxiformis (Bonnemaisoniales) in the Mediterranean, explored with microsatellite alleles and multilocus genotypes. Mol. Ecol. 2009, 18, 212–226. [Google Scholar] [CrossRef]
- Cordeiro, G.M.; Pan, Y.B.; Henry, R.J. Sugarcane microsatellites for the assessment of genetic diversity in sugarcane germplasm. Plant Sci. 2003, 165, 181–189. [Google Scholar] [CrossRef]
- Pinto, L.R.; Oliveira, K.M.; Marconi, T.; Garcia, A.A.F.; Ulian, E.C.; De Souza, A.P. Characterization of novel sugarcane expressed sequence tag microsatellites and their comparison with genomic SSRs. Plant Breed. 2006, 125, 378–384. [Google Scholar] [CrossRef]
- Babaei, A.; Tabaei-Aghdaei, S.R.; Khosh-Khui, M.; Omidbaigi, R.; Naghavi, M.R.; Esselink, G.D.; Smulders, M.J.M. Microsatellite analysis of Damask rose (Rosa damascena Mill.) accessions from various regions in Iran reveals multiple genotypes. BMC Plant Biol. 2007, 7, 1–6. [Google Scholar] [CrossRef]
- Veasey, E.A.; Silva, J.R.D.Q.; Rosa, M.S.; Borges, A.; Bressan, E.D.A.; Peroni, N. Phenology and morphological diversity of sweet potato (Ipomoea batatas) landraces of the Vale do Ribeira. Sci. Agric. 2007, 64, 416–427. [Google Scholar] [CrossRef]
- Yada, B.; Tukamuhabwa, P.; Alajo, A.; Mwanga, R.O.M. Morphological characterization of Ugandan sweetpotato germplasm. Crop Sci. 2010, 50, 2364–2371. [Google Scholar] [CrossRef]
- Elameen, A.; Larsen, A.; Klemsdal, S.S.; Fjellheim, S.; Sundheim, L.; Msolla, S.; Masumba, E.; Rognli, O.A. Phenotypic diversity of plant morphological and root descriptor traits within a sweet potato, Ipomoea batatas (L.) Lam., germplasm collection from Tanzania. Genet. Resour. Crop Evol. 2011, 58, 397–407. [Google Scholar] [CrossRef]
- Karuri, H.; Ateka, E.; Amata, R.; Nyende, A.; Muigai, A.; Mwasame, E.; Gichuki, S. Evaluating diversity among Kenyan sweet potato genotypes using morphological and SSR markers. Int. J. Agric. Biol. 2010, 12, 33–38. [Google Scholar]
- Som, K.; Vernon, G.; Isaac, A.; Eric, Y.D.; Jeremy, T.O.; Tignegre, J.B.; Belem, J.; Tarpaga, M.V. Diversity analysis of sweet potato (Ipomoea batatas [L.] Lam) germplasm from Burkina Faso using morphological and simple sequence repeats markers. Afr. J. Biotechnol. 2014, 13, 729–742. [Google Scholar] [CrossRef]
- Perelli, M.; Graziano, P.L.; Calzavara, R. Nutrire le Piante; ARVAN: Venice, Italy, 2009. [Google Scholar]
- Hu, J.; Nakatani, M.; Mizuno, K.; Fujimura, T. Development and Characterization of Microsatellite Markers in Sweetpotato. Breed. Sci. 2004, 54, 177–188. [Google Scholar] [CrossRef]
- Schuelke, M. An economic method for the fluorescent labeling of PCR fragments. Nat. Biotechnol. 2000, 18, 233. [Google Scholar] [CrossRef]
- Palumbo, F.; Galla, G.; Vitulo, N.; Barcaccia, G. First draft genome sequencing of fennel (Foeniculum Vulgare Mill.): Identification of simple sequence repeats and their application in marker-assisted breeding. Mol. Breed. 2018, 38, 1–17. [Google Scholar] [CrossRef]
- Botstein, D.; White, R.L.; Skolnick, M.; Davis, R.W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 1980, 32, 314–331. [Google Scholar]
- Dice, L.R. Measures of the Amount of Ecologic Association Between Species. Ecology 1945, 26, 297–302. [Google Scholar] [CrossRef]
- Rohlf, F.J. NTSYS-pc: Numerical Taxonomy and Multivariate Analysis System; Applied Biostatistics Inc.: Setauket, NY, USA, 2008. [Google Scholar]
- Hammer, Ø.; Harper, D.A.; Ryan, P.D. PAST: Paleontological Statistics Software Package for Education and Data Analysis. Palaeontol. Electron. 2001, 4, 9. [Google Scholar]
- Peakall, R.; Smouse, P.E. GenALEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar] [PubMed]
- Porras-Hurtado, L.; Ruiz, Y.; Santos, C.; Phillips, C.; Carracedo, Á.; Lareu, M.V. An overview of STRUCTURE: Applications, parameter settings, and supporting software. Front. Genet. 2013, 4, 1–13. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- IBPGR Descriptors for Sweet Potato; Huamán, Z., Ed.; International Board for Plant Genetic Resources: Rome, Italy, 1991. [Google Scholar]
- Singleton, V.L.; Orthofer, R.; Lamuela-Raventós, R.M. Analysis of total phenols and other oxidation substrates and antioxidants by means of folin-ciocalteu reagent. Methods Enzymol. 1999, 299, 152–178. [Google Scholar]
- Benzie, I.F.F.; Strain, J.J. The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: The FRAP assay. Anal. Biochem. 1996, 239, 70–76. [Google Scholar] [CrossRef]
- Roullier, C.; Benoit, L.; McKey, D.B.; Lebot, V. Historical collections reveal patterns of diffusion of sweet potato in Oceania obscured by modern plant movements and recombination. Proc. Natl. Acad. Sci. USA 2013, 110, 2205–2210. [Google Scholar] [CrossRef]
- Nicolle, C.; Simon, G.; Rock, E.; Amouroux, P.; Rémésy, C. Genetic Variability Influences Carotenoid, Vitamin, Phenolic, and Mineral Content in White, Yellow, Purple, Orange, and Dark-orange Carrot Cultivars. J. Am. Soc. Hortic. Sci. 2004, 129, 523–529. [Google Scholar] [CrossRef]
- Tsegaye, E.; Dechassa, N.; Sastry, D.E.V. Genetic Variability for Yield and other agronomic traits in sweet potato. J. Agron. 2007, 6, 94–99. [Google Scholar]
- Solankey, S.S.; Singh, P.K.; Singh, R.K. Genetic Diversity and Interrelationship of Qualitative and Quantitative Traits in Sweet Potato. Int. J. Veg. Sci. 2015, 21, 236–248. [Google Scholar] [CrossRef]
- Lim, S.; Xu, J.; Kim, J.; Chen, T.-Y.; Su, X.; Standard, J.; Carey, E.; Griffin, J.; Herndon, B.; Katz, B.; et al. Role of Anthocyanin-enriched Purple-fleshed Sweet Potato P40 in Colorectal Cancer Prevention. Mol. Nutr. Food Res. 2013, 57, 1908–1917. [Google Scholar] [CrossRef] [PubMed]
- Grace, M.H.; Yousef, G.G.; Gustafson, S.J.; Truong, V.-D.; Yencho, G.C.; Lila, M.A. Phytochemical changes in phenolics, anthocyanins, ascorbic acid, and carotenoids associated with sweetpotato storage and impacts on bioactive properties. Food Chem. 2014, 145, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Park, J.S.; Choi, D.S.; Jung, M.Y. Characterization and quantitation of anthocyanins in purple-fleshed sweet potatoes cultivated in Korea by HPLC-DAD and HPLC-ESI-QTOF-MS/MS. J. Agric. Food Chem. 2013, 61, 3148–3158. [Google Scholar] [CrossRef] [PubMed]
- Gorusupudi, A.; Bernstein, P.S. Macular Carotenoids: Human Health Aspects. In Carotenoids: Nutrition, Analysis, and Technology; Kaczor, A., Baranska, M., Eds.; Wiley Blackwell: Pondicherry, India, 2016; pp. 59–74. [Google Scholar]
- Park, S.Y.; Lee, S.Y.; Yang, J.W.; Lee, J.S.; Oh, S.D.; Oh, S.; Lee, S.M.; Lim, M.H.; Park, S.K.; Jang, J.S.; et al. Comparative analysis of phytochemicals and polar metabolites from colored sweet potato (Ipomoea batatas L.) tubers. Food Sci. Biotechnol. 2016, 25, 283–291. [Google Scholar] [CrossRef]
- Simonne, A.H.; Kays, S.J.; Koehler, P.E.; Eitenmiller, R.R. Assessment of β-Carotene content in sweetpotato breeding lines in relation to dietary requirements. J. Food Compos. Anal. 1993, 6, 336–345. [Google Scholar] [CrossRef]
- Wei, S.; Lu, G.; Cao, H. Effects of cooking methods on starch and sugar composition of sweetpotato storage roots. PLoS ONE 2017, 12, e0182604. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Genetic Material | Plant Type | Country of Origin | Flesh Color | Skin Color | Root Shape |
|---|---|---|---|---|---|
| BR_1 | Extremely spreading | Brazil | Purple | Dark purple | Elliptical |
| BR_11 | Spreading | Brazil | Cream | Pink | Round elliptical |
| BR_13 | Extremely spreading | Brazil | White | Cream | Elliptical |
| BR_25 | Semi-erect | Brazil | Purple | Cream | Long oblong |
| BR_30 | Spreading | Brazil | White | Pink | Long irregular |
| BR_32 | Semi-erect | Brazil | Pale orange | Pink | Oblong |
| BR_33 | Semi-erect | Brazil | Purple | Cream | Oblong |
| BR_51 | Extremely spreading | Brazil | White | Cream | Long elliptical |
| BR_53 | n.a. | Brazil | Purple | White | Oblong |
| BR_54 | Extremely spreading | Brazil | Intermediate orange | Yellow | Elliptical |
| BR_66 | n.a. | Brazil | White | White | Irregular |
| BR_78 | Semi-erect | Brazil | Cream | Pink | Long irregular |
| BR_79 | Spreading | Brazil | Purple | Pink | Obovate |
| BR_80 | n.a. | Brazil | Purple | Dark purple | Obovate |
| IT_41 | Semi-erect | Italy | Cream | Cream | Long irregular |
| IT_43 | Spreading | Italy | Pale yellow | Pink | Obovate |
| IT_44 | Semi-erect | Italy | White | Cream | Elliptical |
| IT_49 | Semi-erect | Italy | Pale yellow | Pink | Round elliptical |
| alIT_81 | Erect | Italy | Cream | Cream | Obovate |
| US_45 | Semi-erect | USA | Purple | Dark purple | Long oblong |
| US_85 | n.a. | USA | Pale yellow | Cream | Round elliptical |
| HO_86 | n.a. | Honduras | Deep Orange | Purple red | Round elliptical |
| Locus Name | Tm (°C) * | Primer Sequence (5’–3’) | SSR Motif | Alleles | Length (bp) | Type | Multiple × | Source |
|---|---|---|---|---|---|---|---|---|
| IBSSR04 | 62 | GAGGTAGTTATTGTGGAGGACCTCCTTTGCCTCCTTTCATGC | (GA)11 | 7 | 216 | nSSR | 1 | [30] |
| 62 | CCTTGCTCCCCATTTTCTTCTTG | |||||||
| J263 | 62 | GGAATTAACCGCTCACTAAAGCTCTGCTTCTCCTGCTGCTT | (AAC)6 | 7 | 156–171 | nSSR | 1 | [5] |
| 61 | GTGCGGCACTTGTCTTTGATA | |||||||
| J544b | 61 | TTGTAAAACGACGGCCAGTAGCAGTTGAGGAAAGCAAGG | (TCT)6 | 8 | 174–194 | nSSR | 1 | [5] |
| 59 | CAGGATTTACAGCCCCAGAA | |||||||
| Ib318 | 60 | GAGGTAGTTATTGTGGAGGACAGAACGCATGGGCATTGA | n.a. | 5 | 125-135 | nSSR | 1 | [4] |
| 60 | CCCACCGTGTAAGGAAATCA | |||||||
| Ib-255F1 | 61 | GGAATTAACCGCTCACTAAAGCGTCCATGCTAAAGGTGTCAA | n.a. | 8 | 210-245 | nSSR | 2 | [4] |
| 59 | ATAGGGGATTGTGCGTAATTTG | |||||||
| Ib297 | 59 | GAGGTAGTTATTGTGGAGGACGCAATTTCACACACAAACACG | (CT)13 | 24 | 129–167 | nSSR | 2 | [5] |
| 60 | CCCTTCTTCCACCACTTTCA | |||||||
| Ib286 | 62 | TTGTAAAACGACGGCCAGTAGCCACTCCAACAGCACATA | n.a. | 10 | 90–122 | nSSR | 2 | [4] |
| 57 | GGTTTCCCAATCAGCAATTC | |||||||
| IbS11 | 58 | GGAATTAACCGCTCACTAAAGCCCTGCGAAATCGAAATCT | (TTC)10 | 13 | 218–248 | nSSR | 3 | [5] |
| 61 | GGACTTCCTCTGCCTTGTTG | |||||||
| J116a | 57 | GAGGTAGTTATTGTGGAGGACTCTTTTGCATCAAAGAAATCCA | (CCT)7 | 15 | 187–227 | nSSR | 3 | [5] |
| 60 | CCTCAGCTTCTGGGAAACAG | |||||||
| J206A | 59 | TTGTAAAACGACGGCCAGTATCAGGGAGAGAGGACAGTAA | (GAT)6 | 9 | 103–121 | nSSR | 3 | [5] |
| 57 | TAGGCAAACCATAAACAGAGA | |||||||
| GDAAS0615 | 56 | TGTAGAAAGACGAAGGGAAGGCCACATACAGACTACAACTTAC | (GA)10 | 7 | 230 | EST-SSR | 4 | [10] |
| 57 | GGAGGAGCGTATTATGAACA | |||||||
| GDAAS0757 | 56 | TTGTAAAACGACGGCCAGTGAGATGATGACGATAGTGTTG | (GAA)11 | 9 | 293 | EST-SSR | 4 | [10] |
| 56 | GGAAGATTCATTGGCAGAAG | |||||||
| IBSSR27 | 56 | GGAATTAACCGCTCACTAAAGGTGTTTATCACATCGTTTTCTG | (TA)6(CA)16 | 9 | 149 | nSSR | 4 | [30] |
| 55 | GGCTCGTACAATTTTCAAAG | |||||||
| GDAAS0156 | 54 | GAGGTAGTTATTGTGGAGGACTCCAAATACCATACCCAAC | (TC)10 | 8 | 118 | EST-SSR | 4 | [10] |
| 55 | CGCTTTCAAATAGAATCGTC |
| IBSSR04 | J544b | Ib-255F1 | Ib297 | Ib286 | ibS11 | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Size | F.o. | F.f. | F.i. | PIC | Size | F.o. | F.f. | F.i. | PIC | Size | F.o. | F.f. | F.i. | PIC | Size | F.o. | F.f. | F.i. | PIC | Size | F.o. | F.f. | F.i. | PIC | Size | F.o. | F.f. | F.i. | PIC |
| 216 | 9% | 12% | 0% | 0.75 | 184 | 91% | 88% | 100% | 0.63 | 233 | 18% | 24% | 0% | 0.87 | 137 | 9% | 6% | 20% | 0.87 | 97 | 5% | 6% | 0% | 0.77 | 224 | 9% | 12% | 0% | 0.87 |
| 218 | 18% | 24% | 0% | 178 | 18% | 24% | 0% | 237 | 23% | 24% | 20% | 147 | 5% | 6% | 0% | 99 | 5% | 6% | 0% | 227 | 55% | 41% | 100% | ||||||
| 220 | 59% | 59% | 60% | 189 | 18% | 24% | 0% | 239 | 5% | 6% | 0% | 149 | 59% | 53% | 80% | 101 | 18% | 24% | 0% | 230 | 18% | 24% | 0% | ||||||
| 222 | 45% | 65% | 0% | 192 | 18% | 24% | 0% | 243 | 5% | 6% | 0% | 151 | 14% | 18% | 0% | 105 | 32% | 41% | 0% | 233 | 9% | 6% | 20% | ||||||
| 224 | 18% | 12% | 40% | 195 | 59% | 47% | 100% | 251 | 50% | 53% | 40% | 155 | 64% | 71% | 40% | 107 | 14% | 18% | 0% | 236 | 55% | 53% | 60% | ||||||
| 226 | 68% | 71% | 60% | 198 | 82% | 76% | 100% | 253 | 45% | 53% | 20% | 159 | 14% | 18% | 0% | 109 | 100% | 100% | 100% | 239 | 32% | 29% | 40% | ||||||
| 228 | 50% | 47% | 60% | 209 | 82% | 76% | 100% | 255 | 73% | 65% | 100% | 161 | 55% | 41% | 100% | 113 | 77% | 71% | 100% | 242 | 27% | 18% | 60% | ||||||
| 230 | 82% | 76% | 100% | 257 | 14% | 18% | 0% | 163 | 64% | 65% | 60% | 115 | 45% | 41% | 60% | 245 | 64% | 71% | 40% | ||||||||||
| 259 | 64% | 53% | 100% | 167 | 27% | 35% | 0% | 121 | 59% | 71% | 20% | 248 | 41% | 53% | 0% | ||||||||||||||
| 261 | 9% | 12% | 0% | 169 | 5% | 0% | 20% | 123 | 9% | 12% | 0% | 251 | 18% | 24% | 0% | ||||||||||||||
| 263 | 32% | 41% | 0% | 171 | 9% | 0% | 40% | 254 | 59% | 59% | 60% | ||||||||||||||||||
| 265 | 27% | 35% | 0% | 175 | 5% | 6% | 0% | 257 | 9% | 12% | 0% | ||||||||||||||||||
| 267 | 9% | 0% | 40% | 177 | 27% | 35% | 0% | 260 | 14% | 18% | 0% | ||||||||||||||||||
| 187 | 32% | 41% | 0% | 263 | 9% | 12% | 0% | ||||||||||||||||||||||
| J116a | J206A | GDAAS0615 | GDAAS0757 | IBSSR27 | |||||||||||||||||||||||||
| Size | F.o. | F.f. | F.i. | PIC | Size | F.o. | F.f. | F.i. | PIC | Size | F.o. | F.f. | F.i. | PIC | Size | F.o. | F.f. | F.i. | PIC | Size | F.o. | F.f. | F.i. | PIC | |||||
| 194 | 23% | 29% | 0% | 0.69 | 118 | 82% | 76% | 100% | 0.61 | 214 | 36% | 29% | 60% | 0.93 | 272 | 18% | 24% | 0% | 0.89 | 139 | 24% | 29% | 0% | 0.88 | |||||
| 200 | 77% | 71% | 100% | 124 | 55% | 59% | 40% | 220 | 14% | 18% | 0% | 281 | 23% | 12% | 60% | 155 | 5% | 6% | 0% | ||||||||||
| 203 | 14% | 12% | 20% | 127 | 100% | 100% | 100% | 226 | 9% | 12% | 0% | 284 | 18% | 24% | 0% | 161 | 14% | 0% | 60% | ||||||||||
| 206 | 91% | 88% | 100% | 130 | 5% | 6% | 0% | 230 | 68% | 76% | 60% | 290 | 9% | 12% | 0% | 163 | 10% | 12% | 0% | ||||||||||
| 209 | 32% | 41% | 0% | 133 | 59% | 53% | 80% | 232 | 5% | 6% | 0% | 293 | 14% | 6% | 40% | 165 | 95% | 88% | 100% | ||||||||||
| 212 | 55% | 47% | 80% | 136 | 9% | 12% | 0% | 234 | 9% | 12% | 0% | 296 | 41% | 53% | 0% | 167 | 10% | 12% | 0% | ||||||||||
| 215 | 82% | 76% | 100% | 236 | 23% | 24% | 20% | 299 | 5% | 6% | 0% | 169 | 5% | 6% | 0% | ||||||||||||||
| 218 | 9% | 12% | 0% | 246 | 9% | 12% | 0% | 302 | 50% | 41% | 80% | 171 | 5% | 0% | 20% | ||||||||||||||
| 221 | 50% | 65% | 0% | 248 | 14% | 18% | 0% | 305 | 27% | 35% | 0% | 177 | 29% | 29% | 20% | ||||||||||||||
| 250 | 18% | 18% | 20% | 308 | 5% | 6% | 0% | ||||||||||||||||||||||
| 256 | 5% | 0% | 20% | 311 | 14% | 12% | 20% | ||||||||||||||||||||||
| 314 | 36% | 29% | 60% | ||||||||||||||||||||||||||
| 317 | 95% | 100% | 80% | ||||||||||||||||||||||||||
| 320 | 9% | 12% | 0% | ||||||||||||||||||||||||||
| 323 | 27% | 24% | 40% | ||||||||||||||||||||||||||
| 326 | 18% | 6% | 60% | ||||||||||||||||||||||||||
| Genotype | K (mg/kg dw) | Mg (mg/kg dw) | Ca (mg/kg dw) | Total Soluble Solids (°Brix) | TP (mg GAE kg−1 fw) | TAA (mg Fe2+E kg−1 fw) | β-Carotene (mg/kg fw) | Vit C (mg/kg dw) | Sucrose (mg/kg dw) | Glucose (mg/k dw) | Fructose (mg/kg dw) | Starch (%) | Dry Matter (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| US_45 | 7381 klm | 2426 bcde | 1810 abc | 9 bc | 6442 a | 6706 a | n.d. | 658 c | 36430 l | 35280 cd | 33561 b | 81 a | 36 ab |
| BR_79 | 12151 b | 1841 def | 2481 ab | 12 a | 2875 bc | 2563 cd | n.d. | 2967 ab | 109542 defg | 12903 jklm | 11357 fghi | 71 ab | 37 ab |
| BR_80 | 5907 n | 2181 cde | 2405 ab | 11 ab | 2369 bc | 2599 cd | n.d. | 3966 ab | 93156 fghi | 9487 klmn | 8501 fghi | 74 ab | 43 a |
| BR_33 | 6828 lm | 1460 ef | 1582 c | 8 c | 1753 c | 2095 cd | n.d. | 3887 ab | 37464 l | 26310 de | 29451 bcd | 83 a | 36 ab |
| BR_25 | 7074 lm | 1580 ef | 2104 ab | 9 bc | 3240 bc | 3578 bc | n.d. | 2988 ab | 55692 kl | 31784 cd | 31623 bc | 72 ab | 36 ab |
| BR_53 | 8965 g | 1732 def | 2308 ab | 11 ab | 3358 bc | 2869 cd | n.d. | 1933 bc | 116776 cde | 21199 efg | 22460 de | 70 b | 34 b |
| BR_13 | 7328 klm | 2640 abc | 1981 abc | 9 bc | 1839 c | 2210 cd | n.d. | 2950 b | 106360 def | 8561 lmn | 6028 hij | 65 bc | 32 b |
| BR_1 | 5316 no | 1484 ef | 1908 abc | 11 ab | 4425 ab | 4651 b | n.d. | 4682 a | 81680 hij | 21667 efg | 15816 ef | 69 b | 36 ab |
| BR_66 | 10647 de | 2270 bcd | 1795 bc | 13 a | 2078 bc | 2156 cd | 23.8 | 2305 b | 149539 a | 6364 mn | 6803 ij | 64 bc | 42 a |
| IT_44 | 8045 ij | 2658 abc | 1922 abc | 9 bc | 875 c | 834 e | n.d. | 1690 bc | 109205 def | 4396 lmn | 3763 j | 79 ab | 37 ab |
| IT_41 | 10626 de | 2156 cde | 1709 bc | 10 b | 1595 c | 1335 de | n.d. | 2112 b | 99035 fg | 8673 lmn | 7189 hij | 83 a | 37 ab |
| IT_81 | 12176 b | 2697 abc | 1925 abc | 10 b | 1067 c | 862 e | n.d. | 2544 b | 124873 bcd | 5007 n | 3671 j | 72 ab | 36 ab |
| BR_51 | 9403 g | 2056 bcd | 1816 abc | 9 bc | 1186 c | 2019 cd | 34.2 | 3856 ab | 113505 def | 46730 a | 46568 a | 70 b | 30 b |
| IT_43 | 15129 a | 1729 cde | 1954 abc | 11 ab | 2085 bc | 1906 de | n.d. | 1867 bc | 144240 ab | 12253 ijklm | 10429 fgh | 75 ab | 34 b |
| IT_49 | 7855 ijk | 2302 bcd | 1563 c | 10 b | 1453 c | 1607 de | 208 | 1389 bc | 92224 ghi | 36301 bc | 24108 cd | 80 a | 30 b |
| BR_30 | 6766 lm | 2118 cde | 1848 bc | 8 c | 1657 c | 1743 de | 90.8 | 1427 bc | 68226 jk | 45548 a | 33478 b | 70 b | 31 b |
| BR_54 | 6666 m | 2619 abc | 2624 a | 9 bc | 1317 c | 1577 de | 571 | 2285 b | 138292 bc | 13282 hijk | 10709 fgh | 78 ab | 37 ab |
| HO_86 | 11914 bc | 2192 bcd | 2282 ab | 9 bc | 1189 c | 1179 de | 512 | 2477 b | 95520 fgh | 42100 ab | 45446 a | 52 c | 36 ab |
| US_85 | 8970 g | 2023 cde | 2264 ab | 8 c | 720 c | 799 e | n.d. | 1595 c | 69641 jk | 25645 def | 30837 bc | 60 c | 36 ab |
| BR_32 | 8096 hi | 1645 def | 2298 ab | 11 ab | 1551 c | 1516 de | 811 | 4709 a | 138295 bc | 17282 ghij | 14933 efg | 72 ab | 32 b |
| BR_11 | 10431 f | 1452 f | 1763 bc | 9 bc | 2477 bc | 1073 e | n.d. | 3087 ab | 88841 ghi | 19294 efgh | 14664 efg | 72 ab | 35 b |
| BR_78 | 6850 lm | 2352 bcd | 1808 bc | 10 b | 2119 bc | 1809 de | n.d. | 1256 c | 100268 efg | 17742 fghi | 13919 fg | 70 b | 42 a |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palumbo, F.; Galvao, A.C.; Nicoletto, C.; Sambo, P.; Barcaccia, G. Diversity Analysis of Sweet Potato Genetic Resources Using Morphological and Qualitative Traits and Molecular Markers. Genes 2019, 10, 840. https://doi.org/10.3390/genes10110840
Palumbo F, Galvao AC, Nicoletto C, Sambo P, Barcaccia G. Diversity Analysis of Sweet Potato Genetic Resources Using Morphological and Qualitative Traits and Molecular Markers. Genes. 2019; 10(11):840. https://doi.org/10.3390/genes10110840
Chicago/Turabian StylePalumbo, Fabio, Aline Carolina Galvao, Carlo Nicoletto, Paolo Sambo, and Gianni Barcaccia. 2019. "Diversity Analysis of Sweet Potato Genetic Resources Using Morphological and Qualitative Traits and Molecular Markers" Genes 10, no. 11: 840. https://doi.org/10.3390/genes10110840
APA StylePalumbo, F., Galvao, A. C., Nicoletto, C., Sambo, P., & Barcaccia, G. (2019). Diversity Analysis of Sweet Potato Genetic Resources Using Morphological and Qualitative Traits and Molecular Markers. Genes, 10(11), 840. https://doi.org/10.3390/genes10110840