Deregulated Adhesion Program in Palatal Keratinocytes of Orofacial Cleft Patients

 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Subjects and Tissue Sampling
2.2. Cell Culture
2.3. RNA Isolation and Microarray Expression Analysis
2.4. Reverse Transcription and Real-Time Quantitative PCR
2.5. Immunostaining of Tissue Samples
2.6. Scratch Assay
3. Results
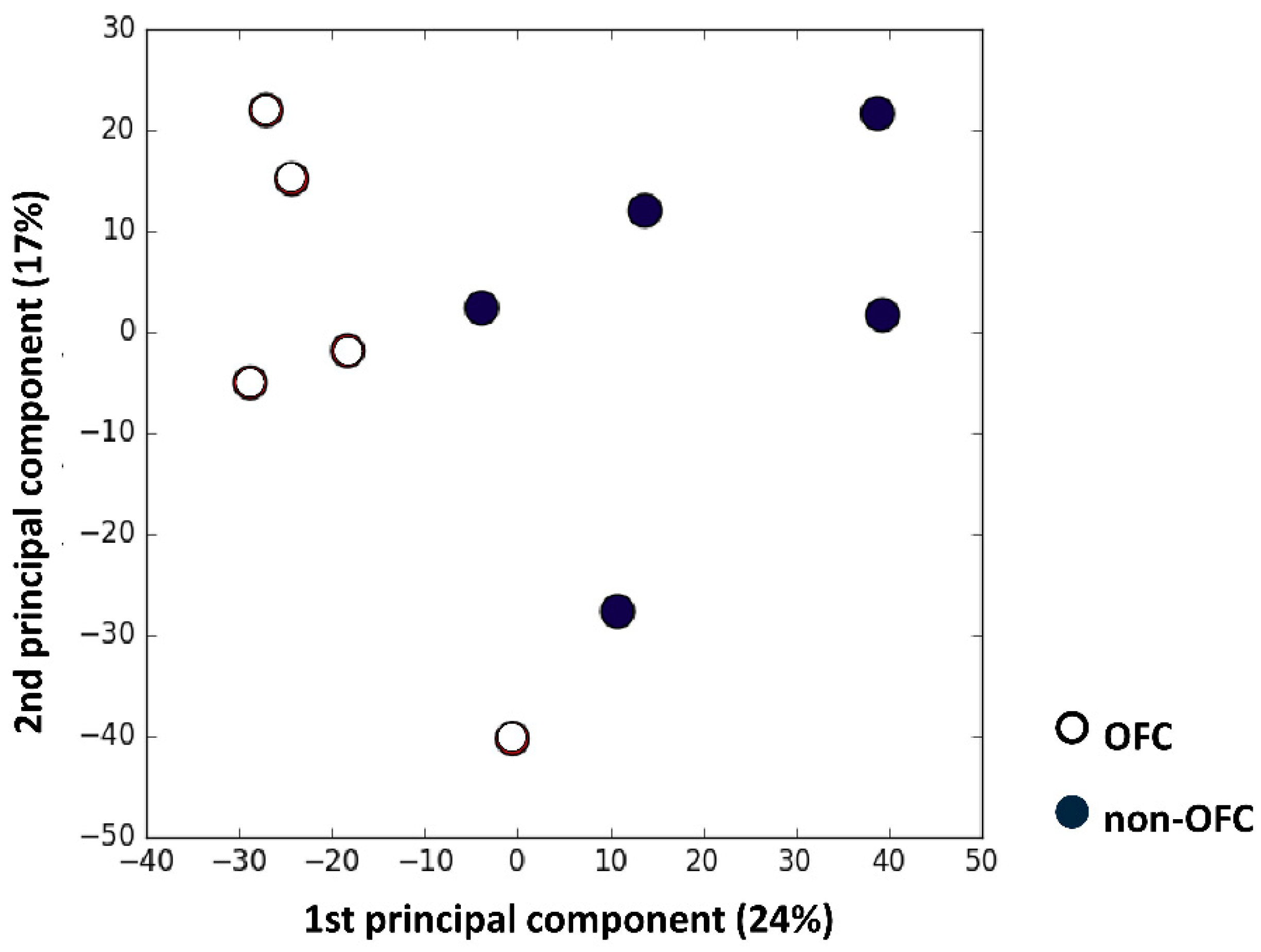
3.1. Microarray Analysis
3.2. Gene Ontology Analysis
3.3. Validation of the Prioritized Genes
3.4. Immunostaining of Tissue Samples
3.5. Functional Validation: Scratch Assay
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
References
- Tanaka, S.A.; Mahabir, R.C.; Jupiter, D.C.; Menezes, J.M. Updating the epidemiology of cleft lip with or without cleft palate. Plast. Reconstr. Surg. 2012, 129, 511e–518e. [Google Scholar] [CrossRef]
- Dixon, M.J.; Marazita, M.L.; Beaty, T.H.; Murray, J.C. Cleft lip and palate: Understanding genetic and environmental influences. Nat. Rev. Genet. 2011, 12, 167–178. [Google Scholar] [CrossRef]
- Lan, Y.; Xu, J.; Jiang, R. Cellular and molecular mechanisms of palatogenesis. Curr. Top. Dev. Biol. 2015, 115, 59–84. [Google Scholar]
- Chai, Y.; Maxson, R.E., Jr. Recent advances in craniofacial morphogenesis. Dev. Dyn. 2006, 235, 2353–2375. [Google Scholar] [CrossRef]
- Richardson, R.J.; Hammond, N.L.; Coulombe, P.A.; Saloranta, C.; Nousiainen, H.O.; Salonen, R.; Berry, A.; Hanley, N.; Headon, D.; Karikoski, R.; et al. Periderm prevents pathological epithelial adhesions during embryogenesis. J. Clin. Investig. 2014, 124, 3891–3900. [Google Scholar] [CrossRef]
- Hu, L.; Liu, J.; Li, Z.; Ozturk, F.; Gurumurthy, C.; Romano, R.A.; Sinha, S.; Nawshad, A. TGFbeta3 regulates periderm removal through DeltaNp63 in the developing palate. J. Cell. Physiol. 2015, 230, 1212–1225. [Google Scholar] [CrossRef]
- Yoshida, M.; Shimono, Y.; Togashi, H.; Matsuzaki, K.; Miyoshi, J.; Mizoguchi, A.; Komori, T.; Takai, Y. Periderm cells covering palatal shelves have tight junctions and their desquamation reduces the polarity of palatal shelf epithelial cells in palatogenesis. Genes Cells Devoted Mol. Cell. Mech. 2012, 17, 455–472. [Google Scholar] [CrossRef]
- Cuervo, R.; Covarrubias, L. Death is the major fate of medial edge epithelial cells and the cause of basal lamina degradation during palatogenesis. Development 2004, 131, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Vaziri Sani, F.; Hallberg, K.; Harfe, B.D.; McMahon, A.P.; Linde, A.; Gritli-Linde, A. Fate-mapping of the epithelial seam during palatal fusion rules out epithelial-mesenchymal transformation. Dev. Biol. 2005, 285, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lewis, A.E.; Singh, V.; Ma, X.; Adelstein, R.; Bush, J.O. Convergence and extrusion are required for normal fusion of the mammalian secondary palate. PLoS Biol. 2015, 13, e1002122. [Google Scholar] [CrossRef] [PubMed]
- Palecek, S.P.; Loftus, J.C.; Ginsberg, M.H.; Lauffenburger, D.A.; Horwitz, A.F. Integrin-ligand binding properties govern cell migration speed through cell-substratum adhesiveness. Nature 1997, 385, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Bodo, M.; Baroni, T.; Carinci, F.; Becchetti, E.; Bellucci, C.; Pezzetti, E.; Conte, C.; Evangelisti, R.; Carinci, P. TGFbeta isoforms and decorin gene expression are modified in fibroblasts obtained from non-syndromic cleft lip and palate subjects. J. Dent. Res. 1999, 78, 1783–1790. [Google Scholar] [CrossRef] [PubMed]
- Baroni, T.; Bellucci, C.; Lilli, C.; Pezzetti, F.; Carinci, F.; Becchetti, E.; Carinci, P.; Stabellini, G.; Calvitti, M.; Lumare, E.; et al. Retinoic acid, GABA-ergic, and TGF-beta signaling systems are involved in human cleft palate fibroblast phenotype. Mol. Med. 2006, 12, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Bosi, G.; Evangelisti, R.; Valeno, V.; Carinci, F.; Pezzetti, F.; Calastrini, C.; Bodo, M.; Carinci, P. Diphenyihydantoin affects glycosaminoglycans and collagen production by human fibroblasts from cleft palate patients. J. Dent. Res. 1998, 77, 1613–1621. [Google Scholar] [CrossRef]
- Beyeler, J.; Schnyder, I.; Katsaros, C.; Chiquet, M. Accelerated wound closure in vitro by fibroblasts from a subgroup of cleft lip/palate patients: Role of transforming growth factor-alpha. PLoS ONE 2014, 9, e111752. [Google Scholar] [CrossRef]
- Liu, J.; Lamme, E.N.; Steegers-Theunissen, R.P.M.; Krapels, I.P.C.; Bian, Z.; Marres, H.; Spauwen, P.H.M.; Kuijpers-Jagtman, A.M.; Von den Hoff, J.W. Cleft Palate Cells Can Regenerate a Palatal Mucosa In Vitro. J. Dent. Res. 2008, 87, 788–792. [Google Scholar] [CrossRef]
- Jansen, B.J.; Gilissen, C.; Roelofs, H.; Schaap-Oziemlak, A.; Veltman, J.A.; Raymakers, R.A.; Jansen, J.H.; Kogler, G.; Figdor, C.G.; Torensma, R.; et al. Functional differences between mesenchymal stem cell populations are reflected by their transcriptome. Stem Cells Dev. 2010, 19, 481–490. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- de Jongh, G.J.; Zeeuwen, P.L.J.M.; Kucharekova, M.; Rolph Pfundt, R.; van der Valk, P.G.; Blokx, W.; Dogan, A.; Hiemstra, P.S.; van de Kerkhof, P.C.; Schalkwijk, J. High expression levels of keratinocyte antimicrobial proteins in psoriasis compared with atopic dermatitis. J. Investig. Dermatol. 2005, 125, 1163–1173. [Google Scholar] [CrossRef]
- Aerts, S.; Lambrechts, D.; Maity, S.; Van Loo, P.; Coessens, B.; De Smet, F.; Tranchevent, L.C.; De Moor, B.; Marynen, P.; Hassan, B.; et al. Gene prioritization through genomic data fusion. Nat. Biotechnol. 2006, 24, 537–544. [Google Scholar] [CrossRef]
- Tranchevent, L.C.; Barriot, R.; Yu, S.; Van Vooren, S.; Van Loo, P.; Coessens, B.; De Moor, B.; Aerts, S.; Moreau, Y. ENDEAVOUR update: A web resource for gene prioritization in multiple species. Nucleic Acids Res. 2008, 36, W377–W384. [Google Scholar] [CrossRef]
- DiPersio, C.M.; Zheng, R.; Kenney, J.; Van De Water, L. Integrin-mediated regulation of epidermal wound functions. Cell Tissue Res. 2016, 365, 467–482. [Google Scholar] [CrossRef]
- Grose, R.; Hutter, C.; Bloch, W.; Thorey, I.; Watt, F.M.; Fässler, R.; Brakebusch, C.; Werner, S. A crucial role of β1 integrins for keratinocyte migration in vitro and during cutaneous wound repair. Development 2002, 129, 2303–2315. [Google Scholar]
- Choma, D.P.; Milano, V.; Pumiglia, K.M.; DiPersio, C.M. Integrin alpha3beta1-dependent activation of FAK/Src regulates Rac1-mediated keratinocyte polarization on laminin-5. J. Investig. Dermatol. 2007, 127, 31–40. [Google Scholar] [CrossRef]
- Etienne-Manneville, S.; Alan Hall, A. Rho GTPases in cell biology. Nature 2002, 420, 629–635. [Google Scholar] [CrossRef]
- Kiritsi, D.; Has, C.; Bruckner-Tuderman, L. Laminin 332 in junctional epidermolysis bullosa. Cell Adhes. Migr. 2013, 7, 135–141. [Google Scholar] [CrossRef]
- Aumailley, M.; Pesch, M.; Tunggal, L.; Gaill, F.; Fässler, R. Altered synthesis of laminin 1 and absence of basement membrane component deposition in β1 integrin-deficient embryoid bodies. J. Cell Sci. 2000, 113, 259–268. [Google Scholar]
- Häkkinen, L.; Hildebrand, H.C.; Berndt, A.; Kosmehl, H.; Larjava, H. Immunolocalization of tenascin-c, a9 integrin subunit, and avb6 integrin during wound healing in human oral mucosa. J. Histochem. Cytochem. 2000, 48, 985–998. [Google Scholar] [CrossRef]
- Sweetwyne, M.T.; Murphy-Ullrich, J.E. Thrombospondin1 in tissue repair and fibrosis: TGF-beta-dependent and independent mechanisms. Matrix Biol. J. Int. Soc. Matrix Biol. 2012, 31, 178–186. [Google Scholar] [CrossRef]
- Plutoni, C.; Bazellieres, E.; Le Borgne-Rochet, M.; Comunale, F.; Brugues, A.; Seveno, M.; Planchon, D.; Thuault, S.; Morin, N.; Bodin, S.; et al. P-cadherin promotes collective cell migration via a Cdc42-mediated increase in mechanical forces. J. Cell Biol. 2016, 212, 199–217. [Google Scholar] [CrossRef]
- Bazellieres, E.; Conte, V.; Elosegui-Artola, A.; Serra-Picamal, X.; Bintanel-Morcillo, M.; Roca-Cusachs, P.; Munoz, J.J.; Sales-Pardo, M.; Guimera, R.; Trepat, X. Control of cell-cell forces and collective cell dynamics by the intercellular adhesome. Nat. Cell Biol. 2015, 17, 409–420. [Google Scholar] [CrossRef]
- Ng, M.R.; Besser, A.; Danuser, G.; Brugge, J.S. Substrate stiffness regulates cadherin-dependent collective migration through myosin-II contractility. J. Cell Biol. 2012, 199, 545–563. [Google Scholar] [CrossRef]
- Geletu, M.; Guy, S.; Arulanandam, R.; Feracci, H.; Raptis, L. Engaged for survival: From cadherin ligation to STAT3 activation. Jak-Stat 2013, 2, e27363. [Google Scholar] [CrossRef]
- Ridley, A.J.; Schwartz, M.A.; Burridge, K.; Firtel, R.A.; Ginsberg, M.H.; Borisy, G.; Parsons, J.T.; Horwitz, A.R. Cell migration: Integrating signals from front to back. Science 2003, 302, 1704–1709. [Google Scholar] [CrossRef]
- Sanz-Moreno, V.; Gaggioli, C.; Yeo, M.; Albrengues, J.; Wallberg, F.; Viros, A.; Hooper, S.; Mitter, R.; Feral, C.C.; Cook, M.; et al. ROCK and JAK1 signaling cooperate to control actomyosin contractility in tumor cells and stroma. Cancer Cell 2011, 20, 229–245. [Google Scholar] [CrossRef]
- Ikeda, W.; Nakanishi, H.; Miyoshi, J.; Mandai, K.; Ishizaki, H.; Tanaka, M.; Togawa, A.; Takahashi, K.; Nishioka, H.; Yoshida, H.; et al. Afadin: A key molecule essential for structural organization of cell–cell junctions of polarized epithelia during embryogenesis. J. Cell Biol. 1999, 146, 1117–1131. [Google Scholar] [CrossRef]
- Suzuki, K.; Hu, D.; Bustos, T.; Zlotogora, J.; Richieri-Costa, A.; Helms, J.A.; Spritz, R.A. Mutations of PVRL1, encoding a cell-cell adhesion molecule/herpesvirus receptor, in cleft lip/palateectodermal dysplasia. Nat. Genet. 2000, 25, 427–430. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GENE | Non-OFC | OFC | Fold Change | p | ||
|---|---|---|---|---|---|---|
| mean | SD | mean | SD | |||
| THBS1* | 601.2 | 212.2 | 284.8 | 74.2 | 2.111 | 0.0057 |
| ARHGAP26 | 289.5 | 59.0 | 156.5 | 45.3 | 1.850 | 0.0059 |
| TNC* | 3192 | 593.3 | 1797 | 234.0 | 1.776 | 0.0006 |
| FBLIM1 | 384.9 | 28.0 | 257.2 | 25.8 | 1.496 | 0.0001 |
| CDH3* | 5647 | 476.3 | 3979 | 413.8 | 1.419 | 0.0004 |
| DST | 228.8 | 16.6 | 163.6 | 16.5 | 1.398 | 0.0003 |
| ITGA3 | 696.1 | 29.8 | 500.5 | 68.4 | 1.391 | 0.0007 |
| EGFR | 480.9 | 32.6 | 347.1 | 31.2 | 1.385 | 0.0002 |
| ITGB4 | 2554 | 106.0 | 1847 | 208.9 | 1.383 | 0.0003 |
| DOCK5 | 88.24 | 9.1 | 64.72 | 4.5 | 1.363 | 0.0006 |
| LAMA3* | 5886 | 354.0 | 4324 | 817.0 | 1.361 | 0.0045 |
| MLLT4 | 827.1 | 72.9 | 613 | 51.2 | 1.349 | 0.0008 |
| SMAD7 | 126.4 | 10.1 | 95.05 | 9.3 | 1.330 | 0.0009 |
| LPP | 111.9 | 12.1 | 85.6 | 3.3 | 1.307 | 0.0007 |
| ITGB1* | 5418 | 215.1 | 4338 | 213.4 | 1.249 | 0.0001 |
| JAK1* | 196.8 | 8.5 | 157.7 | 13.4 | 1.248 | 0.0007 |
| PARVA | 419.8 | 42.9 | 342.3 | 11.5 | 1.226 | 0.0026 |
| PXN | 650.7 | 25.4 | 537.8 | 53.4 | 1.210 | 0.0031 |
| MICAL2 | 42.61 | 5.1 | 35.71 | 3.4 | 1.193 | 0.0330 |
| LAMC1 | 1263 | 42.0 | 1066 | 84.2 | 1.185 | 0.0026 |
| TNS3 | 101.9 | 31.7 | 98.31 | 44.8 | 1.037 | 0.7275 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mammadova, A.; Carels, C.E.L.; Zhou, J.; Gilissen, C.; Helmich, M.P.A.C.; Bian, Z.; Zhou, H.; Von den Hoff, J.W. Deregulated Adhesion Program in Palatal Keratinocytes of Orofacial Cleft Patients. Genes 2019, 10, 836. https://doi.org/10.3390/genes10110836
Mammadova A, Carels CEL, Zhou J, Gilissen C, Helmich MPAC, Bian Z, Zhou H, Von den Hoff JW. Deregulated Adhesion Program in Palatal Keratinocytes of Orofacial Cleft Patients. Genes. 2019; 10(11):836. https://doi.org/10.3390/genes10110836
Chicago/Turabian StyleMammadova, Aysel, Carine E.L. Carels, Jie Zhou, Christian Gilissen, Maria P.A.C. Helmich, Zhuan Bian, Huiqing Zhou, and Johannes W. Von den Hoff. 2019. "Deregulated Adhesion Program in Palatal Keratinocytes of Orofacial Cleft Patients" Genes 10, no. 11: 836. https://doi.org/10.3390/genes10110836
APA StyleMammadova, A., Carels, C. E. L., Zhou, J., Gilissen, C., Helmich, M. P. A. C., Bian, Z., Zhou, H., & Von den Hoff, J. W. (2019). Deregulated Adhesion Program in Palatal Keratinocytes of Orofacial Cleft Patients. Genes, 10(11), 836. https://doi.org/10.3390/genes10110836